

Abstract
Potato (Solanum tuberosum L.) is the third most important food crop, and breeding drought-tolerant varieties is vital research goal. However, detailed molecular mechanisms in response to drought stress in potatoes are not well known. In this study, we developed EMS-mutagenized potatoes that showed significant tolerance to drought stress compared to the wild-type (WT) ‘Desiree’ cultivar. In addition, changes to transcripts as a result of drought stress in WT and drought-tolerant (DR) plants were investigated by de novo assembly using the Illumina platform. One-week-old WT and DR plants were treated with −1.8 Mpa polyethylene glycol-8000, and total RNA was prepared from plants harvested at 0, 6, 12, 24, and 48 h for subsequent RNA sequencing. In total, 61,100 transcripts and 5,118 differentially expressed genes (DEGs) displaying up- or down-regulation were identified in pairwise comparisons of WT and DR plants following drought conditions. Transcriptome profiling showed the number of DEGs with up-regulation and down-regulation at 909, 977, 1181, 1225 and 826 between WT and DR plants at 0, 6, 12, 24, and 48 h, respectively. Results of KEGG enrichment showed that the drought tolerance mechanism of the DR plant can mainly be explained by two aspects, the ‘photosynthetic-antenna protein’ and ‘protein processing of the endoplasmic reticulum’. We also divided eight expression patterns in four pairwise comparisons of DR plants (DR0 vs DR6, DR12, DR24, DR48) under PEG treatment. Our comprehensive transcriptome data will further enhance our understanding of the mechanisms regulating drought tolerance in tetraploid potato cultivars.
Keywords: differentially expressed genes, drought stress, ethyl-methanesulfonate-induced mutation, potato breeding, RNA-Seq
INTRODUCTION
Potato (Solanum tuberosum L.), which is the third most important food crop after rice and wheat in terms of human consumption, is cultivated throughout the world in temperate and continental areas including North China, North America, and the highlands of South America (https://cipotato.org/crops/potato). This plant is generally considered to be rather sensitive to drought stress due to its sparse and shallow root system of about 50–100 cm depth (Dalla Costa et al., 1997; Vos and Groenwold, 1986). Varieties with diverse traits have been developed through traditional breeding, most of which incorporate traits such as, disease resistance, increasing productivity and quality, and the control of maturity. Although developing new cultivars exhibiting drought tolerance is a key aim of potato breeders, the development of drought-tolerant cultivars by conventional breeding strategies is a very complicated process which takes an extended period of time. Traditionally, potato breeding for crop improvement has relied on phenotypic selection with a limited emphasis on genotypic isolation (Hirsch et al., 2014).
The production of mutants, either naturally or artificially, has been considered a very useful method to develop cultivars with new desired traits within defined germplasm pools (Pathirana, 2012; Shu et al., 2012). In potatoes, which are difficult to breed by conventional methods, such mutant production is an indispensable part of gene discovery for molecular breeding as well as breeding of new cultivars (Fischer et al., 2008; Muth et al., 2008). Treatment of ethyl methanesulfonate (EMS), an alkylating chemical mutagen, has successfully assisted the development of new cultivars in both seed and vegetatively propagated plants (Parry et al., 2009). This mutagen is easy to use without specialized equipment and can provide a very high random point mutation frequency (Sikora et al., 2011). Many cases of the development of varieties by EMS mutagenesis have been reported, but potatoes are not relatively common (Behera et al., 2012; Jabeen and Mirza, 2004; Luan et al., 2007; Taheri et al., 2017).
Next-generation sequencing (NGS) platforms that are relatively rapid and cost effective, such as Roche/454, ABI SOLiD, Illumina/Solexa, and the Helicos Genetic Analysis System, have made it possible to complete functional genomics studies to improve crop genetics at the whole-genome level (Cloonan et al., 2008; Vera et al., 2008; Wang et al., 2009). This NGS technology has been used for RNA sequencing and de novo transcriptome assembly of non-model organisms with or without a reference genome (Haas and Zody, 2010; Li and Dewey, 2011), particularly RNA sequencing of minor crops such as sorghum (Sorghum bicolor) (Mizuno et al., 2012), sunflower (Helianthus annuus) (Livaja et al., 2013), Jerusalem artichoke (Helianthus tuberosus) (Jung et al., 2014), red clover (Trifolium pretense) (Yates et al., 2014), and Camelina sativa (Mudalkar et al., 2014) have been reported. In potatoes, the whole-genome has been sequenced via the Potato Genome Sequence Consortium (PGSC) using the S. tuberosum group Phureja clone DM1-3 516 R 44 (doubled monoploid) and the S. tuberosum group Tuberosum RH89-039-16 (heterozygous diploid), released in 2011 (Xu et al., 2011). This information is provided to biologists, breeders, and geneticists as reference genomes (www.solgenomics.org). Massa et al (2011; 2013) has reported a reference for the potato transcriptome using the reference accession (Massa et al., 2011; 2013), and since then, many transcriptomes corresponding to specific conditions have been published (Gálvez et al., 2016; Gong et al., 2015). Nevertheless, since most potato cultivars are autotetraploid, there is a limit to utilize the completed genome sequencing for breeding.
In the current study, we developed EMS-mutagenized potatoes that showed significant tolerance to drought stress compared to the wild-type (WT) ‘Desiree’ cultivar. The RNA-seq approach was applied to generate time-course transcript expression profiles 48 h after PEG treatment. The sequences are analyzed by both de novo assembly of transcripts and alignment to the published diploid potato genome to identify differentially expressed genes (DEGs) related to drought tolerance at the initial stage of stress. In total, 304,976,315 high-quality reads were retained and 61,100 transcripts and 5,118 DEGs showing up- or down-regulation under drought conditions were identified. Those DEGs were then annotated and assigned putative functions, classifications or pathways by alignment with public databases. We considered this approach to be very meaningful for gene expression studies on the initial response of potato plants under drought stress, using this tolerant mutant. The comprehensive transcriptome data obtained from this study may be useful for further characterizing drought stress responses in potatoes and may enable the breeding of new drought-tolerant potato cultivars.
MATERIALS AND METHODS
Generation of potato mutants
Nodal cuttings with one leaf node were harvested from 4-week-old in vitro ‘Desiree’ plantlets, a representative tetraploid commercial potato cultivar, and cultured on a Murashige and Skoog (MS; (Murashige and Skoog, 1962)) basal medium containing 9% sucrose and 8 g l−1 agar in a growth room (17 ± 1°C, 24 h dark) to produce microtubers, which were harvested after 3 months. Media used in this study were autoclaved at 121°C for 20 min and the pH was adjusted to 6.0 before autoclaving. All macro and microelements for tissue culture media, sucrose, and agar were purchased at Duchefa (Haaelem, Netherlands), and other chemicals were purchased at Sigma-Aldrich (USA). To induce mutations, 1,000 microtubers were submerged in a freshly prepared 0.8% (v/v) EMS for 18 h at room temperature under a vacuum hood (following the safety instructions of the manufacturer’s Material Safety Data Sheet), then rinsed in water and kept at room temperature for germination.
Characterization of potato mutants
Mutagenized microtubers were grown to plants in pots (30 × 30 × 21 cm3) containing bed soil in the greenhouse. The water content of each pot was measured three times a week, and the water lost was supplemented to keep equivalent levels according to treatment requirements. After six weeks, plant height and phenotypes were measured before water was withheld from the plants for four weeks to screen for drought tolerance. Plants were re-watered, and tubers were harvested at 100 days total cultivation. WT plants and tubers were cultivated and harvested at normal conditions as a negative control. Germinated sprouts from M1 tubers were surface sterilized with 10% NaOCl and transferred into in vitro culture systems on a MS basal medium. One-week-old in vitro shoots were treated with −1.8 Mpa polyethylene glycol (PEG)-8000 in MS medium for two days. All experiments were performed in three replicates.
Leaf and root samples were fixed in 2.5% paraformaldehyde-glutaraldehyde mixture buffered with 0.1 M phosphate (pH 7.2) for 2 h, post-fixed in 1% osmium tetroxide in the same buffer for 1 h, dehydrated in graded ethanol, and substituted by isoamyl acetate. They were then dried at the critical point in CO2. Finally, the samples were sputtered with gold in a sputter coater (SC502, Polaron) and observed using a scanning electron microscope, FEI Quanta 250 FEG installed in KRIBB.
Total RNA isolation
PEG-treated in vitro DR and WT plants were sampled at 0, 6, 12, 24, and 48 h post treatment for RNA sequencing. Total RNA was extracted from samples using the TRIzol Reagent (Invitrogen, USA), and were then treated with DNase I (Fermentas, USA) according to the manufacturer’s instructions. Three biological replicates for each treatment were used to prepare pooled total RNA samples. The RNA quality was assessed according to optical density ratios (260 nm:280 nm and 260 nm:230 nm) determined using a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, USA).
cDNA library construction and transcriptome sequencing
We prepared RNA-Seq paired-end libraries for the pooled total RNA of each treatment using the Illumina TruSeq RNA Sample Preparation Kit v2 (Illumina, USA). The mRNA was purified using poly (A) selection. The RNA was chemically fragmented and synthesized into single-stranded cDNA using random hexamer primers. The second-strand cDNA was synthesized to create double-stranded blunt-ended cDNA fragments. The cDNA fragments were extended by adenylating the 3′ blunt-end to enable the ligation of sequencing adapters. The adapter-containing cDNA fragments (approximately 200 bp) underwent agarose gel electrophoresis, and fragments were isolated based on size. The cDNA fragments were amplified by PCR using adapter-specific primers. The cDNA library was quantified using the KAPA Library Quantification Kit (Kapa Biosystems, USA) according to the manufacturer’s instructions. The library was used for high-throughput sequencing with the Illumina NextSeq platform, as well as a paired-end sequencing system, to generate raw sequence data.
De novo assembly and sequence analysis
Raw read data were acquired by image analysis and base calling using the Illumina Pipeline software. Raw reads were quality checked with the quality assessment software FastQC (v0.11.5) (Andrews, 2010). Raw reads were trimmed, and clean reads were obtained by removing low quality reads (Phred quality score: Q ≥ 30 for all bases), and adapter sequences were eliminated using Skewer (v0.2.2) (Jiang et al., 2014). De novo sequences were assembled using Velvet (v1.2.07) (Zerbino and Birney, 2008) and Oases (v0.2.08) (Schulz et al., 2012) based on the de Bruijn graph algorithm, as described in Kim et al (Kim et al., 2016). Velvet-Oases were used to assemble short read sequences, and they obtained a higher quality assembly using reads with long N50 lengths. The optimization of program parameters to the N50 length and the trimming of low-quality reads could improve the assembly output significantly (Garg et al., 2011). Velvet was used to assemble contigs from the clean reads with various k-mer lengths (i.e., 51, 53, 55, 57, 59, 61, 63, 65, 69, 71, 75, 79, 81, 85, and 89). The contigs were assembled into transcripts for each k-mer using Oases. After transcripts were assembled for the 57 and 59 k-mer lengths (i.e., optimal k-mer lengths), a final assembly was completed for the hash length (BioProject Accession number: PRJNA476484). Putative transcripts assembled from the total reads of each mRNA sample were confirmed with sequences available in the Phytozome database (http://www.phytozome.net/) using the BLASTx algorithm (E-value ≤ 1E-10).
Functional annotations of differentially expressed genes
Paired-end clean reads were aligned with the assembled loci and expression was quantified by counting the number of mapped clean reads using Kallisto (v0.43.1), which is based on the novel idea of pseudoalignment for rapidly determining the compatibility of reads with targets (Bray et al., 2016). Genes whose expression levels were affected during the PEG-treatment period were identified using NOISeq (v2.16.0) for normalization (TMM normalization of the TPM from Kallisto output) and differentially expressed gene (DEG) analysis (Tarazona et al., 2015). The probability ≥ 0.95 and log2 values ≥ 2 were used to calculate the significance of the altered expression levels with NOIseq.
Functional annotations were completed using the BLASTP algorithm (E-value ≤ 1.0E-10). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Moriya et al., 2007) were analyzed using DAVID (http://david.abcc.ncifcrf.gov/tools.jsp) (Huang et al., 2008). The enrichment p-values were derived from an EASE score, a modified Fisher’s exact p-value, for gene-enrichment analysis. The group enrichment score was calculated from the geometric mean (in −log scale) of all the enrichment p-values. Visualization of the analysis results was shown using NOIseq (hierarchical clustering of samples and various plots of gene expression profile) and In-house R script (heatmap of clustering).
Validation of differentially expressed genes by quantitative real-time PCR
The relative mRNA expression levels of DEGs in drought conditions were analyzed by qRT-PCR using the SYBR Green Master Mix (Enzynomics Co., Korea) and the CFX Connect Real-Time PCR System (Bio-Rad, USA) according to the manufacturer’s instructions. Total RNA (1 μg) was used to synthesize cDNA with the PrimeScript RT Reagent Kit (Takara, Shiga Japan). The qRT-PCR was completed using 1 μl cDNA, gene specific primers (Supplementary Table S1), and the following program: 95°C for 15 min; 45 cycles of 95°C for 10 s, 55°C for 15 s, and 72°C for 30 s. Relative expression levels were determined by normalizing the data for the target transcripts against the Actin transcript data (GenBank accession number: XM_006345899) according to a normalized expression method. Three independent biological replicates for each sample and three technical replicates for each biological replicate were analyzed.
RESULTS
Isolation and characterization of the drought-tolerant potato mutants
The 344th M1 plants of the 1,000 EMS-mutagenized population survived under the no watering condition for 4 wks. This surviving plant was healthy with no drought-related symptoms (Fig. 1A) and tubers were harvested as normal. To verify the plants response to drought stress in detail, in vitro plants of WT and the 344th plant were cultured in the MS basal medium containing PEG as an osmotic pressure agent. After culturing for 10 days, growth rates including the stems and roots of both types of plant were not different under the normal condition (Fig. 1B). The growth rate of the WT plants began to decrease in the drought stress condition. Additionally, root growth was not observed, while chlorotic leaf lesions developed and eventually became necrotic (Fig. 1C). In contrast, the 344th plant exhibited normal leaf and shoot growth, and root growth was especially vigorous. This plant was regarded as drought tolerant, and we referred to it as the DR plant. Interestingly, more stomata were closed in the DR plant than in the wild-type plant under the same culture conditions, otherwise no significant differences in the root surface in both types of plant were observed (Figs. 1D–1E).
Fig. 1. Comparison of WT and DR plants to drought stress.
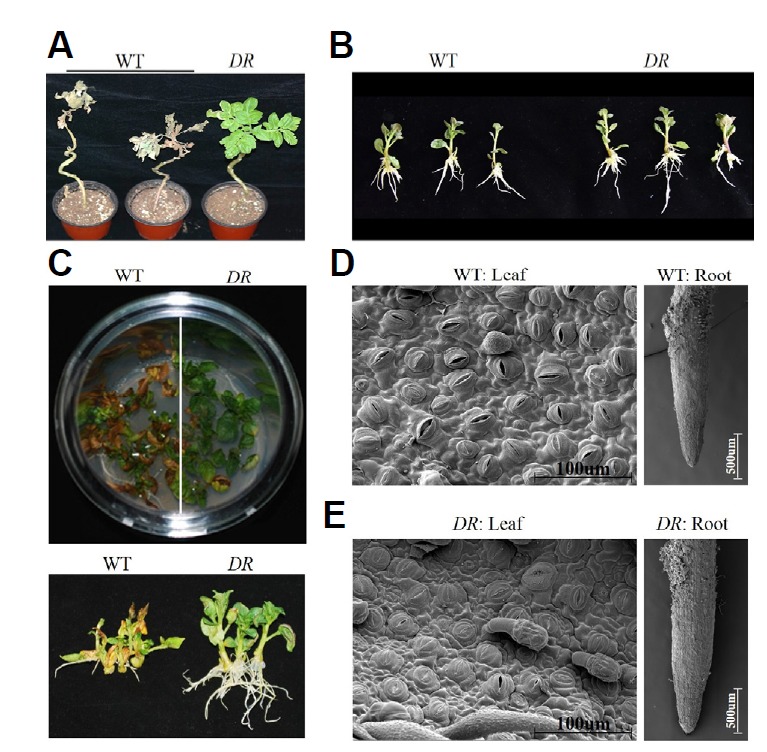
(A) a pot test and (B), (C) in vitro test. (A) Six-week-old plants were not watered for 4 weeks to screen for drought resistance. (B) In vitro plants of WT and DR were grown in MS medium and show normal root growth. (C) In vitro plants of WT and DR were grown in the 1.8 Mpa PEG-8000 medium. The bottom panel shows the shoot morphology with roots. (D), (E) Stomatal and root phenotypes of WT and DR plants.
RNA-Seq analysis and de novo transcriptome assembly
The expression profiling of WT and DR plants in response to a PEG-induced drought was performed. In total, 306,308,385 raw reads were produced from the cDNA libraries of 10 samples of these two types of potato at five time points. After trimming adapters and low-quality reads, we obtained 304,976,315 clean reads with a mean quality score of Q34.73 (Phred quality score: Q ≥ 30). Of the 10 samples, WT reads were typically mapped to the potato reference sequence using Bowtie2 software for reference CDSs (PGSC_DM_v4.03) (Xu et al., 2011). The total mapping rate was only 51.94%, with 32.73% of the reads aligned once, and 19.21% of the reads aligned more than once (Supplementary Table S2). Furthermore, the reference transcript data has 38,982 predicted gene models. In this study, we identified expression of 61,100 transcripts that were difficult to cover with reference transcripts data. Thus, the remaining 48% transcripts were uncharacterized owing to their uniqueness or incompatibility with the current reference database. Clean reads were de novo assembled into the transcriptome containing 61,100 transcripts with over 200-bp transcript lengths and N50 length of 1,667 bp transcripts. The size distribution of the assembled transcripts are shown in Supplementary Fig. S1.
Analysis of differentially expressed genes in response to drought stress
To identify the differentially expressed gene levels between WT and DR under drought stress, clean reads were mapped to the assembled potato transcriptome sequences using Kallisto. The total average mapping rate was 85.09% with 258,649,240 read pairs in the 306,308,385 read pairs of the WT and DR plants. Transcriptome profiling revealed a total number of 909, 977, 1181, 1225, and 826 DEGs (absolute log2 fold change ≥ 2) between WT and DR plants at 0, 6, 12, 24, and 48 h, respectively. Venn diagram data is expressed using DEGs between wild-type and mutant corresponding to all conditions including normal condition (Fig. 2A). To understand the mutant-specific expression profiles, differences in transcript expression were determined by pairwise comparisons between WT and DR plants without drought stress (at 0 h), a total of 909 DEGs were found to be significantly (absolute log2 fold change ≥ 2, probability ≥ 0.95) differentially expressed with 299 up-regulated and 610 down-regulated DEGs (Fig. 2B). An increase in the total number of DEGs between WT and DR plants was observed within one day of the drought stress condition (at 6–24 h). A total of 977 DEGs (518 up-regulated and 459 down-regulated) were detected after 6 h of PEG treatment in WT and DR plants (WT6 vs. DR6), and 1181 DEGs (848 up-regulated and 333 down-regulated) were detected after 12 h (WT12 vs. DR12). Interestingly, the largest number of up-regulated DEGs and the least number of down-regulated DEGs were observed at 12 h of drought stress. To compare gene expression of each pairwise comparison, log10 (FPKM) values of DEGs are shown as scatter plots (Supplementary Fig. S2A). For these plots, ‘significant’ (marked as red color spots) represents a probability ≥ 0.95. The total number of DEGs in WT0 vs. DR0 was similar to other pairwise comparisons, but there were a few genes with significantly high expression levels. In addition, the highest expressed gene of WT0 vs. DR0 was found to be unknown, expressed significantly higher (7.958 log2 fold change) than other DEGs. The gene expression levels of WT and DR plants at 6 and 24 h after PEG treatment were significantly higher than other conditions.
Fig. 2. Comparison of the assembled transcripts.
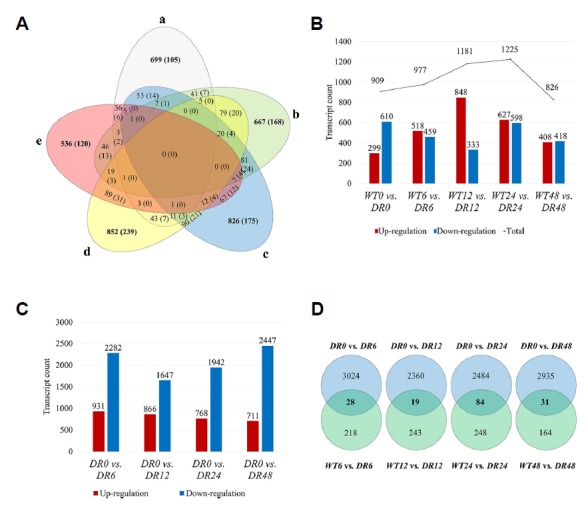
(A) Venn diagram of DEGs mapping results. Areas of overlap show the number of DEGs successfully mapped to all overlapping databases. Results are shown only for WT0 vs. DR0 (a), WT6 vs. DR6 (b), WT12 vs. DR12 (c), WT24 vs. DR24 (d), and WT48 vs. DR48 (e) DEGs. The numbers in parentheses are annotated DEG against phytozome database. Numbers of sequence matches at absolute log2 fold change ≥ 2, probability ≥ 0.95 are shown. (B) Pairwise comparison of up-regulated and down-regulated DEGs in WT and DR plants in the time-course under PEG treatment. (C) Number of up-regulated and down-regulated DEGs following PEG treatment in DR plant. (D) Venn diagram of number of DEGs with combined analysis of results from WT vs. Mutant and DR(drought)_control vs DR.
To understand the transcriptional changes of DR plants in response to drought stress, tolerant-specific expression genes were determined by comparing the PEG-treated to non-treated DR plant (at 0 h). Transcriptome profiling revealed a total number of 3213, 2513, 2710, and 3158 DEGs in the drought tolerant DR plants at 6, 12, 24, and 48 h, respectively (Fig. 2C). We found that the number of DEGs was the highest in DR plants at 6 h (3213 DEGs; 931 up-regulated and 2282 down-regulated), compared to other time course DEGs. In addition, the highest number of up-regulated DEGs occurred at 6 h and gradually decreased until 48 h. A large amount of down-regulated DEGs were observed at 6 h, but the number of DEGs significantly dropped at 12 h (2513 DEGs; 866 up-regulated and 1647 down-regulated). Conversely, a gradual increase in DEGs was observed after 12 h and the highest number of DEGs was observed at 48 h (3158 DEGs; 711 up-regulated and 2447 down-regulated). Overall, the number of down-regulated genes in DR plants was greater than the number of up-regulated genes during drought stress, but the level of each gene expression was significantly higher in up-regulated genes (Supplementary Fig. S2B). The combined analysis were carried out to show the genes involved in the DEGs of DR plants under each drought conditions against identified DEGs between wild-type and DR plant in the same conditions (Fig. 2D). According to the drought conditions, 28, 19, 84, and 31 DEGs were matched between DR_control (DR0) vs. DR and wild-type vs. mutant, respectively (Supplementary Table S3).
Gene Ontology classification analysis of differentially expressed genes
To investigate the potential DEGs activated in response to drought stress in the tolerant plant compared to the wild-type, GO classification was performed on DEGs identified in each pairwise comparison (Fig. 3 and Supplementary Fig. S3). GO information in the assembled potato transcripts was generated by DAVID with a cut-off probability ≥ 0.95 and absolute log2 fold change ≥ 2. In the GO enrichment analysis of total DEGs including up-regulated and down-regulated, three GO categories (Defense response; GO:0006952 in Biological Process, Integral component of membrane; GO: 0016021 in Cellular Component, ADP binding; GO:0043531 in Molecular Function) were found to be related to drought tolerance during all drought conditions (Fig. 3A). Interestingly, these three GO categories included over 75% of up-regulated DEGs in the early hours (0–12 h), and over 50% down-regulated DEGs in the late hours (24–48 h) (Supplementary Fig. S4). A total of 9 GO categories with up-regulated and down-regulated DEGs were identified in two plant types in the normal condition (WT0 vs. DR0). Of these, the four GO categories (ATP binding, ADP binding, integral component of membrane, and defense response) were found to be rich not only among up-regulated DEGs, but also among down-regulated DEGs. However, there are no significant enriched categories (p-value ≤ 0.05) in these 9 GO categories between WT and DR plants under normal conditions (Supplementary Fig. S3A). On the other hand, significantly up-regulated DEGs were observed in the two GO categories (‘Terpene synthase activity; GO:0010333’ and ADP binding in Molecular Function) in DR plants at 6 h in the drought condition. The biggest change in DR plants was observed in GO enriched analysis at 24 h (WT24 vs. DR24) with the highest amount of DEGs. In the WT24 vs. DR24 pairwise comparison, a total of 45 GO categories with 15 significant enriched GO categories (p-value ≤ 0.05) were identified compared to other pairwise comparisons (Supplementary Fig. S3D). The water response (GO:0009415) of the GO biological process containing dehydrin (LOC102599708), a subfamily of LEA protein known to be involved in drought stress, showed a significant increase (5.387 log2 fold change, 1.00 probability) in DR24 compared to WT24 (Fig. 3B). Regarding plant photosynthesis, there were significant differences between WT and DR plants after 24 h of drought stress. A category of Molecular Function (Chlorophyll binding; GO:0016168), three categories of Cellular Component (Chloroplast thylakoid membrane; GO:0009535, Photosys-tem II; GO:0009523, and Photosystem I; GO:0009522), and two categories of Biological Process (Protein chromophore linkage; GO:0018298, and Photosynthesis, light harvesting; GO:0009765) had a highly significant association (−log10 (p-value) ≥ 10) in the down-regulated DEGs (Fig. 3B). Chlorophyll a–b binding proteins (LOC102603980, LOC102586284, LOC102583646, and LOC102589865 were expressed at −3.532, −2.618, −2.504, and −2.357 log2 fold change, respectively) enriched in the photosynthetic GO categories including photosystem I, photosystem II, photosynthesis, light harvesting, protein~chromophore linkage, and chlorophyll binding were found to have low expression in the DR plant at 24 h.
Fig. 3. Gene Ontology classification of differentially expressed genes.
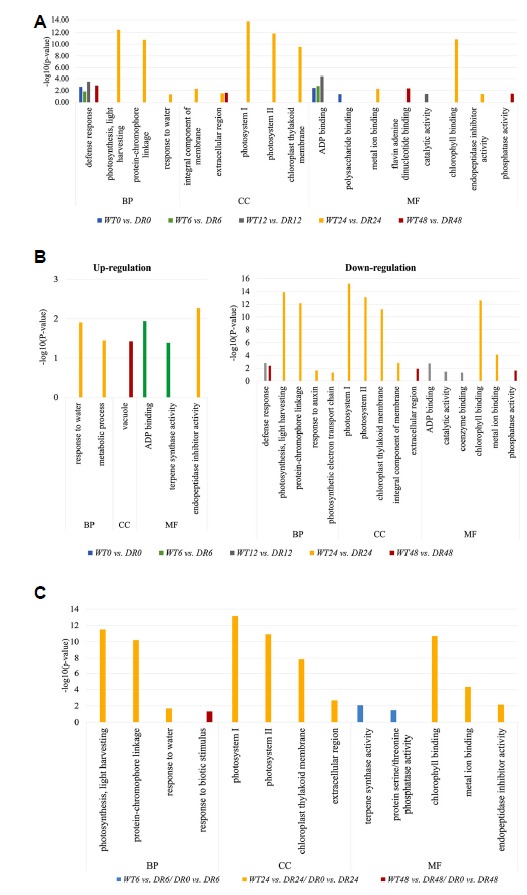
These figures show the distribution of GO terms exhibiting significant differences (p-value ≤ 0.05 and absolute log2 fold change ≥ 2). GO analysis of the total DEGs (A), up-regulated and down-regulated DEGs (B) by time-course of WT and DR plants in drought stress in three main classes (‘Biological Process’, ‘Cellular Component’, and ‘Molecular Function’). (C) The significant biological process, cell compartment and molecular function GO terms are shown in each combined analysis “DR_control (DR0) vs. DR and WT vs. DR.
We also performed GO classification on DEG identified in each combined analysis “DR_control (DR0) vs. DR and WT vs. DR” (Fig. 3C). WT6 vs. DR6/DR0 vs. DR6 containing 28 DEGs was enriched in “terpene synthase activity” and “protein serine”, and only one category “response to biotic stimulus” was enriched in WT48 vs. DR48/DR0 vs. DR48 containing 31 DEGs. In WT24 vs. DR24/DR0 vs. DR24 containing 84 DEGs, it was enriched in 10 categories. In particular, as in the comparison of WT24 vs. DR24, the photosynthesis-related GO categories showed considerably higher enrichment.
Metabolic pathway analysis by KEGG
When plants are affected by drought stress, many genes are expressed or inhibited to protect plants through associated pathways. To identify the biological pathway of DEG associated with drought, all DEGs were compared against the KEGG using DAVID with a cut-off probability ≥ 0.95 and absolute log2 fold change ≥ 2 (Fig. 4 and Supplementary Fig. S5). In total, all DEGs were enriched in 33 different functional pathway categories with 20 categories of up-regulation and 18 categories of down-regulation in all pairwise comparisons. Of these, 12 different functional pathway categories are significantly enriched categories (p-value ≤ 0.05). In particular, the most important biochemical metabolic pathway was identified by the KEGG pathway. KEGG pathway analysis indicated that ‘Photosynthesis-antenna protein, sot00196’ was enriched most significantly (−log10 (p-value): 17.82) in WT24 vs. DR24 (Fig. 4B). Next, ‘protein processing in endoplasmic reticulum, sot04141’ showed a significant association (−log10 (p-value): 8.86) in WT12 vs. DR12 (Fig. 4A). These two different functional pathway categories were the most relevant compared to the other categories of WT and DR plants under PEG treatment, but WT0 vs. DR0 had no significant enrichment category, as with the GO analysis. Interestingly, two different functional pathways (‘Biosynthesis of secondary metabolites’ and ‘Metabolic pathways’) were found to be enriched not only among up-regulated DEGs, but also among down-regulated DEGs of all PEG treatment groups. Results of KEGG enrichment in this study indicate that the mechanism for drought tolerance in the DR plant can be mainly explained in two aspects, photosynthesis-antenna protein and protein processing in the endoplasmic reticulum.
Fig. 4. KEGG ontology (KO) enrichment analysis of DEGs related to metabolism pathway.

All DEGs were enriched in different functional pathway categories with up-regulated DEGs (A) and down-regulated DEGs (B) in all pairwise comparison of WT and DR plants under drought stress. (C) 162 DEGs from combined analysis of DR_control (DR0) vs. DR and WT vs. DR were enriched in 2 KEGG pathway categories. p-value ≤ 0.05 and absolute log2 fold change ≥ 2 were used as thresholds to select significant KEGG pathways.
In the combined analysis, KEGG pathway analysis of the DEGs of DR_control (DR0) vs. DR and WT vs. DR indicated that only “Photosynthesis–antenna proteins” and “Metabolic pathways” were enriched in WT24 vs. DR24/DR0 vs. DR24 (Fig. 4C). In particular, the “Photosynthesis-antenna proteins” pathway were significantly related in the drought condition. This result was similar to the down-regulation result of the KEGG analysis of WT24 vs. DR24, indicating that the analysis of DEGs between WT and DR in each drought conditions involves the results of a combined analysis with DR_control vs. DR under each drought conditions.
To investigate the functional significance of 84 DEGs of WT24 vs. DR24/DR0 vs. DR24, we focused on playing a role in the well-characterized pathway of photosynthesis using the MapMan software (Fig. 5A) (Thimm et al., 2004). Among these DEGs, photosynthesis was affected by drought stress, with down-regulated 10 DEGs involved in the light reaction, down-regulated 2 DEGs in the Calvin cycle, and down-regulated 1 DEG in the photorespiration under drought condition after 24 h. In the KEGG map of the “photosynthesis-antenna protein” pathway, down-regulated 10 DEGs corresponded to the Light-harvesting chlorophyll protein complex (LHC) (Fig. 5B). Among these 10 DEGs, 3 DEGs corresponded to light-harvesting complex I chlorophyll a/b binding protein 2-4 (Lhca2-4) of light-harvesting complex I, and 7 DEGs corresponded to light-harvesting complex II chlorophyll a/b binding protein 1-3 (Lhcb1-3) of the light-harvesting complex II. The KEGG analyses of WT24 vs. DR24 and combined analysis (WT24 vs. DR24/DR0 vs. DR24) showed the same results that expression of these LHC genes decreased.
Fig. 5. MapMan analysis of photosynthesis and KEGG map of the photosynthesis-antenna protein between WT24 vs. DR24 and DR0 vs. DR6.
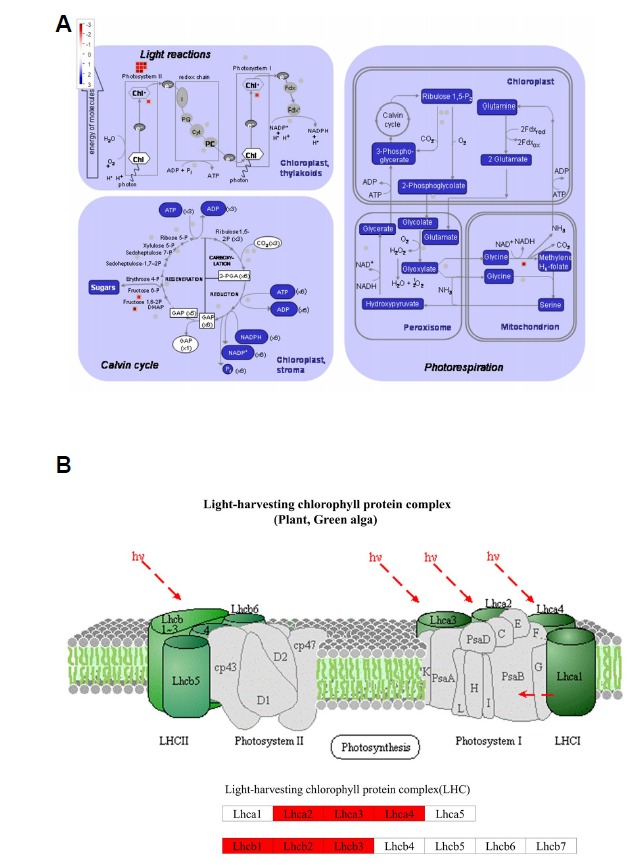
(A) Light reactions, Calvin cycle, and Photorespiration of Photosynthesis pathway in MapMan. Red boxes indicate down-regulation. (B) The pathway of photosynthesis-antenna protein in KEGG map. Red boxes indicate that the corresponding DEGs were down-regulated in DR24.
DEG clustering analysis of DR plants and validation by qRT-PCR
To identify different expression patterns of DR plants under drought conditions, four pairwise comparisons (DR0 vs DR6, DR12, DR24, DR48) were performed using five libraries. Then, 4,395 DEGs identified in these pairwise comparisons were grouped using a clustering algorithm with Pearson’s correlation, and a heatmap and eight clusters were identified (Fig. 6). Cluster 1 and 2 represent up-regulated clusters, which contain 519 and 566 genes, respectively. Cluster 3 included 247 genes which showed up-regulation until 24 h but was not up-regulated in DR48. Cluster 4 included 81 genes, which were only down-regulated in DR6, whereas Cluster 7 included 60 genes that were up-regulated in DR6. Cluster 5 and 6 represented down-regulated clusters, which contained 1830 and 980 genes, respectively. These clusters contained over 60% of the total DEGs. Representative genes of each cluster are shown in Table 1. In this result, the families of LEA proteins, including dehydrin related to the water response mentioned in the GO analysis, showed an up-regulated expression pattern of Cluster 1. In addition, genes involved in the photosystem (e.g. Photosystem I subunit), which had a large impact on the DR plant under drought stress, showed a down-regulated expression pattern of Cluster 5. These results suggest that the genes involved in the water response and photosystem play a major role in the DR plant in the early stages of the drought environment.
Fig. 6. Heatmap, line plot, and GO enrichment from clustering analysis of DEGs.
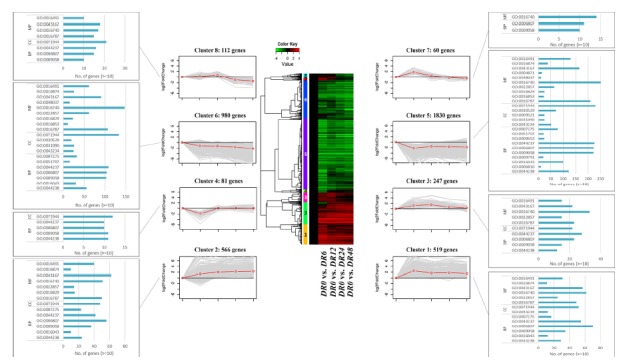
The 4,395 DEGs identified in four pairwise comparisons (DR0 vs DR6, DR12, DR24, DR48) of DR plants were grouped using a clustering algorithm with Pearson’s correlation, and a heatmap and eight clusters were identified. The GO enrichment analysis of genes in each cluster, with p-values.
Table 1.
Differentially expressed genes of DR plants under drought conditions
| TAIR ID | Gene description | Log2FoldChangea | Gene ID | Cluster | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| 0 h vs 06 h | 0 h vs 12 h | 0 h vs 24 h | 0 h vs 48 h | ||||
| AT5G52300 | CAP160 protein | 10.52 | 8.89 | 9.41 | 8.80 | PGSC0003DMP400025183 | 1 |
| AT2G42560 | Late embryogenesis abundant domain-containing protein/LEA domain-containing protein | 10.24 | 8.74 | 9.04 | 8.19 | PGSC0003DMP400034666 | 1 |
| AT4G17030 | Expansin-like B1 | 9.63 | 9.01 | 10.66 | 11.00 | Solyc06g060970.1.1 | 2 |
| AT5G09360 | Laccase 14 | 7.40 | 9.84 | 9.81 | 10.96 | PGSC0003DMP400038026 | 2 |
| AT3G48700 | Carboxyesterase 13 | 3.48 | 4.72 | 3.48 | 2.89 | PGSC0003DMP400055127 | 3 |
| AT2G38470 | WRKY DNA-binding protein 33 | 1.51 | 3.30 | 0.12 | 0.21 | Solyc09g014990.2.1 | 3 |
| AT1G01060 | Homeodomain-like superfamily protein | −5.66 | −0.03 | 0.33 | 0.03 | Solyc10g005080.2.1 | 4 |
| AT1G53540 | HSP20-like chaperones superfamily protein | −3.35 | −0.11 | 0.14 | 0.39 | PGSC0003DMP400020626 | 4 |
| AT2G18570 | UDP-Glycosyltransferase superfamily protein/Anthocyanidin 3-O-glucosyltransferase 5-like | −9.50 | −2.21 | −2.68 | −3.21 | PGSC0003DMP400007969 | 5 |
| AT5G20630 | Germin 3/Auxin-binding protein ABP 19a-like | −9.40 | −6.54 | −9.25 | −9.52 | Solyc07g041720.1 | 5 |
| AT3G01500 | Carbonic anhydrase 1 | −5.95 | −3.95 | −7.22 | −7.98 | PGSC0003DMP400000965 | 6 |
| AT5G09530 | Hydroxyproline-rich glycoprotein family protein | −4.38 | −4.74 | −4.92 | −7.77 | PGSC0003DMP400041258 | 6 |
| AT2G44800 | 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily protein | 3.54 | 0.19 | −0.27 | −0.37 | Solyc07g054930.2.1 | 7 |
| AT3G12120 | Fatty acid desaturase 2 | 3.39 | 0.99 | −1.58 | −1.24 | PGSC0003DMP400056120 | 7 |
| AT5G45650 | Subtilase family protein | −0.44 | −0.12 | −3.73 | −3.92 | Solyc02g071560.2.1 | 8 |
| AT4G16260 | Glycosyl hydrolase superfamily protein | 0.91 | −0.04 | −2.25 | −3.12 | PGSC0003DMP400018565 | 8 |
Log2 fold change of differentially expressed genes in four pairwise comparisons (DR0 vs. DR6, DR0 vs. DR12, DR0 vs. DR24, and DR0 vs DR48).
To confirm the reproducibility of the RNA-seq by qRT-PCR validations, eight DEGs were randomly selected based on the time-course clustering analysis data in the DR plant (Fig. 7). CAP160 and LEA proteins belong to Cluster 1 of the RNA-seq expression pattern, which showed the highest expression at 6 h, and also showed the highest expression in the qRT-PCR result at 6 h. In addition, expansin-like B1 and laccase 14 proteins belong to Cluster 2, which showed the highest expression pattern at 48 h, and also showed the same expression pattern in qRT-PCR. As a result, the selected candidate genes determined by qRT-PCR were closely matched with the RNA-Seq results. This suggests that the data obtained from the DEGs analysis were credible.
Fig. 7. Quantitative reverse transcription PCR validation of DEGs in WT and DR plants following drought treatment.

Comparison of Log2 fold changes of eight differentially expressed genes. Relative expression levels were determined by normalizing the DEGs data with the data for the actin reference gene. LEA, late embryogenesis abundant domain-containing protein (PGSC0003DMP400034666); Expansin, expansin-like B1 protein (Solyc06g060970.1.1); UDP-G(antho), UDP-glycosyltransferase superfamily protein/anthocyanidin 3-O-glucosyltransferase 5-like protein (PGSC0003DMP400007969); GER3(ABP), germin 3/auxin-binding protein ABP 19a-like protein (Solyc07g041720.1); Carbonic, carbonic anhydrase 1 (PGSC0003DMP400000965); HPRG, Hydroxyproline-rich glycoprotein (PGSC0003DMP400041258). DR plants were sampled at five time points, 0 h (before treatment), 6 h, 12 h, 24 h, and 48 h (after treatment).
DISCUSSION
Many studies to increase drought tolerance in potatoes have been performed (Kikuchi et al., 2015), and most of the work has been undertaken using transgenic techniques. However, the degree of tolerance to abiotic stress is not easily controlled due to the complexity of the genetic regulatory mechanism. Traditionally, mutants are not only very useful for the development of new varieties but are also a suitable method of research widely used in gene function studies. Genotypic variations of salt-tolerant potato mutants developed by irradiation were characterized using a Random Amplified Polymorphic DNA-PCR analysis (Yaycili and Alikamanoglu, 2012), and a network-based transcriptome analysis related to biological pathway modifications in irradiated rice mutants was performed based on microarray transcriptional profiling (Hwang et al., 2015). We found that DR plants obtained through EMS treatment had obvious drought tolerance under severe drought conditions, although there was no significant difference in plant growth and tuber yield compared to WT under the normal condition. In addition, transcriptome analysis was performed based on RNA-seq profiling to identify changes in the expression of drought-related genes using DR plants. Our potato (WT0) sequencing data was mapped to the published reference potato genome (DM1-3 516 R44) prior to de novo assembly, resulting in a slightly lower mapping rate. The published reference potato genome was analyzed against the doubled-monoploid potato genome, and many researchers have used this potato genome sequence since (Anithakumari et al., 2012; Gong et al., 2015; Uitdewilligen et al., 2013; Zhang et al., 2014). Using Illumina sequencing technology, we generated sequence data (61,100 transcripts) by de novo transcriptome assembly and identified drought response-related genes in DR plants. Based on the assembled transcriptome data, 909, 977, 1181, 1225, and 826 DEGs were identified in the comparison of WT and DR plants by time conditions (0, 6, 12, 24, 48 h after PEG treatment), respectively.
In the GO annotation in WT and DR plants, DEGs enrichment was observed in the photosynthesis-related category. Photosynthesis is one of the main processes affected by drought stress (Chaves, 1991). Among them, it has been reported that chlorophyll concentration, which is one of the evaluation indexes of environments, is decreased in drought stress conditions and it can be considered as a nonstomatal limiting factor (Khayatnezhad and Gholamin, 2012). Moreover, the decrease in expression of chlorophyll a–b binding proteins was consistent with the results reported previously in tomatoes under drought stress (Iovieno et al., 2016). The antenna protein present in light-harvesting chlorophyll protein complexes of photosynthetic plant act as a peripheral antenna system, allowing more efficient absorption of light energy. MapMan analysis and KEGG map analysis showed that the DR plant had decreased expression of the LHC subunits (Lhca2-4, Lhcb1-3) in drought stress than in the WT plant (Figs. 5A and 5B). In order words, the expression of chlorophyll a–b binding proteins was decreased under drought stress in DR plants, meaning that the synthesis of factors related to photosynthesis is reduced, and this reaction affects the photosynthesis system. Next, the reduction of photosynthesis limits water consumption through plant growth inhibition, thereby maintaining the water status of the plant and assisting the plant in carbon assimilation to survive under stress. Overall, the DEGs of DR plants after 24 h of PEG treatment showed significant down-regulation patterns in six enriched GO categories associated with photosynthesis-related pathways and the same down-regulation pattern in the ‘photosynthesis-antenna proteins’ functional pathway of KEGG pathway analysis. This suggests that the mutation of the photosynthetic genes mentioned in the GO analysis led to changes in the photosynthetic metabolism of the DR plant under drought stress. These results are again consistent with previous reports that photosynthesis related genes are down-regulated under drought stress conditions. These results also suggest that drought stress affects plant photosynthesis, as it occurs in other plant species including S. moorcroftiana (Li et al., 2015), C. morifolium (Xu et al., 2013), B. nivea (Liu et al., 2013b), and A. mongolicus (Liu et al., 2013a).
Inhibition of photosynthesis in a drought environment leads to a decrease in stomatal conductance, thereby improving water use efficiency. Especially, inhibition of CO2 supply to rubisco causes down-regulation of photosynthesis when plants are exposed to high light and temperature. It leads to a decrease in stomatal conductance as shown in our results (Figs. 1D–1E), which can protect plants against stress by improving the efficiency of water consumption utilization (Chaves et al., 2009). Our result which showed that the rubisco activase gene induced significant down-regulation (−2.408 log2 fold change) in DR plants after 48 h of PEG treatment again supports the above-mentioned results. On the other hand, up-regulated DEGs in DR plants were observed in the water response (GO:0009415) category of the GO biological process (Fig. 3B). The LEA proteins, which are protection factors for macromolecules showing protein kinase and phosphatase activity under osmotic stress, are related to tolerance against water deficiency, as well as osmotic, salt, and cold stresses (Sivamani et al., 2000; Zhang et al., 2014). In our results, dehydrin (LOC102599708), a subfamily of LEA protein, showed a significant increase (5.387 log2 fold change) in DR compared to WT, again providing further evidence that the LEA protein is related to drought tolerance in potatoes.
Potato multicystatin, a member of the cystatin family and a subfamily of phytocystatin, is known to act as an inhibitor of Cys-proteases involved in the degradation of storage proteins, senescence, programmed cell death, and stress signaling. Thus, cystatin plays an important role in endogenous process regulation and plant protection from various environmental stresses by regulating the activity of Cys-proteases, but the underlying mechanisms are limited (Tan et al., 2017). In addition, overexpression of cystatins in transformed Arabidopsis (A. thaliana) showed enhanced tolerance to oxidative stresses, high salt, and drought stress (Zhang et al., 2008). In the DR plant, the multicystatin (Gi number: 6671196) gene was significantly higher (4.163 log2 fold change) than in the WT plant after 48 h under drought stress.
Conversely, several drought-related genes in DR plants showed different expression patterns than previously known gene information. The polygalacturonase-1 non-catalytic subunit beta (Gi Number: 565399375) and GDSL esterase/lipase 2 (Gi Number: 565344594) showed the same expression patterns in WT and DR plants for drought stress and induced down-regulation in all time conditions of PEG treatment (data not shown). PG-1 beta is a subunit of polygalacturonase (PG), one of the hydrolytic enzymes involved in cell wall pectin degradation. Overexpression of PG-1 beta induced pectin degradation and affected the integrity and proliferation of the cell wall, thereby reducing tolerance to abiotic stress (Liu et al., 2014). The GDSL esterase/lipase 2 gene involved in plant development, morphogenesis, synthesis of secondary metabolites, and defense responses have been reported to play an important role in abiotic stress including drought in rice (Chepyshko et al., 2012). These results demonstrate that these genes had no effect on the drought tolerance of DR plants, although the PG1 beta gene may have benefited both plants. Another case is that the Defense response (GO: 0006952) category of Biological Process in the DR plant was significantly down-regulated at 12 h after drought stress. Many defense related genes including NB-LRR were significantly increased after drought and salt treatments (Li et al., 2017), which is in contradiction to our results. This inconsistency needs to be confirmed by conducting the disease tolerance test in the future, whether the drought-tolerant DR plants we obtained have weaker disease tolerance than WT plants.
In comparative analysis of gene expression in DR plants by time course under drought stress, increased expression of the LEA family protein, expansin-like B1, and laccase 14 affected the enhanced drought tolerance in the DR plant (Fig. 7 and Table 1). The LEA family protein has already been mentioned above. Expansins are a family of proteins involved in cell wall loosening and cell enlargement during plant growth and development (Sampedro and Cosgrove, 2005; Zhao et al., 2012). In Jatropha subjected to drought stress, two kinds of expansins were highly expressed during the recovery phase (Cartagena et al., 2014). Although the process of cell wall loosening and expansion involves other genes or gene complexes, the up-regulation of expansin transcripts during recovery from stress is understandable. Laccase transcript levels are enhanced in non-lignifying cell types under abiotic stresses, such as drought (Cho et al., 2014). Arabidopsis plants containing three laccase gene mutations showed no root growth under PEG-induced drought conditions (Cai et al., 2006). Here, the expansin-like B1 and laccase 14 genes were continuously highly induced during 48 h of PEG treatment. Based on the up- and down-regulated gene analyses, some genes are consistent with previous reports, but others showed distinctly different expression patterns. For example, UDP-glucuronosyl-transferase (UGT), involved in flavonol glycoside biosynthesis and defense roles against stress, was strongly up-regulated under drought stress in potato leaves (Zhang et al., 2014), but in our results, UGT was the most down-regulated gene. Germin proteins, which have enzymatic activities, including oxalate oxidase and superoxide dismutase, that are involved in drought tolerance mechanisms (Lane et al., 1993), were found in a gene expression analysis of germination in wheat (Lane et al., 1991). The germin 3 protein shows a high sequence homology to auxin binding protein 19a, and it has a role in changing the cell wall characteristics associated with normal growth. Germin 3 is repressed by drought stress in Arabidopsis leaves (Bray, 2004). However, germin3/auxin-binding protein ABP 19a-like was down-regulated here, in contrast to our expectations. Because drought tolerance is a complex trait that involves intricate molecular mechanisms related to many genes, more detailed studies focused on these inconsistencies are needed in the near future.
Supplementary data
ACKNOWLEDGMENTS
This work was supported by a grant from the KRIBB Research Initiative Program and Bio & Medical Technology Development Program of the NRF.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. [Google Scholar]
- Anithakumari A, Nataraja KN, Visser RG, van der Linden CG. Genetic dissection of drought tolerance and recovery potential by quantitative trait locus mapping of a diploid potato population. Mol Breeding. 2012;30:1413–1429. doi: 10.1007/s11032-012-9728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera M, Panigrahi J, Mishra RR, Rath SP. Analysis of EMS induced in vitro mutants of Asteracantha longifolia (L.) Nees using RAPD markers. Indian J Biotechnol. 2012;11:39–47. [Google Scholar]
- Bray EA. Genes commonly regulated by water-deficit stress in Arabidopsis thaliana. J Exp Bot. 2004;55:2331–2341. doi: 10.1093/jxb/erh270. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Cai X, Davis EJ, Ballif J, Liang M, Bushman E, Haroldsen V, Torabinejad J, Wu Y. Mutant identification and characterization of the laccase gene family in Arabidopsis. J Exp Botany. 2006;57:2563–2569. doi: 10.1093/jxb/erl022. [DOI] [PubMed] [Google Scholar]
- Cartagena JA, Seki M, Tanaka M, Yamauchi T, Sato S, Hirakawa H, Tsuge T. Gene expression profiles in jatropha under drought stress and during recovery. Plant Mol Bio Rep. 2014:1–13. [Google Scholar]
- Chaves M. Effects of water deficits on carbon assimilation. J Exp Botany. 1991;42:1–16. [Google Scholar]
- Chaves MM, Flexas J, Pinheiro C. Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann Bot. 2009;103:551–560. doi: 10.1093/aob/mcn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepyshko H, Lai C-P, Huang L-M, Liu J-H, Shaw J-F. Multifunctionality and diversity of GDSL esterase/lipase gene family in rice (Oryza sativa L. japonica) genome: new insights from bioinformatics analysis. BMC Genomics. 2012;13:309. doi: 10.1186/1471-2164-13-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Lee C, Hwang S-G, Park YC, Lim HL, Jang CS. Overexpression of the OsChI1 gene, encoding a putative laccase precursor, increases tolerance to drought and salinity stress in transgenic Arabidopsis. Gene. 2014;552:98–105. doi: 10.1016/j.gene.2014.09.018. [DOI] [PubMed] [Google Scholar]
- Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- Dalla Costa L, Delle Vedove G, Gianquinto G, Giovanardi R, Peressotti A. Yield, water use efficiency and nitrogen uptake in potato: influence of drought stress. Potato Res. 1997;40:19–34. [Google Scholar]
- Fischer L, Lipavska H, Hausman J-F, Opatrny Z. Morphological and molecular characterization of a spontaneously tuberizing potato mutant: an insight into the regulatory mechanisms of tuber induction. BMC Plant Biol. 2008;8:117. doi: 10.1186/1471-2229-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gálvez JH, Tai HH, Lagüe M, Zebarth BJ, Strömvik MV. The nitrogen responsive transcriptome in potato (Solanum tuberosum L.) reveals significant gene regulatory motifs. Sci Rep. 2016;6:26090. doi: 10.1038/srep26090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Patel RK, Tyagi AK, Jain M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18:53–63. doi: 10.1093/dnares/dsq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Zhang H, Gan X, Zhang L, Chen Y, Nie F, Shi L, Li M, Guo Z, Zhang G. Transcriptome Profiling of the Potato (Solanum tuberosum L.) Plant under Drought Stress and Water-Stimulus Conditions. PloS One. 2015;10:e0128041. doi: 10.1371/journal.pone.0128041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Zody MC. Advancing RNA-seq analysis. Nat Biotechnol. 2010;28:421–423. doi: 10.1038/nbt0510-421. [DOI] [PubMed] [Google Scholar]
- Hirsch CD, Hamilton JP, Childs KL, Cepela J, Crisovan E, Vaillancourt B, Hirsch CN, Habermann M, Neal B, Buell CR. Spud DB: A resource for mining sequences, genotypes, and phenotypes to accelerate potato breeding. The Plant Genome. 2014;7 [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hwang S-G, Kim DS, Hwang JE, Park HM, Jang CS. Identification of altered metabolic pathways of γ-irradiated rice mutant via network-based transcriptome analysis. Genetica. 2015;143:635–644. doi: 10.1007/s10709-015-9861-2. [DOI] [PubMed] [Google Scholar]
- Iovieno P, Punzo P, Guida G, Mistretta C, Van Oosten MJ, Nurcato R, Bostan H, Colantuono C, Costa A, Bagnaresi P. Transcriptomic changes drive physiological responses to progressive drought stress and rehydration in tomato. Front Plant Sci. 2016;7:371. doi: 10.3389/fpls.2016.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabeen N, Mirza B. Ethyl methane sulfonate induces morphological mutations in Capsicum annuum. Int J Agric Biol. 2004;6:340–345. [Google Scholar]
- Jiang H, Lei R, Ding S-W, Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014;15:182. doi: 10.1186/1471-2105-15-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung WY, Lee SS, Kim CW, Kim H-S, Min SR, Moon JS, Kwon S-Y, Jeon J-H, Cho HS. RNA-Seq analysis and de novo transcriptome assembly of jerusalem artichoke (Helianthus tuberosus linne) PLoS One. 2014;9:e111982. doi: 10.1371/journal.pone.0111982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayatnezhad M, Gholamin R. The effect of drought stress on leaf chlorophyll content and stress resistance in maize cultivars (Zea mays) Afr J Microbiol Res. 2012;6:2844–2848. [Google Scholar]
- Kikuchi A, Huynh HD, Endo T, Watanabe K. Review of recent transgenic studies on abiotic stress tolerance and future molecular breeding in potato. Breeding Sci. 2015;65:85. doi: 10.1270/jsbbs.65.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HA, Shin AY, Lee MS, Lee HJ, Lee HR, Ahn J, Nahm S, Jo SH, Park JM, Kwon SY. De novo transcriptome analysis of Cucumis melo L. var makuwa Mol Cells. 2016;39:141. doi: 10.14348/molcells.2016.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane B, Bernier F, Dratewka-Kos E, Shafai R, Kennedy T, Pyne C, Munro J, Vaughan T, Walters D, Altomare F. Homologies between members of the germin gene family in hexaploid wheat and similarities between these wheat germins and certain Physarum spherulins. J Biol Chem. 1991;266:10461–10469. [PubMed] [Google Scholar]
- Lane B, Dunwell JM, Ray J, Schmitt M, Cuming A. Germin, a protein marker of early plant development, is an oxalate oxidase. J Biol Chem. 1993;268:12239–12242. [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yao W, Fu Y, Li S, Guo Q. De novo assembly and discovery of genes that are involved in drought tolerance in Tibetan Sophora moorcroftiana. PloS One. 2015;10:e111054. doi: 10.1371/journal.pone.0111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang Y, Yin L, Lu J. Overexpression of pathogen-induced grapevine TIR-NB-LRR gene VaRGA1 enhances disease resistance and drought and salt tolerance in nicotiana benthamiana. Protoplasma. 2017;254:957–969. doi: 10.1007/s00709-016-1005-8. [DOI] [PubMed] [Google Scholar]
- Liu H, Ma Y, Chen N, Guo S, Liu H, Guo X, Chong K, Xu Y. Overexpression of stress-inducible OsBURP16, the β subunit of polygalacturonase 1, decreases pectin content and cell adhesion and increases abiotic stress sensitivity in rice. Plant Cell Environ. 2014;37:1144–1158. doi: 10.1111/pce.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Shi J, Lu C. Identification of stress-responsive genes in Ammopiptanthus mongolicus using ESTs generated from cold-and drought-stressed seedlings. BMC Plant Biol. 2013a;13:88. doi: 10.1186/1471-2229-13-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Zhu S, Tang Q, Yu Y, Tang S. Identification of drought stress-responsive transcription factors in ramie (Boehmeria nivea L. gaud) BMC Plant Biology. 2013b;13:130. doi: 10.1186/1471-2229-13-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livaja M, Wang Y, Wieckhorst S, Haseneyer G, Seidel M, Hahn V, Knapp SJ, Taudien S, Schön CC, Bauer E. BSTA: a targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genomics. 2013;14:628. doi: 10.1186/1471-2164-14-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan YS, Zhang J, Gao XR, An LJ. Mutation induced by ethylmethanesulphonate (EMS), in vitro screening for salt tolerance and plant regeneration of sweet potato (Ipomoea batatas L.) Plant Cell Tiss Organ Cult. 2007;88:77–81. [Google Scholar]
- Massa AN, Childs KL, Buell CR. Abiotic and biotic stress responses in Solanum tuberosum group Phureja DM1-3 516 R44 as measured through whole transcriptome sequencing. The Plant Genome. 2013;6 [Google Scholar]
- Massa AN, Childs KL, Lin H, Bryan GJ, Giuliano G, Buell CR. The transcriptome of the reference potato genome Solanum tuberosum Group Phureja clone DM1-3 516R44. Plos One. 2011;6:e26801. doi: 10.1371/journal.pone.0026801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Kawahigashi H, Kawahara Y, Kanamori H, Ogata J, Minami H, Itoh T, Matsumoto T. Global transcriptome analysis reveals distinct expression among duplicated genes during sorghum-Bipolaris sorghicola interaction. BMC Plant Biol. 2012;12:121. doi: 10.1186/1471-2229-12-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudalkar S, Golla R, Ghatty S, Reddy AR. De novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. using Illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol Biol. 2014;84:159–171. doi: 10.1007/s11103-013-0125-1. [DOI] [PubMed] [Google Scholar]
- Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plantarum. 1962;15:473–497. [Google Scholar]
- Muth J, Hartje S, Twyman RM, Hofferbert HR, Tacke E, Prüfer D. Precision breeding for novel starch variants in potato. Plant Biotechnol J. 2008;6:576–584. doi: 10.1111/j.1467-7652.2008.00340.x. [DOI] [PubMed] [Google Scholar]
- Parry MA, Madgwick PJ, Bayon C, Tearall K, Hernandez-Lopez A, Baudo M, Rakszegi M, Hamada W, Al-Yassin A, Ouabbou H. Mutation discovery for crop improvement. J Exp Bot. 2009;60:2817–2825. doi: 10.1093/jxb/erp189. [DOI] [PubMed] [Google Scholar]
- Pathirana R. Plant mutation breeding in agriculture. Plant Sci Rev. 2012;2011:107. [Google Scholar]
- Sampedro J, Cosgrove DJ. The expansin superfamily. Genome Biology. 2005;6:242. doi: 10.1186/gb-2005-6-12-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Q, Forster B, Nakagawa H. Principles and applications of plant mutation breeding. Plant Mutation Breeding and Biotechnology. 2012:301–325. [Google Scholar]
- Sikora P, Chawade A, Larsson M, Olsson J, Olsson O. Mutagenesis as a tool in plant genetics, functional genomics, and breeding. Int J Plant Genomics. 2011;2011 doi: 10.1155/2011/314829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivamani E, Bahieldin A, Wraith JM, Al-Niemi T, Dyer WE, Ho T-HD, Qu R. Improved biomass productivity and water use efficiency under water deficit conditions in transgenic wheat constitutively expressing the barley HVA1 gene. Plant Sci. 2000;155:1–9. doi: 10.1016/s0168-9452(99)00247-2. [DOI] [PubMed] [Google Scholar]
- Taheri S, Abdullah TL, Jain SM, Sahebi M, Azizi P. TILLING, high-resolution melting (HRM), and next-generation sequencing (NGS) techniques in plant mutation breeding. Mol Breeding. 2017;37:40. [Google Scholar]
- Tan Y, Li M, Yang Y, Sun X, Wang N, Liang B, Ma F. Overexpression of MpCYS4, a phytocystatin gene from Malus prunifolia (Willd.) Borkh., enhances stomatal closure to confer drought tolerance in transgenic Arabidopsis and apple. Front Plant Sci. 2017;8:33. doi: 10.3389/fpls.2017.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazona S, Furió-Tarí P, Turrà D, Pietro AD, Nueda MJ, Ferrer A, Conesa A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43:e140–e140. doi: 10.1093/nar/gkv711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O, Blaesing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37:914–939. doi: 10.1111/j.1365-313x.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- Uitdewilligen JG, Wolters A-MA, Bjorn B, Borm TJ, Visser RG, van Eck HJ. A next-generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS One. 2013;8:e62355. doi: 10.1371/journal.pone.0062355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol Ecol. 2008;17:1636–1647. doi: 10.1111/j.1365-294X.2008.03666.x. [DOI] [PubMed] [Google Scholar]
- Vos J, Groenwold J. Root growth of potato crops on a marine-clay soil. Plant Soil. 1986;94:17–33. [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Pan S, Cheng S, Zhang B, Bachem C, de Boer J, Borm T, Kloosterman B, van Eck H, Datema E. Genome sequence and analysis of the tuber crop potato. Nature. 2011;475:189–195. doi: 10.1038/nature10158. [DOI] [PubMed] [Google Scholar]
- Xu Y, Gao S, Yang Y, Huang M, Cheng L, Wei Q, Fei Z, Gao J, Hong B. Transcriptome sequencing and whole genome expression profiling of chrysanthemum under dehydration stress. BMC Genomics. 2013;14:662. doi: 10.1186/1471-2164-14-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates SA, Swain MT, Hegarty MJ, Chernukin I, Lowe M, Allison GG, Ruttink T, Abberton MT, Jenkins G, Sk⊘t L. De novo assembly of red clover transcriptome based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genomics. 2014;15:453. doi: 10.1186/1471-2164-15-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaycili O, Alikamanoglu S. Induction of salt-tolerant potato (Solanum tuberosum L.) mutants with gamma irradiation and characterization of genetic variations via RAPD-PCR analysis. Turk J Biol. 2012;36:405–412. [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Liu B, Ma C, Zhang G, Chang J, Si H, Wang D. Transcriptome characterization and sequencing-based identification of drought-responsive genes in potato. Mol Biol Rep. 2014;41:505–517. doi: 10.1007/s11033-013-2886-7. [DOI] [PubMed] [Google Scholar]
- Zhang X, Liu S, Takano T. Two cysteine proteinase inhibitors from Arabidopsis thaliana, AtCYSa and AtCYSb, increasing the salt, drought, oxidation and cold tolerance. Plant Mol Biol. 2008;68:131–143. doi: 10.1007/s11103-008-9357-x. [DOI] [PubMed] [Google Scholar]
- Zhao MR, Han YY, Feng YN, Li F, Wang W. Expansins are involved in cell growth mediated by abscisic acid and indole-3-acetic acid under drought stress in wheat. Plant Cell Rep. 2012;31:671–685. doi: 10.1007/s00299-011-1185-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.