

Abstract
Background
The C9orf72 hexanucleotide expansion is one of the latest discovered repeat expansion disorders related to neurodegeneration. Its association with the FTD/ALS spectrum disorders is well established, and it is considered to be one of the leading related genes. It has also been reported as a possible cause of several other phenotypes, including parkinsonism and other movement disorders. Its significance, though outside the FTD/ALS spectrum, is not well defined.
Methods
A comprehensive search of the literature was performed. All relevant papers, including reviews and case series/reports on movement disorder phenotypes reported with the C9orf72 repeat expansion, were reviewed. Data on frequency, natural history, phenotype, genetics, and possible underlying mechanisms were assessed.
Results and Discussion
In a number of studies, C9orf72 accounts for a small fraction of typical PD. Atypical parkinsonian syndromes, including CBS, PSP, and MSA have also been reported. Features that increase the probability of positive testing include early cognitive and/or behavioral symptoms, positive family history of ALS or FTD, and the presence of UMN and LMN signs. Furthermore, several studies conclude that C9orf72 is the most common cause of HD‐phenocopies. Interestingly, many cases with the parkinsonian phenotype that bear an intermediate range of repeats are also reported, questioning the direct causal role of C9orf72 and suggesting the possibility of being a susceptibility factor, while the presence of the expansion in normal controls questions its clinical significance. Finally, studies on pathology reveal a distinctive broad range of C9orf72‐related neurodegeneration that could explain the wide phenotypic variation.
Keywords: C9orf72, movement disorders, parkinsonism, Parkinson's disease, repeat expansion disorders
Background
Discovery and Clinical Significance of C9orf72
The chromosome 9 open read frame 72 (C9orf72) hexanucleotide repeat expansion was first discovered and reported simultaneously in 2011 by two separate groups1, 2 after several families with multiple‐affected members with amyotrophic lateral sclerosis (ALS) and/or frontotemporal dementia (FTD) had been previously linked to chromosome 9p21. ALS and FTD, or the related pathologically defined frontotemporal lobe degeneration (FTLD), have been long considered part of the same spectrum of neurodegenerative disorders, sharing several common clinicopathological features, and are commonly termed as the FTD/A LS (or FTLD/ALS) spectrum.3, 4, 5 The significance of C9orf72 within the FTD/ALS spectrum has been well established.6, 7 It is the most common cause of familial ALS and FTD and one of the major genetic causes of apparently sporadic cases. Reported frequencies seem to be higher in populations of European origin. A large Pan‐European study on the association of C9orf72 to FTD reported frequencies of 10% in all FTD cases, and 18.5% and 6.2% in familial and sporadic patients, respectively.8 A recent meta‐analysis on the genetic epidemiology of ALS estimated that 33.7% of familial cases and 5.1% of sporadic cases in European populations are related to C9orf72. 9
There has been an increasing number of reports that the C9orf72 expansion could be responsible for a range of other phenotypes, including neuropsychiatric and dementia presentations, movement disorders, and other complex neurodegenerative syndromes.10, 11 The early observation that several C9orf72‐positive patients with FTD/ALS present additional symptoms, more often parkinsonism and psychotic features,12, 13, 14 led to the assumption that C9orf72 repeat expansion could be responsible for a wider and more heterogeneous range of clinical syndromes. Therefore, a number of studies have examined the possible association of the expansion to different phenotypes.
Such phenotypes include psychiatric manifestations and dementia syndromes other than FTD. An increased prevalence of psychotic symptoms in FTD patients with C9orf72 expansion has been reported. However, a recent comprehensive review concluded that the prevalence of C9orf in primary psychiatric presentations is estimated to be around 0.1%15 and not larger than its prevalence in healthy controls. A possible association with Alzheimer's disease (AD) has also been examined. The results indicate a small prevalence of <1%, even in studies where a positive association was detected in clinically diagnosed or pathologically confirmed cases.16, 17, 18 Additionally, studies on pathology indicate the presence of TDP‐43‐related pathology in these patients, raising the possibility of misdiagnosing cases of amnesic FTD for AD.
For quite some time, the clinical relevance of C9orf in movement disorders has been debated. Initial studies reported the increased prevalence of parkinsonian symptoms in FTD/ALS patients who carry the expansion.12, 19, 20 An increased incidence of parkinsonism and Parkinson's disease (PD) in relatives of ALS/FTD patients who carry the C9orf72 expansion has also been reported.12, 20, 21 However, the connection among the expansion and parkinsonism or other movement disorders, as well as its clinical significance beyond the FTD/ALS spectrum, have not been clarified in the literature to date.
Basic Genetics and Molecular Mechanisms
The repeat expansion refers to a GGGGCC hexanucleotide in the non‐coding region of the first exon of C9orf72.1, 2 The normal repeat size seems to be variable, and 90% of the European population has a small number of two to 10 repeat units.1, 22 Therefore, normal size of <20 to 23 repeats was originally defined, while the pathogenic range was considered to be of >30 repeat units.1, 2, 23 The estimated expansion range in most confirmed cases is much larger and usually falls within the range of 700 to 4,400 repeat units.2, 11 Large expansions, defined as >30 to 32 repeats, have also been detected in the 0.15 to 0.6% of the controls in some large case–control studies of ALS and FTD,11, 20, 24 but their clinical significance has been debated. Recently, smaller expansions of 45 to 80 units have been confirmed as pathogenic in some cases of FTD/ALS.25
The expansion is associated, with a risk haplotype that has increased frequency in some European populations, supporting the theory of a common founder effect.6, 24, 26 Finland has the highest prevalence of C9orf72 in sporadic cases of ALS and FTD, 21.1% and 18.8%, respectively,1 and is considered to be the origin of this risk haplotype. Smaller frequencies (4–8%) are reported in populations of African and Hispanic origin,24, 27 while the C9orf72 repeat expansion is considered quite rare in Asians. Population studies in various Asian populations calculated an estimated prevalence of C9orf72 in sporadic ALS at around 0 to 2%.28
The size of expansion could be inversely correlated with the age of disease onset, although earlier reports did not support such a correlation.29 A recent study concluded that patients who carry smaller expansions (45 to 80 units) seem to have almost a 10‐year‐later age of onset.25 Incomplete penetrance and anticipation are also observed.25, 30 This disease anticipation results in earlier age of onset, but not in more severe phenotypes. Intergenerational variability of the length of the expansion could be implicated, although exact effect and clinical significance have not been completely clarified.11, 31 Interestingly, anticipation and intergenerational variability do not seem to apply for smaller expansions.8, 25
The significant degree of clinical heterogeneity that can occur even within the same family is commonly reported.32 This observation doesn't seem to be completely explained by the genetic variability of the expansion. A combination of additional genetic, epigenetic, and environmental factors is suggested to be implicated, though not completely understood.31, 33 An interesting modifier that seems to play an important role is the reported hypermethylation of the promoter region and the expansion itself.25, 34 This seems to affect larger repeats of >90 units, while smaller expansion is completely unmethylated,34 or much less methylated.25 Somatic instability of the repeat expansion has also been observed and could contribute to the phenotypic variability.35, 36
The molecular basis of the C9orf72 neurodegeneration has not been completely clarified, although a combination of gain‐ and loss‐of‐function mechanisms is considered to cause disease.37 Studies of the related pathology have revealed a characteristic pattern of cellular changes that seems to be defined by the presence of cytoplasmic ubiquitin/p62‐positive, TDP‐43‐negative inclusions38, 39 that make C9orf72‐related neurodegeneration quite distinctive, even within the FTD/ALS spectrum.
Methods
A comprehensive search of the literature was performed in PubMed, up to the June 10, 2018. We used a combination of key words such as “C9orf72” and “parkinsonism”, “movement disorders”, “Parkinson's disease”, “progressive supranuclear palsy”, “multiple system atrophy”, “corticobasal syndrome”, “dementia with Lewy bodies”, “chorea”, “dystonia”, and “ataxia” to identify possibly relevant papers. Additionally, we performed a manual search through the citations of the identified papers to detect further related studies. Only papers written in English were considered.
Our target was to include all relevant identified papers, encompassing reviews, case series, and case reports that refer to parkinsonian and other movement disorder phenotypes in relation to the C9orf72 expansion, either in the commonly accepted pathogenic range of >30 repeats, or the intermediate range as defined by the authors.
In the following paragraphs, we will try to summarize and critically acclaim the reviewed papers. We focused on papers that reported data on the frequency of C9orf72 in typical and atypical parkinsonism and other movement disorders, the natural history of the disease, considerations on inheritance pattern, possible correlations of the phenotypic variability to the size of expansion, and finally, data on C9orf72‐related pathology and the underlying mechanisms in relation to the different phenotypes.
Results and Discussion
Prevalence of C9orf72 in Parkinson's Disease
We identified several papers that studied the prevalence of C9orf72 expansion in typical PD. We noticed that the studies used different methods and a variable threshold of copy repeat units to identify normal and positive subjects. In most cases, positives are identified by either determining a pathogenic threshold of >30 or > 60 copy repeats, or by the classic sawtooth pattern that is observed in the repeat‐primed PCR assays, as reported in the original research studies.1, 2, 24 It is worth mentioning that this method is not considered reliable to determine the size of larger expansion of >60 repeat units and require the concomitant use of complex Southern blot techniques to determine the number of repeats. The characteristics and the findings of the studies are summarized in Table 1.
Table 1.
Characteristics and results of reviewed cohorts that included clinically diagnosed patients with Parkinson's disease
| Study | Country/ethnicity | Population description | Subjects (n=) | Mean AAO ± SD [range], in years | Diagnostic criteria | Definition of pathogenic expansions | Number of positive cases (repeat count) | Prevalence | Family history of positive cases | Number of intermediate expansion carriers (repeat count); features & history |
|---|---|---|---|---|---|---|---|---|---|---|
| Xi et al., 2012 | United States & Europe (Caucasians) | Sporadic & familial PD | 289 (incl. 116 familial) | 52.6 ± 13 | n.m. | >30 repeats | 2 (RC: 32, 39) | 0.7% (0.9% in familial PD) | 1/2 positive for PD, 1/2 negative | 4 (RC: 20–29); 2/4 early severe dementia |
| Lesage et al., 2013 | France | Sporadic & familial PD | 1225 | 48.3 ± 12.5 [6–86] | UKPDBB | ≥60 repeats | 3 | 0.2% | 3/3 positive family history of complex phenotypes | 0 |
| Akimoto et al., 2013 | Sweden | Sporadic PD | 135 | 70.5 ± 9.5 [41–90] | UKPDBB | Sawtooth pattern | 0 | 0% | n.a. | 2.2%, no further details given |
| Majounie et al., 2015 | United States (Caucasians) | Sporadic & familial PD | 781 (incl. 254 familial) | 60 [12–91] | n.m. | Sawtooth pattern, >30 repeats | 0 | 0% | n.a. | 4 (RC:21, 23, 24, 38) |
| Dejesus‐Hernandez et al., 2013 | United States (Caucasians) | PD | 676 | 69 ± 11 | n.m. | Sawtooth pattern | 0 | 0% | n.a. | n.m. |
| Nuytemans et al., 2013 | United States (Caucasians) | PD | 889 | 53.6 [10–85] | n.m. | >30 repeats | 0 | 0% | n.a. | 13 (RC: 21–30+); typical dopa‐responsive PD |
| Harms et al., 2013 | United States (99.8% Caucasians) | PD | 478 | 61.3 ± 10.6 | UKPDBB | Sawtooth pattern, >30 repeats | 0 | 0% | n.a. | n.m. |
| Daoud et al., 2013 | Canada (French‐Canadians) | Sporadic PD | 285 | 57.5 [24–83] | Ward & Gibb | Sawtooth pattern, >30 repeats | 0 | 0% | n.a. | 1 (RC: 24) |
| Alavi et al., 2017 | Iran | Sporadic & familial PD | 186 | 49.9 ± 13.2 [7–85] | n.m. | Sawtooth pattern, >20 repeats | 0 | 0% | n.a. | n.m. |
| Yeh et al., 2013 | Taiwan | Sporadic PDD | 71 | 60.1 ± 9.3 | n.m. | Sawtooth pattern | 0 | 0% | n.a. | n.m. |
| Chen et al., 2016 | China | Sporadic PD | 619 | 58.1 ± 10.9 | UKPDBB | >30 repeats | 0 | 0% | n.a. | n.m. |
| Jiao et al., 2013 | China | Sporadic PD | 911 | 55.1 ± 11.7 | UKPDBB | >30 repeats | 0 | 0% | n.a. | 5 (RC:18, 19, 22, 23, 27) |
| Lin et al., 2014 | Taiwan | Sporadic EOPD | 201 | 42.5 ± 5.2 | UKPDBB | >30 repeats | 0 | 0% | n.a. | 1 (RC: 25); typical dopa‐responsive PD |
| Lin et al., 2014 | Taiwan | Familial PD | 109 | 66.5 ± 12.2 | UKPDBB | >30 repeats | 0 | 0% | n.a. | n.m. |
Abbreviations: PD, Parkinson's disease; PDD, Parkinson's disease with dementia; EOPD, early onset Parkinson's disease; UKPDBB, UK Parkinson's disease brain bank; AAO, age at onset; SD, standard deviation; RC, repeat count; n.m., not mentioned; n.a., not applicable.
A couple of studies that have been carried in mostly Caucasian populations from Europe and North America reported small frequencies of C9orf72 in PD cohorts, smaller than 1%.40, 41 All other studies in Caucasian populations, including European and North American patients, did not report any positive C9orf72 expansion carriers.22, 42, 43, 44, 45, 46, 47 Finally, many studies that have been carried out in populations of Asian origin were also negative for the presence of expansion carriers,48, 49, 50, 51, 52 a fact that is coherent with the small frequency of the expansion in Asian populations.
A single worldwide study and meta‐analysis of typical PD populations that included more than 7,000 patients from 12 different counties in four continents was performed by the Genetic Epidemiology of Parkinson's disease (GEo‐PD) consortium.53 This study reported a worldwide prevalence of 0.06% (n = 4/7,232) using a cutoff of >60 repeat units to define positives. All positives were of European origin; one from Germany and three from France. The French cases have been previously reported elsewhere.41 They all had a positive family history of ALS, dementia, or atypical parkinsonism. Interestingly, the same study reports one control of Asian origin harboring a positive >60 repeats expansion, bringing the relevant frequency of the expansion in the controls to 0.02% (n = 1/5,478). A subsequent meta‐analysis of the data failed to find statistically significant differences in the attributable risk for disease among the two groups of PD patients and controls, using a cutoff of ≥17 repeat units (P = 0.03, with P ≤ 0.002 being the limit for statistical significance after Bonferroni correction for the number of comparisons).
Some of the previous studies reported cases that harbor intermediate repeat alleles, usually defined in the range of 20 to 30 repeat units.24, 40, 44, 46, 47, 52 A case with 38 repeats and a positive family history of PD was also reported, although the expansion did not segregate in the family and therefore was not considered pathogenic.24 One of the studies reported that 2% of the studied PD cases were carrying intermediate expansions of 21 to >30 copy repeat units.44 Subsequent statistical analysis suggested that intermediate repeats could be a risk factor for PD with an odds ratio of 9.6 (95% CI: 1.32, 421). The large reported confidence intervals, due to the small number of cases, seem to limit the clinical significance of this finding. A study of the Swedish population, reported a frequency of intermediate alleles with >20 repeat units, but no typical sawtooth pattern in 2.2% of the PD patients and 2.9% of the healthy controls. The authors did not investigate this finding further.22 Finally, a study of the Chinese population reported an association of intermediate alleles to the risk of PD, with an odds ratio of 1.37 (95% CI: 1.05, 1.79), but the authors used a much smaller cutoff of ≥7 repeats.51
The global study by GEo‐PD also investigated the possible contribution of smaller expansions to disease susceptibility. They reported a slight increment of the risk with increasing number of repeats, but the calculated effect size did not reach the level of statistical significance for expansions larger than nine to 10 repeat units (P = 0.009, with P ≤ 0.002 being the limit for statistical significance after Bonferroni correction).
Notably, almost all previously mentioned studies included patients who had been diagnosed as PD based solely on clinical criteria. Only two papers that have studied the prevalence of C9orf72 expansion in cohorts of pathologically confirmed PD cases were identified.54, 55 These studies report the findings of 377 and 488 respectively autopsied brains from sporadic PD patients. Only the first study reported a prevalence of 0.2% among all studied cases (n = 1/377), or 0.3% among the cases that presented Lewy body pathology (n = 1/338).54 This single positive case had a positive family history of ALS. Further details of the pathological findings will be discussed later.
Prevalence of C9orf72 in Atypical Parkinsonism
The data on the prevalence of C9orf72 in atypical parkinsonian syndromes are much sparser and presented mostly as case reports and small case series. We identified a few cases reported either separately or as part of larger cohorts. The phenotypes include corticobasal syndrome (CBS),41, 56, 57, 58 progressive supranuclear palsy (PSP),41, 58, 59, 60, 61 multiple system atrophy (MSA),57, 62 and dementia with Lewy bodies (DLB)63 The characteristics and results of studies on smaller or larger cohorts of patients with atypical parkinsonism are presented in Table 2.
Table 2.
Characteristics and results of reviewed cohorts that included clinically diagnosed patients with atypical parkinsonism
| Study | Country/ethnicity | Population description | Subjects (n=) | Mean AAO ± SD [range], in years | Definition of pathogenic expansion | Number of positive cases | Prevalence | Family history of positive cases | Additional symptoms of positive cases | Number of intermediate expansion carriers (repeat count); features & history |
|---|---|---|---|---|---|---|---|---|---|---|
| Anor et al., 2015 | Canada | CBS | 39 | 58.9 ± 13.2 | >30 repeats | 1 | 2.6% | Father with possible bvFTD | n.m. | n.m. |
| Lesage et al., 2013 | France | CBS | 21 | 64.2 ± 8.8 [50–80] | ≥60 repeats | 1 | 4.8% | 1/1 positive for dementia | 1/1 pyramidal signs | 0 |
| Schottlaender et al., 2014 | United Kingdom | CBS | 37 | n.m. | >30 repeats | 3 | 8% | 2/3 positive for dementia | 1/3 motor neuron signs, 2/3 dysphagia and dysarthria | 0 |
| Galimberti et al., 2013 | Italy | CBS | 21 | n.m. | >30 repeats | 0 | 0% | n.a. | n.a. | n.m. |
| Yeh et al., 2013 | Taiwan | CBS | 13 | 65.5 ± 12.6 | Sawtooth pattern | 0 | 0% | n.a. | n.a. | n.m. |
| Origone et al., 2013 | Italy | CBS | 27 | 64.3 ± 6.9 | >30 repeats | 0 | 0% | n.a. | n.a. | n.m. |
| Le Ber et al., 2013 | France | CBS | 14 | 56.8 ± 9.9 [41–71] | ≥60 repeats | 0 | 0% | n.a. | n.a. | n.m. |
| Lesage et al., 2013 | France | PSP | 123 | 64.1 ± 8.2 [40–79] | ≥60 repeats | 1 | 0.8% | 1/1 positive for dementia, PSP, and parkinsonism | n.m. | 2 (RC: 20, 26); typical PSP |
| Schottlaender et al., 2014 | United Kingdom | PSP | 22 | n.m. | >30 repeats | 0 | 0% | n.a. | n.a. | 1 (RC: 27); typical PSP, positive family history for dementia and PD |
| Galimberti et al., 2013 | Italy | PSP | 31 | n.m. | >30 repeats | 0 | 0% | n.a. | n.a. | n.m. |
| Yeh et al., 2013 | Taiwan | PSP | 35 | 61.1 ± 7.6 | Sawtooth pattern | 0 | 0% | n.a. | n.a. | n.m. |
| Origone et al., 2013 | Italy | PSP | 12 | 58.8 ± 7.0 | >30 repeats | 1 | 8.3% | Negative | 1/1 muscular wasting | n.m. |
| Le Ber et al., 2013 | France | PSP | 17 | 54.2 ± 10.2 [41–71] | ≥60 repeats | 1 | 5.9% | 1/1 positive | n.m. | n.m. |
| Lesage et al., 2013 | France | MSA‐P | 25 | 56.3 ± 13.2 [35–81] | ≥60 repeats | 0 | 0% | n.a. | n.a. | 0 |
| Chen et al., 2016 | China | MSA | 381 | 56.7 ± 9.6 | >30 repeats | 0 | 0% | n.a. | n.a. | n.m. |
| Sun et al., 2015 | China | MSA | 141 (93 MSA‐c, 48 MSA‐p) | 53.7 ± 7.3 | >30 repeats | 0 | 0% | n.a. | n.a. | 60 with RC ≥7 |
| Hsiao et al., 2014 | Taiwan | MSA‐C | 331 | 55.5 ± 8.1 | >30 repeats | 0 | 0% | n.a. | n.a. | n.m. |
| Snowden et al., 2012 | United Kingdom | DLB | 102 | 67 ± 8 | >30 repeats | 2 | 2% | Negative | 2/2 psychotic symptoms | n.m. |
| Lesage et al., 2013 | France | DLB | 43 | 63.6 ± 11.9 [34–83] | ≥60 repeats | 0 | 0% | n.a. | n.a. | 0 |
| Cannas et al., 2015 | Italy (Sardinia) | Sporadic PD complicated by dementia or psychosis | 19 | n.m. | >30 repeats | 0 | 0% | n.a. | n.a. | 1 (RC: 22); typical PD on onset complicated by psychosis, no family history |
| Cannas et al., 2015 | Italy (Sardinia) | Atypical parkinsonism (unclassified) | 60 | n.m. | >30 repeats | 0 | 0% | n.a. | n.a. | 0 |
| Cannas et al., 2015 | Italy (Sardinia) | NCAP | n.m. | >30 repeats | 0 | 0% | n.a. | n.a. | 3 (RC: 20, 23, 28); 2/3 severe cognitive impairment, 1/3 psychosis, 2/3 positive family history for parkinsonism with dementia |
Abbreviations: AAO, age at onset; bvFTD, behavioral variant frontotemporal dementia; CBS, corticobasal syndrome; MSA, multiple system atrophy; MSA‐C, MSA cerebellar type; MSA‐P, MSA parkinsonian type; n.a., not applicable; NCAP, non‐classical atypical parkinsonism; n.m., not mentioned; PD, Parkinson's disease; PSP, progressive supranuclear palsy; RC, repeat count; SD, standard deviation.
When patients were diagnosed based solely on clinical criteria, the results that were reported in these cohorts seem to vary significantly. Some of the studies reported positive findings for the presence of large expansions of C9orf72 hexanucleotide. These include a prevalence of 2% in a cohort of 102 DLB patients,63 and some positive cases of CBS and PSP in small cohorts of patients.41, 58, 59, 60 The rest of the studies did not report the presence of large C9orf72 expansions in clinically diagnosed patients.47, 49, 50, 64, 65, 66 Finally, a study that failed to identify large expansions in a cohort of patients with atypical parkinsonism, reported a significant frequency of intermediate sizes of 20 to 29 repeats (n = 4/92).47 Occasional cases of intermediate repeats have been reported in other papers as well.41, 58 All reported cases that carried large or intermediate expansions were of European origin. A study of the Chinese population that used a cutoff of ≥7 repeat units to define intermediate expansions reported a frequency of 42.6% in a cohort of MSA patients, but this was not statistically significant compared to controls that were reported to have a higher frequency of 50.5%.65 No further details for the exact repeat counts were given.
An important observation was that all the reported cohorts, which included pathologically confirmed patients, were uniformly negative for the detection of any C9orf72 positive cases. These studies include a cohort of MSA, CBD, and PSP patients (n = 96, 18, and 177, respectively),58 a cohort of 100 MSA patients,67 and two cohorts of 111 and 53 DLB patients, respectively.68, 69 The last and most recent study, examined a large international cohort of 1,398 neuropathologically confirmed DLB cases and a smaller cohort of 126 clinically diagnosed patients.70 Among the pathologically diagnosed patients, three cases carried marginally expanded alleles of 32 to 33 repeat units that were not considered pathogenic. The same study reported two cases of expanded alleles in the clinically diagnosed patients who had been previously reported.63
Only one pathologically diagnosed case of atypical parkinsonism has been reported in the literature so far. This refers to a CBD patient that was previously clinically diagnosed as FTD and had a strong family history of Huntington's disease (HD).71 In the same study, a clinically diagnosed CBS patient was classified as FTLD‐tau CBD after studying the pathology. A recent study that identified three clinically diagnosed DLB patients as C9orf72 carriers eventually reclassified the patients as FTLD based on pathological criteria.72
Prevalence of C9orf72 in Other Movement Disorders
In a few studies, C9orf72 was confirmed as an important genetic cause of HD‐phenocopies and reported as one of the leading genetic causes of such cases in populations of European origin. These studies were conducted on patients who presented with an HD‐like phenotype and tested negative for pathogenic expansions in the huntingtin gene. Reported frequencies range from 1.75% to 5%, and all studies included populations of European origin: British,11, 73 Serbian,74 Greek,75 and Portuguese.76 A study of the French population did not identify any C9orf72 positives.77 The results are summarized in Table 3.
Table 3.
Characteristics and results of reviewed cohorts that included patients diagnosed as Huntington's disease phenocopies
| Study | Country/ethnicity | Subjects (n=) | Mean AAO ± SD [range], in years | Number of positive cases | Prevalence | Family history of positive cases | Number of intermediate expansion carriers (repeat count); features and history |
|---|---|---|---|---|---|---|---|
| Beck et al., 2013 | United Kingdom | 421 | n.m. | 7 | 1.7% | n.m. | n.m. |
| Hensman Moss et al., 2014 | United Kingdom | 514 | 48.8 ± 19.3 | 10 | 1.95% | 7/10 positive | n.m. |
| Kostic et al., 2014 | Serbia | 39 | 47.6 ± 14.7 [24–73] | 1 | 2.6% | Mother with dementia | n.m. |
| Koutsis et al., 2015 | Greece | 40 | 48.5 ± 17.1 [8–83] | 2 | 5% | 1/2 positive | n.m. |
| Martins et al., 2018 | Portugal | 20 | n.m. | 1 | 5% | 1/1 positive for psychiatric disorders | 1 (RC: 27), positive family history for dementia |
| Mariani et al., 2016 | France | 23 | n.m. | 0 | 0% | n.a. | n.m. |
Abbreviations: AAO, age at onset; n.a. = not applicable; n.m., not mentioned; RC, repeat count; SD, standard deviation.
In two studies43, 51 on the presence of C9orf72 repeat expansion in cohorts of patients with essential tremor (ET) and restless legs syndrome (RLS), only one positive case was reported; a 61‐year‐old male who presented symptoms reminiscent of RLS, but soon developed cognitive and behavioral symptoms and was eventually diagnosed as FTD.43 The clinical significance of these studies is limited by the relatively small size of the cohorts.
The relation of C9orf72 expansion to various cases of ataxia has been investigated in a few studies. Three studies in cohorts of patients with sporadic cerebellar ataxia who previously tested negative for other possible genetic causes were reviewed. The first reported one positive case among a cohort of 209 patients (prevalence of 0.5%).78 The second reported no positives in a cohort of 98 Chinese patients diagnosed as idiopathic spinocerebellar ataxia (SCA).66 The third did not identify any positives in a cohort of 440 German patients.79
A study on genetically confirmed SCA patients identified seven cases with concomitant C9orf72 hexanucleotide expansions within the pathogenic range, defined as >30 repeat units, in a cohort of 277 patients diagnosed with pathogenic CAG expansions in SCA1, SCA2, SCA3, and SCA6.80 All were reported to have various degrees of motor neuron signs, but none had ALS. The same study reported that 40% of the patients had intermediate expansions in C9orf72 and studied their phenotype. The authors suggested that intermediate expansions might affect the progression of non‐motor symptoms, including depression, but not the progression of ataxia in SCA patients.
An interesting association has also been reported in C9orf72‐positive patients who carried concomitant intermediate ATXN2 alleles in the range of 27 to 33 repeats. These patients were reported to have an increased risk to present with an ALS phenotype, suggesting that ATXN2 could be a modifier of C9orf72 expansion carriers towards ALS. The frequency of patients who carried larger than normal ATXN2 alleles was small, but statistically significant compared to controls (1.5% vs. 0%, P = 0.029).81 A family with the simultaneous presence of pathogenic expansions both in C9orf72 and ATXN2 was previously reported to present with a complex phenotype that included ataxia, dementia, and parkinsonism, but not ALS.82
Additional ataxia case reports include a patient with pure cerebellar ataxia and a large C9orf72 expansion,83 and a patient with progressive cerebellar ataxia and psychiatric symptoms, who was reported to carry an intermediate expansion of 21 repeat units.84
Natural History and Symptoms
Patients with C9orf72 that manifest parkinsonian symptoms were reported to have a typical presentation that is commonly asymmetric onset akinetic‐rigid syndrome with prominent bradykinesia and little or no tremor.12, 14, 42, 61 Positive family history was commonly reported and included FTD/ALS, parkinsonism, and other than FTD types of dementia.41, 53, 61
A systematic review of C9orf72 positive patients with parkinsonism, including PD and atypical parkinsonism, reported an average age of onset of disease of 52 years and early onset of parkinsonian signs within the first year of the disease in most cases.61 In the same review, the most commonly reported accompanying symptoms included cognitive and behavioral dysfunction (85% and 56%, respectively), followed by psychiatric symptoms (31%), while upper motor neuron (UMN) and lower motor neuron (LMN) signs were also not uncommon (60% and 36%, respectively).61 In the global meta‐analysis by GEoPD, most PD patients who carried pathogenic expansions (n = 3/4) were reported to show signs of cognitive decline within one to eight years after disease onset.53 Another patient with no significant cognitive deterioration even >10 years after disease onset and early dopa‐responsiveness was also reported, as described elsewhere.41
Dopa‐responsiveness was also reported in cases of PD that were reported to carry intermediate expansions,44, 52 but not in most patients with larger expansions that presented parkinsonism as part of more complex phenotypes that included symptoms suggestive of FTD/ALS.12, 85
In most instances, authors were very cautious to suggest genetic testing for C9orf72, especially for typical PD cases. Overall, the most commonly reported features that increased the possibility of positive C9orf72 testing in patients with typical or atypical parkinsonism included early cognitive and/or behavioral symptoms, positive family history of ALS or FTD or other neurodegenerative syndromes, and the presence of UMN and LMN signs.41, 53, 61
Imaging
A few reports presented radiographic features of patients with parkinsonism that carry the C9orf72 expansion. A systematic review concluded that cerebral atrophy is the most commonly reported imaging finding (90%), including frontotemporal atrophy in 53% of the cases, temporal atrophy (37%), parietal atrophy (13%), and generalized cerebral atrophy (30%).61
In some cases, brain MRI findings seemed to correlate with the clinical phenotype. A PSP‐like patient was reported to show significant midbrain atrophy and mild cerebellar atrophy.61 Another patient, diagnosed as CBS, showed asymmetrical frontotemporal atrophy.56 And finally, a patient with pure cerebellar ataxia presented significant cerebellar atrophy, affecting mostly the vermis.83
Genetics
A quite interesting issue is the possible correlation between the phenotypic variation to genetic factors, including the size of the C9orf72 hexanucleotide expansion. Most studies agreed on the observation that there is no correlation between repeat length and risk for PD or the age of onset,42, 43, 45 although a possible association of the size of expansion to the age of onset has also been suggested.53 There are indications that intermediate repeat size >10 to 20 units may have a role in risk for PD, although the reported effect size seems to be small or questionable.44, 51, 53
Due to the small number of reported cases, there has not been enough data to support age‐related penetrance or disease anticipation, as suggested in similar studies with FTD/ALS patients.
The possibility that the C9orf72 hexanucleotide expansion could interact with other repeat expansion disorders, including the CAG expansions disorders of several SCA genes and HD, possibly altering the phenotypic expression of either condition, has been suggested in some papers.74, 80, 81, 86 This finding could mean that C9orf72 expansion, either in the pathogenic or in the intermediate size of repeat units, could act as a genetic modifier of other repeat expansion disorders and vice versa.
Pathology and Underlying Mechanisms of Disease
Studies have been researching the underlying pathology and possible correlations to the phenotypic variations. Most studies of pathologically confirmed cases were negative for the presence of C9orf72 expansion in PD,55 MSA,58, 67 and DLB cases.68, 69, 70 A single reported positive PD patient,54 apart from Lewy body pathology, exhibited signs of FTLD and C9orf72‐related pathology with p62 positive inclusions. Coexistence of Lewy body and p62 pathology has been reported elsewhere, in cases initially diagnosed as DLB, although it is unknown in what degree the two pathologies contributed to the clinical phenotype.72
Notably, it was reported that patients who carry the expansion and show parkinsonian symptoms display more features of C9orf72 pathology instead of typical Lewy body pathology.54, 87 This finding could suggest a distinct mechanism of C9orf72‐related parkinsonism, where the C9orf72‐related pathology could affect the substantia nigra and cause parkinsonian symptoms. The presence of a significant degree of pathology found in the striatum and substantia nigra has been previously reported in ALS/FTD patients.88
Clinical Significance of C9orf72 in Parkinsonism and Movement Disorders
Studies that have confirmed the presence of large expansions in patients diagnosed as PD and atypical parkinsonism have mostly used clinical criteria for diagnosing the patients. Reported frequencies are very small for PD and variable for atypical parkinsonism, as discussed earlier. Studies that have used pathological criteria to define the diagnosis have failed to reproduce findings of studies based on clinical criteria and reclassified many patients.54, 58, 67, 71, 72
The observation that some of the patients present early cognitive symptoms raises the question that these patients may actually belong to the FTD/ALS spectrum and have accompanying parkinsonian signs.53 A study identified a high prevalence of the C9orf72 expansion (> 20%) in a cohort of patients with parkinsonism defined as ALS‐plus syndromes, when they also fulfilled the clinical criteria for ALS.85 This spectrum includes ALS‐PD, ALS‐CBS, ALS‐PSP, and ALS‐MSA patients.
The relatively common presence of parkinsonian symptoms in FTD/ALS patients who carry the expansion is confirmed in several studies. The prevalence is estimated to be as high as 35% within the first two years after disease onset,12 and even higher in familial cases.19 It seems that in patients who present with the phenotype of behavioral variant FTD (bvFTD), the prevalence of parkinsonian symptoms can be as high as 50 to 75% and significantly higher compared to patients who present with ALS phenotype.12, 89
The observation that there is an increased prevalence of parkinsonism in the families of C9orf72‐positive patients has been examined by some studies. A systematic review of patients with typical and atypical parkinsonism that carry the expansion revealed a high prevalence of parkinsonism (38%) in the family history, including a significant prevalence of PD (13%).61 Positive family history seems to include mostly complex phenotypes. In a study that identified five patients carrying large expansions in a cohort of sporadic and familial cases with parkinsonian phenotypes, none of the C9orf72 positive patients had a family history of typical PD.41
Many studies report an intermediate range of repeats in patients with different phenotypes that include: typical PD,40, 42, 44, 46, 52 atypical parkinsonism, including PSP, MSA, and PD complicated by psychosis,41, 47, 58 ET plus parkinsonism,44 or even cerebellar ataxia.84 Previous studies have reported the presence of patients with an intermediate size of repeats in cases with FTD/ALS phenotypes.20, 27, 90, 91 The exact definition of intermediate repeat expansion size is not firmly established though, and its possible role in disease is still being questioned, but cannot be completely disregarded with the current data.92
Conclusions, Limitations, and Suggestions for Further Research
In general, there is a small amount of evidence to support the direct causal relation of the C9orf72 expansion in PD, atypical parkinsonism, and most other movement disorders. On the contrary, there is a satisfying amount of data that the expansion may be one of the leading genetic causes of HD‐phenocopies in populations of European origin. By comparing the segregate data on prevalence from the reviewed studies to the figures reported in the latest published meta‐analysis of ALS and FTD related to C9orf72,93 we noticed a big gap in the observed frequencies and the number of cases, questioning the relevance of C9orf72 to parkinsonian disorders (Fig. 1).
Figure 1.
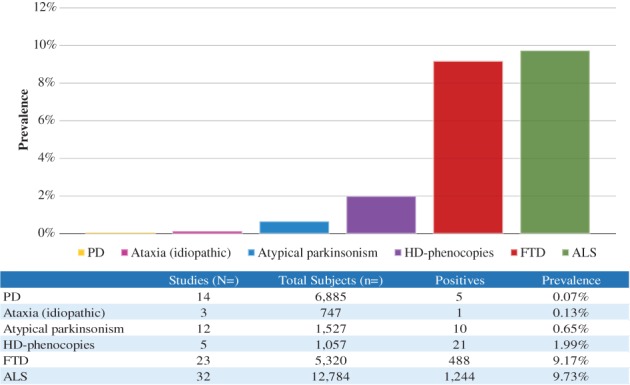
Calculated frequencies of C9orf72 expansion carriers based on aggregate data of reviewed studies. Frequency and number of cases for ALS and FTD as reported elsewhere (see text).
Especially for typical PD, the reported frequencies of large repeat expansions in many sizeable case–control studies are very small and statistically insignificant, suggesting that C9orf72 expansion should not be considered as an important genetic cause for PD. Data on atypical parkinsonism are more variable and mostly based on small cohorts of clinically‐only diagnosed patients. This fact is further complicated by the difficulties often encountered in the phenotypic classification of complex syndromes that present parkinsonian symptoms. Pathological studies on such patients often reconsider the initial clinical diagnosis. Therefore, it is not surprising that all studies on C9orf72 in pathologically confirmed patients with atypical parkinsonism failed to reproduce the findings of previously reported, clinically defined cohorts. Further prospective studies with larger and better‐defined cohorts of patients with atypical parkinsonism are required to address this issue.
Most reported cases in literature are of European origin, a fact that is consistent with the theory of common founder effect. Interestingly, the same populations have a slightly larger average size of copy repeats and infrequent expanded alleles within normal controls when compared to other populations. Since the clinical significance and the contribution of expansion size to disease variability are still unknown, this finding needs to be clarified.
The observation that some patients carry a smaller, or intermediate size of repeat expansion suggest that C9orf72 could be a susceptibility factor for parkinsonism rather than a direct causing gene, although there is not enough evidence to support that theory. The lack of consensus in determining the exact length of repeat units that are considered pathogenic needs to be addressed. The technical difficulties that limit the accuracy in determining the exact size of the expansion, especially in larger expansion sizes, stress the need to use precise Southern blotting techniques that can allow more concrete conclusions of future research studies. The reported somatic instability of the expansion, with larger expansions observed in the brain in comparison to the peripheral blood, complicates, even more, the effort to establish firm phenotype–genotype correlations. It seems that this observation does not affect smaller expansions and needs to be studied further.
Furthermore, the variability of the expansion size does not seem to correlate directly with the clinical heterogeneity. The possibility that additional genetic, epigenetic, and environmental factors could play a significant role in the expression of the expansion has been suggested and could explain the significant degree of observed phenotypical heterogeneity. The role of methylation has already been studied and seems to correlate with the pathogenicity of the expansion. The possible interaction with other repeat expansions has also been suggested. Further research is required to determine the exact role of genetic, epigenetic, and/or environmental modification and its impact on clinical variability.
The relatively high prevalence of parkinsonian signs among patients who present within the FTD/ALS spectrum raises the possibility that parkinsonism is just one of the phenotypes that overlap with the complex syndromes that can be caused by the C9orf72‐related neurodegeneration. Studies of pathology in such patients seem to support that theory, revealing a mixed Lewy body and p62 pathology. Further studies are required to define the significance of such overlap and the contribution of the C9orf72 expansion in the observed findings.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript: A. Writing of the First Draft, B. Review and Critique.
T.B.: 1A, 1B, 1C, 2A, 2B, 3A.
H.H.: 1A, 1B, 2A, 2C, 3B.
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The authors confirm that the approval of an institutional review board and patient consent was not required for this work.
Funding Sources and Conflict of Interest: No specific funding was received for this work. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the previous 12 months: The authors declare that there are no additional disclosures to report.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 2011;72(2):257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p‐Linked FTD and ALS. Neuron 2011;72(2):245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lillo P, Hodges JR. Frontotemporal dementia and motor neurone disease: Overlapping clinic‐pathological disorders. J Clin Neurosci 2009;16(9):1131–1135. [DOI] [PubMed] [Google Scholar]
- 4. Couratier P, Corcia P, Lautrette G, Nicol M, Marin B. ALS and frontotemporal dementia belong to a common disease spectrum. Rev Neurol (Paris) 2017;173(5):273–279. [DOI] [PubMed] [Google Scholar]
- 5. Murphy J, Henry R, Lomen‐Hoerth C. Establishing subtypes of the continuum of frontal lobe impariment in amyotrophic lateral sclerosis. Arch Neurol 2007;64(3):330–334. [DOI] [PubMed] [Google Scholar]
- 6. Smith BN, Newhouse S, Shatunov A, et al. The C9ORF72 expansion mutation is a common cause of ALS+/‐FTD in Europe and has a single founder. Eur J Hum Genet 2013;21(1):102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Rheenen W, Van Blitterswijk M, Huisman MHB, et al. Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology 2012;79(9):878–882. [DOI] [PubMed] [Google Scholar]
- 8. van der Zee J, Gijselinck I, Dillen L, et al. A Pan‐European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 2013;34(2):363–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zou Z, Zhou Z, Che C, Liu C, He R, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta‐analysis. J Neurol Neurosurg Psychiatry 2017;0:1–10. [DOI] [PubMed] [Google Scholar]
- 10. Cooper‐Knock J, Shaw PJ, Kirby J. The widening spectrum of C9ORF72‐related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol 2014;127(3):333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beck J, Poulter M, Hensman D, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet 2013;92(3):345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boeve BF, Boylan KB, Graff‐Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135(3):765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Floris G, Borghero G, Cannas A, et al. Frontotemporal dementia with psychosis, parkinsonism, visuo‐spatial dysfunction, upper motor neuron involvement associated to expansion of C9ORF72: a peculiar phenotype? J Neurol 2012;259(8):1749–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dowd SO, Curtin D, Waite AJ, et al. C9ORF72 expansion in amyotrophic lateral sclerosis/frontotemporal dementia also causes parkinsonism. Mov Disord 2012;27(8):1072–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ducharme S, Bajestan S, Dickerson BC, Voon V. Psychiatric presentations of C9orf72 mutation: what are the diagnostic implications for clinicians? J Neuropsychiatry Clin Neurosci 2017;29(3):195–205. [DOI] [PubMed] [Google Scholar]
- 16. Cacace R, Van Cauwenberghe C, Bettens K, et al. C9orf72 G4C2repeat expansions in Alzheimer's disease and mild cognitive impairment. Neurobiol Aging 2013;34(6):1712.e1–1712.e7. [DOI] [PubMed] [Google Scholar]
- 17. Kohli MA, John‐Williams K, Rajbhandary R, et al. Repeat expansions in the C9ORF72 gene contribute to Alzheimer's disease in Caucasians. Neurobiol Aging 2013;34(5):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harms M, Benitez B, Cairns N, et al. C9ORF72 hexanucleotide repeat expansions in clinical Alzheimer's disease. JAMA Neurol 2013;70(6):736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hsiung GYR, Dejesus‐Hernandez M, Feldman HH, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 2012;135(3):709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simón‐Sánchez J, Dopper EGP, Cohn‐Hokke PE, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 2012;135(3):723–735. [DOI] [PubMed] [Google Scholar]
- 21. Cooper‐Knock J, Hewitt C, Highley JR, et al. Clinico‐pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012;135(3):751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akimoto C, Forsgren L, Linder J, et al. No GGGGCC‐hexanucleotide repeat expansion in C9ORF72 in parkinsonism patients in Sweden. Amyotroph Lateral Scler Front Degener 2013;14(1):26–29. [DOI] [PubMed] [Google Scholar]
- 23. Cerami C, Scarpini E, Cappa SF, Galimberti D. Frontotemporal lobar degeneration: current knowledge and future challenges. J Neurol 2012;259(11):2278–2286. [DOI] [PubMed] [Google Scholar]
- 24. Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol 2012;11(4):323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gijselinck I, Van Mossevelde S, Van Der Zee J, et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry 2016;21(8):1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mok K, Traynor BJ, Schymick J, et al. The chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol Aging 2012;33(1):209.e3–209.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. García‐Redondo A, Dols‐Icardo O, Rojas‐García R, et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mutat 2013;34(1):79–82. [DOI] [PubMed] [Google Scholar]
- 28. He J, Tang L, Benyamin B, et al. C9orf72 hexanucleotide repeat expansions in Chinese sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2015;36:1–8. [DOI] [PubMed] [Google Scholar]
- 29. Rutherford NJ, Heckman MG, DeJesus‐Hernandez M, et al. Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol Aging 2012;33(12):2950.e5–2950.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Mossevelde S, van der Zee J, Cruts M, Van Broeckhoven C. Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev 2017;44(Figure 1):117–124. [DOI] [PubMed] [Google Scholar]
- 31. Dols‐Icardo O, García‐Redondo A, Rojas‐García R, et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum Mol Genet 2014;23(3):749–754. [DOI] [PubMed] [Google Scholar]
- 32. Stewart H, Rutherford NJ, Briemberg H, et al. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 2012;123(3):409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chi S, Jiang T, Tan L, Yu JT. Distinct neurological disorders with C9orf72 mutations: genetics, pathogenesis, and therapy. Neurosci Biobehav Rev 2016;66:127–142. [DOI] [PubMed] [Google Scholar]
- 34. Xi Z, Zhang M, Bruni AC, et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol 2015;129(5):715–727. [DOI] [PubMed] [Google Scholar]
- 35. Van Blitterswijk M, Dejesus‐hernandez M, Niemantsverdriet E, et al. Associations of repeat sizes with clinical and pathological characteristics in C9ORF72 expansion carriers (Xpansize‐72): a cross‐sectional cohort study. Lancet Neurol 2013;12(10):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fratta P, Polke JM, Newcombe J, et al. Screening a UK amyotrophic lateral sclerosis cohort provides evidence of multiple origins of the C9orf72 expansion. Neurobiol Aging 2015;36(1):546.e1–546.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi Y, Lin S, Staats KA, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 2018;24(3):313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mackenzie IRA, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol 2014;127(3):347–357. [DOI] [PubMed] [Google Scholar]
- 39. Al‐Sarraj S, King A, Troakes C, et al. P62 positive, TDP‐43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72‐linked FTLD and MND/ALS. Acta Neuropathol 2011;122(6):691–702. [DOI] [PubMed] [Google Scholar]
- 40. Xi Z, Zinman L, Grinberg Y, et al. Investigation of C9orf72 in 4 neurodegenerative disorders. Arch Neurol 2012;69(12):1583–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lesage S, Le Ber I, Condroyer C, et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 2013;136(2):385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Majounie E, Abramzon Y, Renton AE, Keller MF, Traynor BJ, Singleton AB. Large C9orf72 repeat expansions are not a common cause of Parkinson's disease. Neurobiol Aging 2012;33(10):7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dejesus‐Hernandez M, Rayaprolu S, et al. Analysis of the C9orf72 repeat in Parkinson's disease, essential tremor and restless legs syndrome. Parkinsonism Relat Disord 2013;19(2):198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nuytemans K, Bademci G, Kohli MM, et al. C9orf72 intermediate repeat copies are a significant risk factor for parkinson disease. Ann Hum Genet 2013;77(5):351–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harms MB, Neumann D, Benitez BA, et al. Parkinson disease is not associated with C9ORF72 repeat expansions. Neurobiol Aging 2013;34(5):1519.e1–1519.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Daoud H, Noreau A, Rochefort D, et al. Investigation of C9orf72 repeat expansions in Parkinson's disease. Neurobiol Aging 2013;34(6):1710.e7–1710.e9. [DOI] [PubMed] [Google Scholar]
- 47. Cannas A, Solla P, Borghero G, et al. C9ORF72 intermediate repeat expansion in patients affected by atypical parkinsonian syndromes or Parkinson's disease complicated by psychosis or dementia in a Sardinian population. J Neurol 2015;262(11):2498–2503. [DOI] [PubMed] [Google Scholar]
- 48. Alavi A, Malakouti Nejad M, Shahidi G, Elahi E. Mutations in C19orf12 and intronic repeat expansions in C9orf72 not observed in Iranian Parkinson's disease patients. Neurobiol Aging 2017;54:214.e11–214.e12. [DOI] [PubMed] [Google Scholar]
- 49. Yeh TH, Lai SC, Weng YH, et al. Screening for C9orf72 repeat expansions in parkinsonian syndromes. Neurobiol Aging 2013;34(4):2013–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen X, Chen Y, Wei Q, et al. C9ORF72 repeat expansions in Chinese patients with Parkinson's disease and multiple system atrophy. J Neural Transm 2016;123(11):1341–1345. [DOI] [PubMed] [Google Scholar]
- 51. Jiao B, Guo J, Wang Y, et al. C9orf72 mutation is rare in Alzheimer's disease, Parkinson's disease, and essential tremor in China. Front Cell Neurosci 2013;7(September):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin CH, Chen TF, Chiu MJ, Lin HI, Wu RM. Lack of C9orf72 repeat expansion in Taiwanese patients with mixed neurodegenerative disorders. Front Neurol 2014;5 APR(April):5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Theuns J, Verstraeten A, Sleegers K, et al. Global investigation and meta‐analysis of the C9orf72 (G4C2)n repeat in Parkinson disease. Neurology 2014;83(21):1906–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cooper‐Knock J, Frolov A, Highley JR, et al. C9ORF72 expansions, parkinsonism, and Parkinson disease: a clinicopathologic study. Neurology 2013;81(9):808–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nuytemans K, Inchausti V, Beecham GW, et al. Absence of C9ORF72 expanded or intermediate repeats in autopsy‐confirmed Parkinson's disease. Mov Disord 2014;29(6):827–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Anor CJ, Xi Z, Zhang M, et al. Mutation analysis of C9orf72 in patients with corticobasal syndrome. Neurobiol Aging 2015;36(10):2905.e1–2905.e5. [DOI] [PubMed] [Google Scholar]
- 57. Lindquist SG, Duno M, Batbayli M, et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet 2013;83(3):279–283. [DOI] [PubMed] [Google Scholar]
- 58. Schottlaender LV, Polke JM, Ling H, et al. The analysis of C9orf72 repeat expansions in a large series of clinically and pathologically diagnosed cases with atypical parkinsonism. Neurobiol Aging 2015;36(2):1221.e1–1221.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Origone P, Verdiani S, Ciotti P, et al. Enlarging the clinical spectrum associated with C9orf 72 repeat expansions: findings in an Italian cohort of patients with Parkinsonian syndromes and relevance for genetic counselling. Amyotroph Lateral Scler Front Degener 2013;14(5–6):479–480. [DOI] [PubMed] [Google Scholar]
- 60. Le Ber I, Camuzat A, Guillot‐Noel L, et al. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow‐chart for genetic testing. J Alzheimer's Dis 2013;34(2):485–499. [DOI] [PubMed] [Google Scholar]
- 61. Wilke C, Pomper JK, Biskup S, Puskás C, Berg D, Synofzik M. Atypical parkinsonism in C9orf72 expansions: a case report and systematic review of 45 cases from the literature. J Neurol 2016;263(3):558–574. [DOI] [PubMed] [Google Scholar]
- 62. Goldman JS, Quinzii C, Dunning‐Broadbent J, et al. Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72. JAMA Neurol 2014;71(6):771–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Snowden JS, Rollinson S, Lafon C, et al. Psychosis, C9ORF72 and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2012;83(10):1031–1032. [DOI] [PubMed] [Google Scholar]
- 64. Galimberti D, Fenoglio C, Serpente M, et al. Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: late‐onset psychotic clinical presentation. Biol Psychiatry 2013;74(5):384–391. [DOI] [PubMed] [Google Scholar]
- 65. Sun ZF, Jiang H, Jiao B, et al. C9orf72 hexanucleotide expansion analysis in Chinese patients with multiple system atrophy. Park Relat Disord 2015;21(7):811–812. [DOI] [PubMed] [Google Scholar]
- 66. Hsiao CT, Tsai PC, Liao YC, Lee YC, Soong BW. C9ORF72 repeat expansion is not a significant cause of late onset cerebellar ataxia syndrome. J Neurol Sci 2014;347(1–2):322–324. [DOI] [PubMed] [Google Scholar]
- 67. Scholz SW, Majounie E, Revesz T, et al. Multiple system atrophy is not caused by C9orf72 hexanucleotide repeat expansions. Neurobiol Aging 2015;36(2):1223.e1–1223.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Geiger JT, Arthur KC, Dawson TM, et al. C9orf72 Hexanucleotide repeat analysis in cases with pathologically confirmed dementia with lewy bodies. Neurodegener Dis 2016;16(5–6):370–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Robinson A, Davidson Y, Snowden JS, Mann DMA. C9ORF72 in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2014;85(12):10–12. [DOI] [PubMed] [Google Scholar]
- 70. Kun‐Rodrigues C, Ross OA, Orme T, et al. Analysis of C9orf72 repeat expansions in a large international cohort of dementia with Lewy bodies. Neurobiol Aging 2017;49:214.e13–214.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mann DMA, Rollinson S, Robinson A, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 2013;1(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ramos‐Campoy O, Avila‐Polo R, Grau‐Rivera O, et al. Systematic screening of ubiquitin/p62 aggregates in cerebellar cortex expands the neuropathological phenotype of the C9orf72 expansion mutation. J Neuropathol Exp Neurol 2018;77(8):703–709. [DOI] [PubMed] [Google Scholar]
- 73. Hensman DJ, Poulter M, Beck J, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 2014;82(4):292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kostić VS, Dobričić V, Stanković I, Ralić V, Stefanova E. C9orf72 expansion as a possible genetic cause of Huntington disease phenocopy syndrome. J Neurol 2014;261(10):1917–1921. [DOI] [PubMed] [Google Scholar]
- 75. Koutsis G, Karadima G, Kartanou C, Kladi A, Panas M. C9ORF72 hexanucleotide repeat expansions are a frequent cause of Huntington disease phenocopies in the Greek population. Neurobiol Aging 2015;36(1):547.e13–547.e16. [DOI] [PubMed] [Google Scholar]
- 76. Martins J, Damásio J, Mendes A, et al. Clinical spectrum of C9orf72 expansion in a cohort of Huntington's disease phenocopies. Neurol Sci 2018;39(4):741–744. [DOI] [PubMed] [Google Scholar]
- 77. Mariani L‐L, Tesson C, Charles P, et al. Expanding the spectrum of genes involved in Huntington disease using a combined clinical and genetic approach. JAMA Neurol 2016;73(9):1105–1114. [DOI] [PubMed] [Google Scholar]
- 78. Fogel BL, Pribadi M, Pi S, Perlman SL, Geschwind DH, Coppola G. C9ORF72 expansion is not a significant cause of sporadic spinocerebellar ataxia. Mov Disord 2012;27(14):1835–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aydin G, Dekomien G, Hoffjan S, Gerding WM, Epplen JT, Arning L. Frequency of SCA8, SCA10, SCA12, SCA36, FXTAS and C9orf72 repeat expansions in SCA patients negative for the most common SCA subtypes. BMC Neurol 2018;18(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Figueroa KP, Gan SR, Perlman S, et al. C9orf72 repeat expansions as genetic modifiers for depression in spinocerebellar ataxias. Mov Disord 2018;33(3):497–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. van Blitterswijk M, Mullen B, Heckman MG, et al. Ataxin‐2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol Aging 2014;35(10):2421.e13–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang M, Xi Z, Misquitta K, et al. C9orf72 and ATXN2 repeat expansions coexist in a family with ataxia, dementia, and parkinsonism. Mov Disord 2017;32(1):158–162. [DOI] [PubMed] [Google Scholar]
- 83. Corcia P, Vourch P, Guennoc AM, et al. Pure cerebellar ataxia linked to large C9orf72 repeat expansion. Amyotroph Lateral Scler Front Degener 2016;17(3–4):301–303. [DOI] [PubMed] [Google Scholar]
- 84. Meloni M, Farris R, Solla P, Mascia MM, Marrosu F, Cannas A. C9ORF72 intermediate repeat expansion in a patient with psychiatric disorders and progressive cerebellar ataxia. Neurologist 2017;22(6):245–246. [DOI] [PubMed] [Google Scholar]
- 85. Ticozzi N, Tiloca C, Calini D, et al. C9orf72 repeat expansions are restricted to the ALS‐FTD spectrum. Neurobiol Aging 2014;35(4):936.e13–936.e17. [DOI] [PubMed] [Google Scholar]
- 86. Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012;135(3):693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shinagawa S, Naasan G, Karydas AM, et al. Clinicopathological study of patients with C9ORF72‐associated frontotemporal dementia presenting with delusions. J Geriatr Psychiatry Neurol 2015;28(2):99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Geser F, Martinez‐Lage M, Robinson J, et al. Clinical and pathological continuum of multisystem TDP‐43 proteinopathies. Arch Neurol 2009;66(2):180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Floris G, Borghero G, Di Stefano F, et al. Phenotypic variability related to C9orf72 mutation in a large Sardinian kindred. Amyotroph Lateral Scler Front Degener 2016;17(3–4):245–248. [DOI] [PubMed] [Google Scholar]
- 90. Byrne S, Heverin M, Elamin M, Walsh C, Hardiman O. Intermediate repeat expansion length in C9orf72 may be pathological in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front Degener 2014;15(1–2):148–150. [DOI] [PubMed] [Google Scholar]
- 91. Gómez‐Tortosa E, Gallego J, Guerrero‐López R, et al. C9ORF72 hexanucleotide expansions of 20‐22 repeats are associated with frontotemporal deterioration. Neurology 2013;80(4):366–70. [DOI] [PubMed] [Google Scholar]
- 92. Ng ASL, Tan EK. Intermediate C9orf72 alleles in neurological disorders: Does size really matter. J Med Genet 2017;54(9):591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Curtis AF, Masellis M, Hsiung G‐YR, et al. Sex differences in the prevalence of genetic mutations in FTD and ALS. Neurology 2017;89(15):1633–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]