

Abstract
Traditionally, small-molecule or antibody-based therapies against human diseases have been designed to inhibit the enzymatic activity or compete for the ligand binding sites of pathological target proteins. Despite its demonstrated effectiveness, such as in cancer treatment, this approach is often limited by recurring drug resistance. More importantly, not all molecular targets are enzymes or receptors with druggable ‘hot spots’ that can be directly occupied by active site-directed inhibitors. Recently, a promising new paradigm has been created, in which small-molecule chemicals harness the naturally occurring protein quality control machinery of the ubiquitin-proteasome system to specifically eradicate disease-causing proteins in cells. Such ‘chemically induced protein degradation’ may provide unprecedented opportunities for targeting proteins that are inherently undruggable, such as structural scaffolds and other non-enzymatic molecules, for therapeutic purposes. This review focuses on surveying recent progress in developing E3-guided proteolysis-targeting chimeras (PROTACs) and small-molecule chemical modulators of deubiquitinating enzymes upstream of or on the proteasome.
Keywords: deubiquitinating enzyme, induced proteolysis, PROTAC, small-molecules, ubiquitin-proteasome system, undruggable target
INTRODUCTION
Cellular proteolysis can be achieved through two major pathways: the ubiquitin-proteasome system (UPS) and the lysosomal degradation pathway (Ciechanover, 2005). Ubiquitination, a type of protein post-translational modification, is covalent conjugation of the small, stable protein, ubiquitin through the formation of an isopeptide bond between a Gly of ubiquitin and a Lys on the substrate. This reaction occurs through an enzymatic cascade of ubiquitin-modifying enzymes comprising E1 (ubiquitin activating enzyme), E2 (ubiquitin conjugating enzyme), and E3 (ubiquitin ligase) (Finley and Chau, 1991). Although ubiquitin conjugation may serve non-proteolytic functions, its most recognized function is targeted protein degradation via the 26S proteasome, a multi-protein protease complex (Fig. 1) (Finley, 2009; Komander and Rape, 2012). Ubiquitination process is tightly controlled, reflecting the critical importance of protein turnover rates for cellular function. This requirement strongly suggests that the ubiquitination reaction must be remarkably versatile. Notable in this context, more than 600 E3 ligases are known, helping to explain the observed substrate specificity (Deshaies and Joazeiro, 2009); moreover, deubiquitinating enzymes (DUBs) can reverse this reaction by breaking down ubiquitin polymers (Wilkinson, 1997). E3 ligases transfer ubiquitin from a ubiquitin-charged E2 complex to the substrate and this reaction can be repeated, producing a polyubiquitinated substrate that is recognized by the proteasome. The specificity of E3 ligases suggests that E3 inhibitors could produce more targeted effects and thus might be less harsh than E1- or E2-targeting drugs. In fact, several studies have shown that specifically targeting E3 ligase may be therapeutically effective against certain cancers (Chan et al., 2013; Shangary et al., 2008; Vassilev et al., 2004). In addition to E3 ligases, the human proteome contains approximately 100 DUBs belonging to at least six subfamilies (Komander et al., 2009), which play key roles in ubiquitin-mediated proteolytic pathways as well as other biological processes, including (a) rescue of protein substrates from degradation by ubiquitin removal, (b) editing of ubiquitin chains to regulate the function or half-lives of protein substrates, and (c) ubiquitin recycling into free chains or ubiquitin monomers.
Fig. 1. Mechanisms of induced protein degradation by PROTACs and DUB inhibitors.
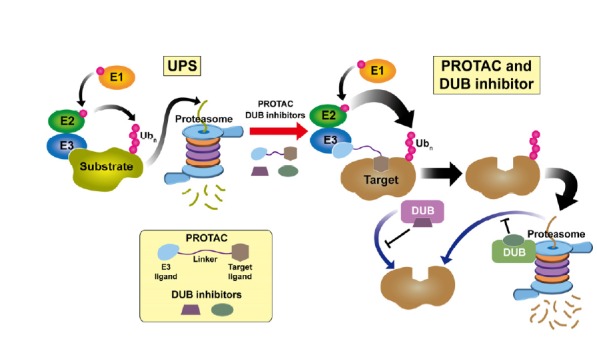
(Left) General scheme of ubiquitin-proteasome system. An enzymatic cascade of E1-E2-E3 transfers ubiquitin to Lys residues on the substrate. As a consequence, the polyubiquitinated substrate is recognized by the 26S proteasome and undergoes degradation. (Right) Induced proteolysis by PROTAC and/or DUB inhibitor. PROTAC links E3 and the target protein, enhances E3-mediated ubiquitination, and promotes degradation of the target molecule. By contrast, DUBs acting upstream of the proteasome (violet) or on the proteasome (olive drab) can inhibit degradation through substrate deubiquitination. Therefore, DUB inhibitors may facilitate the proteasomal degradation pathway by antagonizing deubiquitination.
Since the initial discovery of the proteolytic machinery, a plethora of evidence has shown that the UPS is closely associated with various diseases, such as cancers and neuro-degenerative diseases (Popovic et al., 2014). These findings have garnered immense attention in treating such diseases by targeting the UPS. For example, following a path similar to that for kinase inhibitors, bortezomib and its progeny have been successfully developed as FDA-approved anti-cancer drugs for the treatment of multiple myeloma (MM) and other blood cancers, and in fact, these agents are proteasome inhibitors that block the proteolytic reaction (Manasanch and Orlowski, 2017; Richardson et al., 2005; Stewart et al., 2015). Moreover, a number of E3 ligase inhibitors and DUB inhibitors have been tested in preclinical or clinical phase studies, representing novel strategies for target-directed therapies (Gopinath et al., 2016; Farshi et al., 2015; Weathington and Mallampalli, 2014).
Traditional active site-directed therapies, which generally employ small-molecule inhibitors and antibodies, are designed to inhibit the activity of target proteins. Small-molecule chemicals have proven effective for inhibiting several oncogenic enzymes, including protein kinases (e.g., imatinib and erlotinib), histone deacetylases (e.g., belinostat), and poly (ADP-ribose) polymerase (PARP) (e.g., olaparib) by directly targeting their active sites and thereby disrupting the enzymatic function (Salami and Crews, 2017). Despite being promising, these approaches have inherent limitations. First, there are only about 400 human proteins as possible drug targets among ~3000 disease-related proteins (Rask-Andersen et al., 2011). Furthermore, more than 90% of these possible protein targets fall into the categories of GPCRs, enzymes, CD markers, transporters and ion channels, which are considered “druggable”. This suggests that current therapeutics target only a fraction of the disease-causing proteome. Second, small-molecule and antibody binding is predominantly non-covalent and reversible. This indicates that treatment efficacy requires the inhibitors to be maintained at high concentration. Maintaining a high dose of drug is one of the major challenges in modern drug development, and this may also cause unexpected side effects (Salami and Crews, 2017). Lastly, antibodies induce altered cellular responses by blocking extracellular protein-protein or protein-ligand interactions. Such antibodies, including rituximab, tocilizumab, and belimumab, are effective against tumors and several autoimmune diseases. The main advantage of antibody-based therapy is its high affinity for the target protein and the longer half-life owing to endosomal FcRn-IgG recycling. However, therapeutic antibodies are also highly limited because of their poor cell permeability, oral unavailability, and high cost (Neklesa et al., 2017).
An emerging new paradigm of disease treatment–“targeted protein degradation (TPD)”–promises to help overcoming the limitations of typical active site-directed therapies (Zhou, 2005). TPD aims to induce degradation of target proteins, especially those considered “undruggable”, by harnessing the endogenous protein quality control machinery–the UPS. One elaborate approach is to induce ubiquitin tagging and subsequent target protein degradation through E3-guided proteolysis-targeting chimeras (PROTACs). Small-molecule DUB inhibitors that act upstream of or on the proteasome can also be utilized for antagonizing substrate deubiquitination. This review will focus on recent progress in the development of TPD, specifically PROTACs and DUB inhibitors, and their therapeutic potential. Figure 1 highlights PROTAC and DUB inhibitors as chemical proteolytic inducers in the UPS pathway.
INDUCED PROTEOLYSIS BY PROTACS
PROTAC, an E3-guided proteolysis-targeting chimera, is a bifunctional molecule composed of three parts: an E3 ligase binding ligand, a substrate binding ligand, and a linker between the two. E3 ligase generally requires sequential recognition events through multi-step E1/E2/substrate reactions (Deshaies and Joazeiro, 2009). PROTACs can simplify this process–they transiently recruit target substrate and E3 ligase in close proximity and facilitate the ubiquitination of the substrate (Coleman and Crews, 2018). As a result, target proteins become more efficiently polyubiquitinated and undergo degradation by the 26S proteasome (Fig. 1). This idea originated with the elegant work of Sakamoto and colleagues, in which the researchers developed a chimera linking the E3 ligase SCF-β-TRCP (Skp1/Cullin/F box-β-TRCP) to methionine aminopeptidase 2 (MetAP2) (Sakamoto et al., 2001). This chimeric molecule, PROTAC-1, contains a 10-amino acid IκBα phosphopeptide that can be recognized by β-TRCP at one end, while the other side is an MetAP1 inhibitor, ovalicin. PROTAC-1 indeed promotes degradation of MetAP1 in a ubiquitination-dependent manner. Since first-generation PROTACs had low cell permeability because of the peptidic sequence, next-generations adopted von Hippel–Lindau (VHL) as an E3 ligase instead of SCF-β-TRCP (Schneekloth et al., 2004). This variant, developed by Crews and colleagues, exploited a short hydroxyproline-containing peptide derived from the VHL substrate, hypoxia-inducible factor 1-α (HIF1α), and they applied the resulting chimeras for selective degradation of androgen receptor (AR) and FK506-binding protein 12 (FKBP12) in intact cells. Although the hydroxyproline-containing degron displayed improved cell applicability, this VHL-recognized peptide still lacked ideal drug-like properties. Recent efforts have further validated MDM2/Nutlins, Cereblon (CRBN)/thalidomide, cIAP ligands, and optimized VHL ligands as E3-recruiting moieties for target protein degradation in PROTAC technology (Table 1). Notably, landmark papers from Bradner and Crews groups demonstrated that small-molecule VHL- and CRBN-directed PROTACs are highly effective in degrading estrogen-related receptor α (ERRα) and Bromodomain-containing protein 4 (BRD4) in animal cancer models (Bondeson et al., 2015; Winter et al., 2015). PROTACs can be designed to behave like traditional small-molecules, yet their distinctive mode of action provides innovative therapeutic opportunities. There has also been considerable effort devoted to PROTACs into commercial drugs, exemplified by the recent foundation of Arvinas and C4 Therapeutics (Neklesa et al., 2017).
Table 1.
Representative examples of PROTACs
| E3 ligase/HyTa | E3 binder/HyT (typeb) | Target binder (type) | Target | Reference |
|---|---|---|---|---|
| β-TRCP | IκBα phosphopeptide (P) | Ovalicin (S) | MetAP2 | Sakamoto et al., 2001 |
| cIAP | MeBS (S) | ATRA (S) | CRABP-I, II | Itoh et al., 2010 |
| BE04 (S) | Ch55 / Estrone / DHT (S) | RAR / ER / AR | Itoh et al., 2011 | |
| BE04 (S) | KHS108 (S) | TACC3 | Ohoka et al., 2014 | |
| BE04 (S) | Alkyl chloride analog (S) | HaloTag | Tomoshige et al., 2015 | |
| CRBN | Thalidomide (S) | JQ1 / SLF (S) | BET family / FKBP12 | Winter et al., 2015 |
| Pomalidomide (S) | Bosutinib, Dasatinib (S) | BCR–ABL | Lai et al., 2016 | |
| Thalidomide (S) | Aminopyrazole analog (S) | CDK9 | Robb et al., 2017 | |
| Pomalidomide (S) | Ceritinib (S) | ALK | Zhang et al., 2018 | |
| Lenalidomide (S) | HJB97 (S) | BET family | Zhou et al., 2018 | |
| Pomalidomide (S) | Ibrutinib derivative (S) | BTK (Bruton’s Tyr kinase) | Buhimschi et al., 2018 | |
| Pomalidomide (S) | HDAC inhibiting aldehydes (S) | HDAC6 | Yang et al., 2018 | |
| Thalidomide (S) | Sirtuin Rearranging Ligand (SirReal) (S) | Sirtuin 2 (Sirt2) | Schiedel et al., 2018 | |
| Keap1 | Keap1 binding motif (P) | Tau binding motif (P) | Tau | Lu et al., 2018 |
| MDM2 | Nutlin-3 (S) | SARM (S) | AR | Schneekloth et al., 2008 |
| VHL | HIFα degron (P) | DHT / AP21998 (S) | AR / FKBP12 | Schneekloth et al., 2004 |
| HIFα degron (P) | DHT / Estradiol (S) | AR / ER | Rodriguez-Gonzalez et al., 2008 | |
| HIFα degron (P) | TrkA degron / ErbB3 degron (P) | FRS2α / PI3K | Hines et al., 2013 | |
| HIFα derivative (S) | Vandetanib / Phenoxy TZD analog (S) | RIPK2 / ERRα | Bondeson et al., 2015 | |
| HIFα derivative (S) | JQ1 (S) | BRD4 | Zengerle et al., 2015 | |
| HIFα derivative (S) | Chloroalkane analog (S) | HaloTag | Buckley et al., 2015 | |
| HIFα derivative (S) | Triazolo-diazepine acetamide (S) | BET family | Raina et al., 2016 | |
| HIFα degron (P) | CPP-tri_a (S) | AKT | Henning et al., 2016 | |
| HIFα derivative (S) | Dasatinib (S) | c–ABL | Lai et al., 2016 | |
| HIFα degron (P) | EN300-72284 (S) | Smad3 | Wang et al., 2016a | |
| HIFα degron (P) | Tau binding motif (P) | Tau | Chu et al., 2016 | |
| HIFα derivative (S) | TBK1 ligand (S) | TBK1 (TANK-Binding Kinase 1) | Crew et al., 2018 | |
| HIFα degron (P) | TD-PERM (P) | ERα | Jiang et al., 2018 | |
| HIFα derivative (S) | Lapatinib, Gefitinib, Afatinib (S) | EGFR, HER2, c-Met | Burslem et al., 2018 | |
| HyT | Adamantane (S) | Haloalkane reactive linker (S) | HaloTag | Neklesa et al., 2011 |
| Boc3Arg (S) | Trimethoprim / EA, Thiobenzofurazan (S) | DHFR / GST | Long et al., 2012 | |
| Adamantane (S) | TX1-85-1 (S) | HER3 | Xie et al., 2014 | |
| Adamantane (S) | RU59063 (S) | AR | Gustafson et al., 2015 |
HyT, Hydrophobic tagging;
P, peptide, S, small molecule
PROTACs have several advantages over active site-directed therapies, ideally overcoming the drawbacks of small-molecule inhibitors and therapeutic antibodies. The most prominent feature of PROTAC is that induced protein degradation, in principle, renders conventionally undruggable targets druggable. As previously noted, typical active site-directed drug targets constitute only about 13% of the thousands of disease-related proteins, mostly enzymes or membrane receptors. This clearly indicates that transcription factors, scaffolding proteins, and toxic protein aggregates have remained therapeutically intractable. PROTACs, by linking ligands and appropriate E3 ligases, can induce the proteolysis of traditionally undruggable proteins, including transcriptional regulators (e.g., Myc, Gli, β-catenin, and STAT family) and signaling scaffolds (e.g., ERBB3, KSR, Gab family, β-arrestin, BCL10, and AKAPs) (Coleman and Crews, 2018; Neklesa et al., 2017). Protein aggregates are particularly interesting targets because they are common features of several neurodegenerative diseases, such as tauopathies and amyotrophic lateral sclerosis (ALS) (Ross and Poirier, 2004). Recent studies have shown that the low efficacy problem of early PROTACs can be substantially resolved, as evidenced by the apparent nanomolar range of EC50s of more recently developed ones (Lai and Crews, 2017). Analogous to E3-guided PROTAC, the ‘hydrophobic tagging (HyT)’ technology has also been developed to destabilize target proteins and/or recruit chaperones for induced protein degradation (Huang and Dixit, 2016; Lai and Crews, 2017). The novel repertoire of E3 ligases (e.g., Keap1), HyT binders, and target ligands has rapidly expanded, strongly suggesting that PROTAC may serve as a platform technology (Chu et al., 2016; Lu et al., 2018). Table 1 summarizes representative examples of PROTACs.
SMALL-MOLECULE INHIBITORS TARGETING DEUBIQUITINATING ENZYMES UPSTREAM OF OR ON THE PROTEASOME
Reversing the ubiquitination process is exclusively fulfilled by deubiquitinating enzymes (DUBs), numbering nearly 100 in the human genome (Komander et al., 2009). To date, six subfamilies of DUBs have been identified: (1) ubiquitin carboxyl-terminal hydrolases (UCHs), (2) ubiquitin specific proteases (USPs), (3) ovarian tumor like proteases (OTUs), (4) JAMM/MPN metalloproteases, (5) Machado-Jacob-disease proteases (MJDs), and (6) motif interacting with Ub-containing novel DUB family (MINDY) (Abdul Rehman et al., 2016; Clague et al., 2013). DUBs should represent promising drug targets because they become increasingly implicated in various human diseases (Harrigan et al., 2018). For example, a growing number of DUBs (e.g., USP28, JOSD1, UCHL1, CSN5, USP9x, USP10, USP11, USP22, and USP48) were found to be overexpressed in diverse cancer types. Moreover, genetic alterations of DUBs (e.g., USP6/Tre2 and USP28) can be truly oncogenic in certain cancers (Fraile et al., 2012; Sacco et al., 2010). The isopeptidase activity of DUBs can be selectively targeted by inhibiting the catalytic sites with drug-like compounds, and in this sense, they are typical targets of active site-directed inhibitors. In an interesting twist, however, most cellular proteins are degraded by the ubiquitin-mediated proteasome pathway; thus, DUBs in principle should antagonize the destruction of these proteins (Fig. 1). Indeed, deubiquitination can occur on histones, transcription factors, and other regulatory proteins, so the pool of target substrates must far exceed the number of conventionally druggable targets. Therefore, similar to PROTACs, small-molecule DUB inhibitors may also induce the degradation of previously intractable targets. While of great therapeutic interest, only a handful of DUB inhibitors have been reported, and none has reached in advanced clinical trials, providing a valuable opportunity for developing first-in-class therapeutic DUB inhibitors (Farshi et al., 2015)(Table 2).
Table 2.
Representative examples of deubiquitinating enzyme inhibitors


reported to inhibit both USP7 and USP47;
reported to inhibit both USP14 and UCH37
Analogous to the cell cycle, deubiquitination reactions at two points–upstream of and on the proteasome–may serve as critical checkpoints for ubiquitin-mediated degradation pathways (Fig. 1). DUBs acting upstream of the proteasome may act as a “rescue crew”, salvaging specific substrates by deubiquitination before they are engaged in degradation. Accordingly, chemical blockers of the DUB may accelerate the turnover of the specific substrate for proteasome-mediated degradation. For instance, USP7/HAUSP is the most well-recognized drug target because of its cellular role as a regulator of the tumor suppressor p53. USP7 deubiquitinates MDM2, an E3 ligase of p53, and in turn destabilizes p53 (Colland et al., 2009). Thus, by stabilizing p53, a USP7 inhibitor may be beneficial for the treatment of certain cancers. The USP7 inhibitors, HBX 19,818 and P5091, were previously reported to selectively inhibit USP7 in vitro and in vivo, and importantly, P5091 showed cytotoxicity in relapsed MM cells derived from cancer patients (Chauhan et al., 2012; Reverdy et al., 2012). More recently, a series of USP7 inhibitors have been discovered and among them, a non-covalent inhibitor FT671 and a covalent inhibitor FT827 were identified by molecular-based screening (Turnbull et al., 2017). These compounds were found to be highly selective for USP7 in counter-screening against a panel of DUBs including USP10 and USP47, which are also inhibited by P5091. In vivo experiments in cancer cell lines and MM.1s xenograft animal models demonstrated that FT671 induces p53 stabilization and MDM2 degradation, leading to anti-tumor activity via USP7 blocking. Another NMR and structure-based screening study identified the USP7 inhibitors, GNE-6640 and GNE-6776 (Kategaya et al., 2017). These compounds may selectively interfere with K48 linkage-directed ubiquitin chain cleavage mediated by USP7, suggesting that K48-linked substrates such as MDM2 could be susceptible. More recently, an elegant fragment-based screen combined with structure-guided medicinal chemistry identified a highly potent and selective USP7 inhibitor, “compound 4” (IC50 = 6 nM). This allosteric inhibitor showed strong anti-proliferative effects against several cancer cell lines with equal or even greater efficacy compared to known clinical MDM2 antagonists (Gavory et al., 2018). A mitochondria-localized DUB, USP30 may also represent a promising therapeutic target due to its involvement in mitophagy-related Parkinson’s disease as well as cancers. USP30 antagonizes Parkin-mediated ubiquitination on multiple mitochondrial substrates (Bingol et al., 2014; Liang et al., 2015). Recently, a potent USP30 inhibitor MF-094 was developed through high-throughput screening and subsequent structure-activity relationship (SAR) studies of acyl benzenesulfonamide derivatives, and this compound showed the increased mitophagy in C2C12 cells (Kluge et al., 2018).
Targeting DUBs on the proteasome may also offer an exciting strategy for induced protein degradation. There are three major and distinctive DUBs on human proteasome: USP14, UCH37, and RPN11 (de Poot et al., 2017; Finley, 2009). USP14 and UCH37 may rescue substrates from degradation prior to the proteasome’s commitment step, whereas RPN11 is coupled to degradation. Finley and colleagues have screened out highly selective USP14 inhibitors, IU1 and its derivatives, and showed that their treatment promotes the degradation of proteopathic substrates in neurodegenerative disease models (Boselli et al., 2017; Lee et al., 2010; 2016). USP14 inhibitors may uncheck and bypass the deubiquitination-mediated proteolytic checkpoint on the proteasome under certain conditions of proteotoxic stress. By contrast, the proteasome 19S DUB inhibitors, b-AP15 and VLX1570, were reported to suppress tumor progression by inhibiting both USP14 and UCH37 activities (D’Arcy et al., 2011; Wang et al., 2015; 2016b). b-AP15 treatment leads to accumulation of polyubiquitinated conjugates and inhibition of protein degradation. Recently, capzimin was identified as a potent and specific RPN11 inhibitor (Li et al., 2017). Capzimin, a quinoline-8-thiol (8-TQ) derivative, induced the stabilization of proteasome substrates and inhibited cancer cell proliferation probably through the unfolded protein response (UPR). Unlike IU1, the anti-tumor effects of b-AP15 and capzimin might rely on ‘restrained protein degradation’ rather than induced proteolysis.
FUTURE PERSPECTIVES
Here we described PROTACs and DUB inhibitors–two emerging strategies of chemically induced proteolysis that utilize the endogenous ubiquitinproteasome system to inhibit previously undruggable targets. While certainly bearing tremendous promise for new therapeutic applications, these approaches could also face several challenges. For example, current PROTACs are orally unavailable, probably due to its relatively large size, typically 700–1000 Da. Their pharmacokinetic properties also need to be improved for better drug metabolism. Besides, only a few E3 ligases have been exploited, and not all E3 ligases might be co-expressed with target proteins in specific tissues, which makes diagnostics arduous (Huang and Dixit, 2016). PROTAC optimization–E3 ligase selection, ligand availability, and linker design–is another challenging issue. In this context, ligand screening can be performed by advanced screening tools, such as computer-aided drug design and DNA-encoded small molecule libraries, which can be accomplished on the order of ~109 compounds in a single vial (Chan et al., 2015). Although DUB inhibitors might be more orally bioavailable, their specificity and utility still remain to be explored. Given the smaller pool of DUB members compared to over 600 E3 ligases, DUB inhibitors may target only a subset of substrates with limited specificity. Nevertheless, one can envisage that ‘degradation inducers’ might pioneer the valuable therapeutic strategies and provide more advanced platform technologies, leading to a new era of UPS-related drug development. It will be also interesting to investigate potential combinatorial treatment with E3-guided and DUB-guided degradation inducers or to design new classes of chemically induced proteolysis chimeras recruiting substrates, E3s, DUBs, chaperones, or proteasomes.
ACKNOWLEDGEMENTS
The authors apologize to researchers whose citations are not included due to space limitations. We greatly appreciate HJ Park for help with graphics and other lab members for critical reading. BHL gratefully acknowledges the National Research Foundation of Korea (NRF) grant, DGIST Start-up Fund, and HRHR grant funded by the Ministry of Science, ICT, and Future Planning (2017R1A2B4006671, 2018010095, and 2018010146) for financial support.
REFERENCES
- Abdul Rehman Syed A, Kristariyanto Yosua A, Choi SY, Nkosi PJ, Weidlich S, Labib K, Hofmann K, Kulathu Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol Cell. 2016;63:146–155. doi: 10.1016/j.molcel.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611-U120. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boselli M, Lee BH, Robert J, Prado MA, Min SW, Cheng C, Silva MC, Seong C, Elsasser S, Hatle KM, et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J Biol Chem. 2017;292:19209–19225. doi: 10.1074/jbc.M117.815126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD, Crews CM. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem Biol. 2015;10:1831–1837. doi: 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM, Woyach JA, Johnson AJ, Byrd JC, Crews CM. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry-Us. 2018;57:3564–3575. doi: 10.1021/acs.biochem.8b00391. [DOI] [PubMed] [Google Scholar]
- Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong HQ, Qian YM, Wang J, et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chemical Biology. 2018;25:67–77. doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AI, McGregor LM, Liu DR. Novel selection methods for DNA-encoded chemical libraries. Curr Opin Chem Biol. 2015;26:55–61. doi: 10.1016/j.cbpa.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345–358. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, Maloney DJ, Jadhav A, Simeonov A, Zhuang Z. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18:1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu TT, Gao N, Li QQ, Chen PG, Yang XF, Chen YX, Zhao YF, Li YM. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem Biol. 2016;23:453–461. doi: 10.1016/j.chembiol.2016.02.016. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbe S. Deubiquitylases from genes to organism. Physiol Rev. 2013;93:1289–1315. doi: 10.1152/physrev.00002.2013. [DOI] [PubMed] [Google Scholar]
- Coleman KG, Crews CM. Proteolysis-Targeting Chimeras: Harnessing the Ubiquitin-Proteasome System to Induce Degradation of Specific Target Proteins. Annual Review of Cancer Biology. 2018;2:41–58. [Google Scholar]
- Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN, Camonis J, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther. 2009;8:2286–2295. doi: 10.1158/1535-7163.MCT-09-0097. [DOI] [PubMed] [Google Scholar]
- Crew AP, Raina K, Dong H, Qian Y, Wang J, Vigil D, Serebrenik YV, Hamman BD, Morgan A, Ferraro C, et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J Med Chem. 2018;61:583–598. doi: 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17:1636–1640. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- de Poot SAH, Tian G, Finley D. Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J Mol Biol. 2017;429:3525–3545. doi: 10.1016/j.jmb.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Dexheimer TS, Rosenthal AS, Luci DK, Liang Q, Villamil MA, Chen J, Sun H, Kerns EH, Simeonov A, Jadhav A, et al. Synthesis and structure-activity relationship studies of N-benzyl-2-phenylpyrimidin-4-amine derivatives as potent USP1/UAF1 deubiquitinase inhibitors with anticancer activity against nonsmall cell lung cancer. J Med Chem. 2014;57:8099–8110. doi: 10.1021/jm5010495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshi P, Deshmukh RR, Nwankwo JO, Arkwright RT, Cvek B, Liu J, Dou QP. Deubiquitinases (DUBs) and DUB inhibitors: a patent review. Expert Opin Ther Pat. 2015;25:1191–1208. doi: 10.1517/13543776.2015.1056737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annual Review of Biochemistry. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Chau V. Ubiquitination. Annu Rev Cell Biol. 1991;7:25–69. doi: 10.1146/annurev.cb.07.110191.000325. [DOI] [PubMed] [Google Scholar]
- Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373–2388. doi: 10.1038/onc.2011.443. [DOI] [PubMed] [Google Scholar]
- Gavory G, O’Dowd CR, Helm MD, Flasz J, Arkoudis E, Dossang A, Hughes C, Cassidy E, McClelland K, Odrzywol E, et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat Chem Biol. 2018;14:118–125. doi: 10.1038/nchembio.2528. [DOI] [PubMed] [Google Scholar]
- Gopinath P, Ohayon S, Nawatha M, Brik A. Chemical and semisynthetic approaches to study and target deubiquitinases. Chem Soc Rev. 2016;45:4171–4198. doi: 10.1039/c6cs00083e. [DOI] [PubMed] [Google Scholar]
- Gustafson JL, Neklesa TK, Cox CS, Roth AG, Buckley DL, Tae HS, Sundberg TB, Stagg DB, Hines J, McDonnell DP, et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew Chem Int Ed Engl. 2015;54:9659–9662. doi: 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17:57–77. doi: 10.1038/nrd.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning RK, Varghese JO, Das S, Nag A, Tang G, Tang K, Sutherland AM, Heath JR. Degradation of Akt using protein-catalyzed capture agents. J Pept Sci. 2016;22:196–200. doi: 10.1002/psc.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines J, Gough JD, Corson TW, Crews CM. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. P Natl Acad Sci USA. 2013;110:8942–8947. doi: 10.1073/pnas.1217206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016;26:484–498. doi: 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein Knockdown Using Methyl Bestatin-Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J Am Chem Soc. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Kitaguchi R, Ishikawa M, Naito M, Hashimoto Y. Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg Med Chem. 2011;19:6768–6778. doi: 10.1016/j.bmc.2011.09.041. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Deng Q, Zhao H, Xie M, Chen L, Yin F, Qin X, Zheng W, Zhao Y, Li Z. Development of Stabilized Peptide-Based PROTACs against Estrogen Receptor alpha. ACS Chem Biol. 2018;13:628–635. doi: 10.1021/acschembio.7b00985. [DOI] [PubMed] [Google Scholar]
- Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase Inhibition by Small-Molecule WP1130 Triggers Aggresome Formation and Tumor Cell Apoptosis. Cancer Res. 2010;70:9265–9276. doi: 10.1158/0008-5472.CAN-10-1530. [DOI] [PubMed] [Google Scholar]
- Kategaya L, Di Lello P, Rouge L, Pastor R, Clark KR, Drummond J, Kleinheinz T, Lin E, Upton JP, Prakash S, et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550:534–538. doi: 10.1038/nature24006. [DOI] [PubMed] [Google Scholar]
- Kluge AF, Lagu BR, Maiti P, Jaleel M, Webb M, Malhotra J, Mallat A, Srinivas PA, Thompson JE. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg Med Chem Letters. 2018;28:2655–2659. doi: 10.1016/j.bmcl.2018.05.013. [DOI] [PubMed] [Google Scholar]
- Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, Crews CM. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberto I, Liu X, Seo HS, Schauer NJ, Iacob RE, Hu W, Das D, Mikhailova T, Weisberg EL, Engen JR, et al. Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7. Cell Chem Biol. 2017;24:1490–1500e411. doi: 10.1016/j.chembiol.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179-U163. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lu Y, Prado MA, Shi Y, Tian G, Sun S, Elsasser S, Gygi SP, King RW, Finley D. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature. 2016;532:398–401. doi: 10.1038/nature17433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yakushi T, Parlati F, Mackinnon AL, Perez C, Ma Y, Carter KP, Colayco S, Magnuson G, Brown B, et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat Chem Biol. 2017;13:486–493. doi: 10.1038/nchembio.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JR, Martinez A, Lane JD, Mayor U, Clague MJ, Urbe S. USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. Embo Rep. 2015;16:618–627. doi: 10.15252/embr.201439820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Liu C, Li X, Liao S, Song W, Yang C, Zhao C, Huang H, Guan L, Zhang P, et al. A novel proteasome inhibitor suppresses tumor growth via targeting both 19S proteasome deubiquitinases and 20S proteolytic peptidases. Sci Rep. 2014a;4:5240. doi: 10.1038/srep05240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu NN, Li XF, Huang HB, Zhao C, Liao SY, Yang CS, Liu ST, Song WB, Lu XY, Lan XY, et al. Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget. 2014b;5:5453–5471. doi: 10.18632/oncotarget.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Lashuel HA, Choi S, Xing XC, Case A, Ni J, Yeh LA, Cuny GD, Stein RL, Lansbury PT. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem Biol. 2003;10:837–846. doi: 10.1016/j.chembiol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Long MJ, Gollapalli DR, Hedstrom L. Inhibitor mediated protein degradation. Chem Biol. 2012;19:629–637. doi: 10.1016/j.chembiol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Liu T, Jiao Q, Ji J, Tao M, Liu Y, You Q, Jiang Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018;146:251–259. doi: 10.1016/j.ejmech.2018.01.063. [DOI] [PubMed] [Google Scholar]
- Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14:417. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry H, Hsieh G, Buhrlage SJ, Huang M, Park E, Cuny GD, Galinsky I, Stone RM, Gray NS, D’Andrea AD, et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther. 2013;12:2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, Raina K, Holley SA, Crews CM. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat Chem Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138–144. doi: 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- Ohoka N, Nagai K, Hattori T, Okuhira K, Shibata N, Cho N, Naito M. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20:1242–1253. doi: 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- Raina K, Lu J, Qian YM, Altieri M, Gordon D, Rossi AMK, Wang J, Chen X, Dong HQ, Siu K, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. P Natl Acad Sci USA. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, Harpon J, Battaglia V, Vivat V, Sippl W, et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem Biol. 2012;19:467–477. doi: 10.1016/j.chembiol.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- Robb CM, Contreras JI, Kour S, Taylor MA, Abid M, Sonawane YA, Zahid M, Murry DJ, Natarajan A, Rana S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC) Chem Commun (Camb) 2017;53:7577–7580. doi: 10.1039/c7cc03879h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, Sakamoto KM. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Sacco JJ, Coulson JM, Clague MJ, Urbe S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life. 2010;62:140–157. doi: 10.1002/iub.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. P Natl Acad Sci USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salami J, Crews CM. Waste disposal-An attractive strategy for cancer therapy. Science. 2017;355:1163–1167. doi: 10.1126/science.aam7340. [DOI] [PubMed] [Google Scholar]
- Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D, Olah J, Ovadi J, Sippl W, Jung M. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals) J Med Chem. 2018;61:482–491. doi: 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth JS, Jr, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Spicka I, Oriol A, Hajek R, Rosinol L, Siegel DS, Mihaylov GG, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142–152. doi: 10.1056/NEJMoa1411321. [DOI] [PubMed] [Google Scholar]
- Tian X, Isamiddinova NS, Peroutka RJ, Goldenberg SJ, Mattern MR, Nicholson B, Leach C. Characterization of selective ubiquitin and ubiquitin-like protease inhibitors using a fluorescence-based multiplex assay format. Assay Drug Dev Technol. 2011;9:165–173. doi: 10.1089/adt.2010.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoshige S, Naito M, Hashimoto Y, Ishikawa M. Degradation of HaloTag-fused nuclear proteins using bestatin-HaloTag ligand hybrid molecules. Org Biomol Chem. 2015;13:9746–9750. doi: 10.1039/c5ob01395j. [DOI] [PubMed] [Google Scholar]
- Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL, Tonkin LM, Townsend EC, Buker SM, Lancia DR, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550:481–486. doi: 10.1038/nature24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Wang X, D’Arcy P, Caulfield TR, Paulus A, Chitta K, Mohanty C, Gullbo J, Chanan-Khan A, Linder S. Synthesis and evaluation of derivatives of the proteasome deubiquitinase inhibitor b-AP15. Chem Biol Drug Des. 2015;86:1036–1048. doi: 10.1111/cbdd.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng S, Fan J, Li X, Wen Q, Luo N. New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem Pharmacol. 2016a;116:200–209. doi: 10.1016/j.bcp.2016.07.017. [DOI] [PubMed] [Google Scholar]
- Wang X, Mazurkiewicz M, Hillert EK, Olofsson MH, Pierrou S, Hillertz P, Gullbo J, Selvaraju K, Paulus A, Akhtar S, et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci Rep-Uk. 2016b;6:26979. doi: 10.1038/srep26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weathington NM, Mallampalli RK. Emerging therapies targeting the ubiquitin proteasome system in cancer. J Clin Invest. 2014;124:6–12. doi: 10.1172/JCI71602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock J, Wu J, Cao P, Kingsbury WD, McDermott JL, Kodrasov MP, McKelvey DM, Suresh Kumar KG, Goldenberg SJ, Mattern MR, et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP47. ACS Med Chem Lett. 2012;3:789–792. doi: 10.1021/ml200276j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11:1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Lim SM, Westover KD, Dodge ME, Ercan D, Ficarro SB, Udayakumar D, Gurbani D, Tae HS, Riddle SM, et al. Pharmacological targeting of the pseudokinase Her3. Nat Chem Biol. 2014;10:1006–1012. doi: 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Song Y, Xie H, Wu H, Wu YT, Leisten ED, Tang W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg Med Chem Lett. 2018;28:2493–2497. doi: 10.1016/j.bmcl.2018.05.057. [DOI] [PubMed] [Google Scholar]
- Yue W, Chen Z, Liu H, Yan C, Chen M, Feng D, Yan C, Wu H, Du L, Wang Y, et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014;24:482–496. doi: 10.1038/cr.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. Acs Chem Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Han XR, Yang X, Jiang B, Liu J, Xiong Y, Jin J. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK) Eur J Med Chem. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang CY, Zhao Y, et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J Med Chem. 2018;61:462–481. doi: 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P. Targeted protein degradation. Curr Opin Chem Biol. 2005;9:51–55. doi: 10.1016/j.cbpa.2004.10.012. [DOI] [PubMed] [Google Scholar]