

Corticobasal degeneration (CBD) is a rare neurodegenerative four‐repeat (4R) tauopathy with a wide clinical spectrum.1 Corticobasal syndrome (CBS) is characterized by highly asymmetric parkinsonism combined with dystonia, myoclonus, apraxia, cortical sensory deficits, or alien limb phenomena.2 Whereas asymmetric limb rigidity and bradykinesia are the most common motor features in cases of pathologically confirmed CBD presenting as CBS (CBD‐CBS cases), myoclonus has been recently found to be much less prevalent than previously reported.3 Here, we describe a patient with CBD‐CBS highlighting a myoclonus‐predominant phenotype.
Case Report
A 58‐year‐old man with unremarkable medical and family history was referred to our clinic because of right‐hand clumsiness and action tremor in the past 6 months. Examination revealed a markedly asymmetric upper‐limb akinetic‐rigid syndrome (right > left) and right‐arm action‐induced involuntary movements resembling tremor (Video, Segment 1). Behavioral, cognitive, or sleep disturbances were not present. Laboratory work‐up, brain MRI and dopamine transporter/single‐photon emission computed tomography (DaT‐SPECT; Fig. 1A) were normal.
Figure 1.
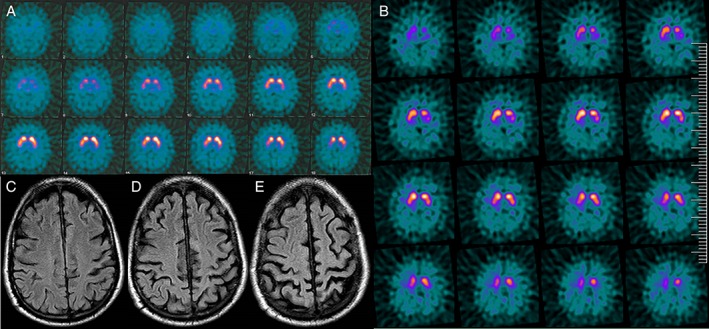
(A) DaT‐SPECT 10 months after first initial symptom demonstrates normal striatal uptake. (B) DaT‐SPECT 3 years after clinical onset shows left putamen uptake. (C–E) Brain MRI 3 years after symptom onset. Axial fluid‐attenuation inversion recovery images demonstrate severe and bilateral left greater than right frontoparietal atrophy.
After 2 years, bradykinesia and involuntary movements were severe despite symptomatic treatment (up to 1,250 mg of levodopa, levetiracetam, and clonazepam). On examination, jerks contaminating all voluntary movements were observed, but cortical signs typical of CBS, including ideomotor apraxia, were absent (Video, Segment 2). By the third year of disease, the patient was wheelchair bound. He had eyelid‐opening apraxia, impaired saccadic eye movements to the right, and generalized prominent action‐induced muscle jerks (Video, Segment 3). Right‐handed dystonic posture and mild cortical sensory deficits were then present whereas stimulus‐sensitive myoclonus was absent. Surface electromyography recording from right‐wrist extensor and flexor muscles showed nonrhythmic, short‐duration (20–40 ms) bursts and silent periods that appeared synchronously in both muscles. Transcranial magnetic stimulation showed normal latency motor evoked potentials (21.5 ms in the right side and 21.8 ms in the left side) and normal duration of contralateral silent period during sustained muscle contraction (162 ms after right‐side stimulation and 180 ms after left‐side stimulation). Cerebrospinal fluid 14‐3‐3 protein was negative, and follow‐up neuroimaging studies showed left putamen uptake reduction in DaT‐SPECT (Fig. 1B) and severe frontoparietal atrophy (Fig. 1C–E). The patient was bed bound 4 years after symptom onset and died 3 years later.
Written informed consent was obtained from the next of kin for brain donation for diagnostic and research purposes. Unfixed brain weight was 1,090 g. There was a moderate atrophy of the pre‐/postcentral region with narrowing of gyri and widening of sulci. In addition, moderate pallor of the SN was observed. Histological examination revealed moderate neuronal loss and gliosis in cortical areas with moderate superficial spongiosis, with preferential involvement of pre‐/postcentral region, basal ganglia, thalamus, and SN (with axonal spheroids). There were frequent ballooned neurons in frontal, temporal, and parietal cortices (Fig. 2A). Immunohistochemistry revealed frequent hyperphosphorylated tau (AT8 antibody)‐positive astrocytic plaques in cortical areas (Fig. 2B), abundant threads in gray and white matter, and frequent oligodendroglial coiled bodies in white matter (Fig. 2C). Neuronal pathology was predominated by pretangles in cortical and subcortical areas. This pathology was composed of 4R tau isoforms and was consistent with the morphological features of CBD.
Figure 2.
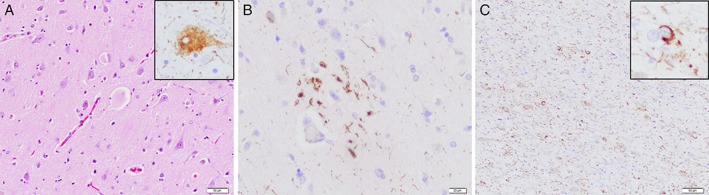
(A) Hematoxylin‐eosin–stained section reveals frequent enlarged or ballooned neurons that are immunoreactive for hyperphosphorylated Tau (inset; AT8). (B) Immunohistochemistry for hyperphosphorylated Tau reveals a characteristic astrocytic plaque in frontal cortex (AT8 immunohistochemistry). (C) Abundant cellular processes in white matter along with frequent oligodendroglial inclusions in the form of coiled bodies (inset; AT8 immunohistochemistry).
Discussion
CBD‐CBS was the suspected clinical diagnosis in this case with progressive and asymmetrical l‐dopa‐unresponsive action myoclonus, parkinsonism, and a “clumsy useless limb.”4 Highlights of the present case are the presence of pronounced action myoclonus and the initially normal DaT‐SPECT, which prompted us to exclude Creutzfeldt–Jakob disease at first, in spite of the long disease duration. Alzheimer's disease is a probable etiology for CBS‐myoclonus.5 However, abnormal DaT‐SPECT with the disease progression in our patient favored CBD‐CBS. According to Ling et al., neuronal loss of the SN occurs in late stages of CBD, hence explaining why DaT‐SPECT was normal into the first year of the disease.6
Myoclonus has been reported to occur in 15% of CDB‐CBS cases at clinical presentation and 27% during the entire course of disease.3 Typically, jerks are prominent with voluntary action or in response to sensory stimulation, located usually in the arms (less frequently in a lower extremity or face) and, at times, superimposed with limb dystonia.2, 7, 8 Electrophysiological studies may point to a cortical origin with enhanced cortical excitability.8 Low‐amplitude action myoclonus may resemble action tremor, and, in some CBD‐CBS patients, myoclonus may be mistaken for tremor.3 Tremor in CBD has been characterized as a mixture of resting, postural, and action tremor, but, unlike the tremor of Parkinson's disease, it is coarse and minimal at rest while it is amplified with activity.9 “Undefined” tremors are also mentioned in CBD cases.3 In a recent report of a CBD‐CBS case with a 10‐month course, episodes of intermittent truncal tremor triggered by limb movement and high‐frequency jerky hand tremor were described.10 In the present case, what initially resembled high‐frequency action tremor showed, in fact, clinical and neurophysiological features of action myoclonus.
The myoclonus‐predominant phenotype with abnormal DaT‐SPECT, along with disease progression, is an uncommon clinical presentation of CBD.
Author Roles
(1) Case Report Project: A. Conception, B. Organization, C. Execution; (2) Manuscript: A. Writing of the First Draft, B. Review and Critique.
N.C.: 1A, 1B, 1C, 2A
J.N.O.: 1A, 1B, 1C, 2B
A.A.: 1B, 1C, 2B
M.B.: 1A, 2B
R.M.F.: 1A, 1B, 2B
O.D.F.: 1A, 2B
E.G.: 1A, 1B, 1C 2B
JVS.: 1A, 1B, 2B
E.T.: 1A, 1B, 1C, 2B
Acknowledgments
We are indebted to patients and families for their generous brain donation for research, and to the Neurological Tissue Bank of the Biobanc‐Hospital Clinic‐IDIBAPS for data procurement. We acknowledge the CERCA Programme of the Generalitat de Catalunya for their support.
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: N.C. has received honoraria from Bial, Italfármaco, Qualigen, Zambon, Teva, and KRKA. A.A. has received honoraria from Zambon, Qualigen, Bial, and Teva and sponsorship from Zambon and Teva for attending conferences. R.M.F. has received payment from Zambon for travel and accommodation to attend scientific meetings and honoraria for lecturing from Teva, Zambon, and Alter. O.D.F. has received speaker honoraria from AbbVie and Zambon. E.G. holds a grant from the Fundació Marató de TV3 and the Fundación Tatiana Pérez de Guzmán el Bueno and served as a member of the scientific advisory board at the Institut du Cerveau et de la Moelle Epiniere (Institute for Brain and Spinal Cord), Hospital Pitie Salpetriere, Paris, and served as a member of the scientific advisory board of the Oxford Parkinson's Disease Center, UK. E.T. received honoraria for consultancy from Novartis, TEVA, Bial, Accorda, Boehringer Ingelheim, UCB, Solvay, Lundbeck, AbbVie, and BIOGEN and has received funding for research from the Spanish Network for Research on Neurodegenerative Disorders (CIBERNED)–Instituto Carlos III (ISCIII) and The Michael J. Fox Foundation for Parkinson's Research (MJFF).
Supporting information
Supplemental Video 1. Segment 1 (20 months after clinical onset): tremor not present at rest. Asymmetric upper‐limb akinetic‐rigid syndrome (right > left). Right arm action‐induced involuntary movements resembling tremor. Segment 2 (2 years after first symptom): normal ocular saccade movements. Severe right‐sided bradykinesia. Jerks interfering with voluntary movements. No ideomotor apraxia. Segment 3 (3 years after initial symptom): eyelid‐opening apraxia. Horizontal saccades: normal to the left, increased latency, and abnormal velocity to the right. Vertical saccades: normal upward and mild impairment downward. Prominent bilateral upper‐limb action‐induced myoclonus (right > left), which is worse in his right hand. The patient is still able to perform gestures with his right hand. Generalized severe muscle jerks.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Kouri N, Whitell JL, Josephs KA, et al. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol 2011;7:263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grijalvo‐Perez AM Litvan I. Corticobasal degeneration. Semin Neurol 2014;34:160–173. [DOI] [PubMed] [Google Scholar]
- 3. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ling H O'Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re‐evaluation. Brain 2010;133:2045–2057. [DOI] [PubMed] [Google Scholar]
- 5. Hu WT, Rippon GW, Boeve BF, et al. Alzheimer's disease and corticobasal degeneration presenting as corticobasal syndrome. Mov Disord 2009;24:1375–1392. [DOI] [PubMed] [Google Scholar]
- 6. Ling H, Kovacs GG, Vonsattel JP, et al. Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain 2016;139:3237–3252. [DOI] [PubMed] [Google Scholar]
- 7. Notturno F, Zappasodi F, Maruotti V, et al. Cortical origin of myoclonus in early stages of corticobassal degeneration. Mov Disord 2011;26:1567–1569. [DOI] [PubMed] [Google Scholar]
- 8. Thompson PD, Day BL, Rothwell JC, et al. The myoclonus in corticobasal degeneration. Evidence for two forms of cortical reflex myoclonus. Brain 1994;117:1197–1207. [DOI] [PubMed] [Google Scholar]
- 9. Litvan I, ed. Atypical Parkinsonian Disorders: Clinical and Research Aspects. Totowa, NJ: Humana; 2005. [Google Scholar]
- 10. Rodriguez‐Porcel F, Lowder l, Rademakers R, et al. Fulminant corticobasal degeneration: agrypnia excitata in corticobasal syndrome. Neurology 2016;86:1164–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video 1. Segment 1 (20 months after clinical onset): tremor not present at rest. Asymmetric upper‐limb akinetic‐rigid syndrome (right > left). Right arm action‐induced involuntary movements resembling tremor. Segment 2 (2 years after first symptom): normal ocular saccade movements. Severe right‐sided bradykinesia. Jerks interfering with voluntary movements. No ideomotor apraxia. Segment 3 (3 years after initial symptom): eyelid‐opening apraxia. Horizontal saccades: normal to the left, increased latency, and abnormal velocity to the right. Vertical saccades: normal upward and mild impairment downward. Prominent bilateral upper‐limb action‐induced myoclonus (right > left), which is worse in his right hand. The patient is still able to perform gestures with his right hand. Generalized severe muscle jerks.