

Abstract
The stepwise development of T cells from a multipotent precursor is guided by diverse mechanisms, including interactions among lineage-specific transcription factors (TFs) and epigenetic changes, such as DNA methylation and hydroxymethylation, which play crucial roles in mammalian development and lineage commitment. To elucidate the transcriptional networks and epigenetic mechanisms underlying T-cell lineage commitment, we investigated genome-wide changes in gene expression, DNA methylation and hydroxymethylation among populations representing five successive stages of T-cell development (DN3, DN4, DP, CD4+, and CD8+) by performing RNA-seq, MBD-seq and hMeDIP-seq, respectively. The most significant changes in the transcriptomes and epigenomes occurred during the DN4 to DP transition. During the DP stage, many genes involved in chromatin modification were up-regulated and exhibited dramatic changes in DNA hydroxymethylation. We also observed 436 alternative splicing events, and approximately 57% (252) of these events occurred during the DP stage. Many stage-specific, differentially methylated regions were observed near the stage-specific, differentially expressed genes. The dynamic changes in DNA methylation and hydroxymethylation were associated with the recruitment of stage-specific TFs. We elucidated interactive networks comprising TFs, chromatin modifiers, and DNA methylation and hope that this study provides a framework for the understanding of the molecular networks underlying T-cell lineage commitment.
Keywords: DNA hydroxymethylation, DNA methylation, T-cell development
INTRODUCTION
The lineage-specific differentiation of immature T cells into CD4+ or CD8+ mature T cells is a stepwise process with well-defined sequential developmental stages (Germain, 2002). Early immature thymocytes, such as CD4 and CD8, lack the expression of T-cell receptors (TCR) and are thus termed double-negative (DN) cells. DN cells can be subdivided into the following four stages of differentiation: DN1 (CD44+CD25−), DN2 (CD44+CD25+), DN3 (CD44−CD25+), and DN4 (CD44−CD25−) (Godfrey et al., 1993). Successive T-cell differentiation produces double positive (DP; CD4+ CD8+) αβ TCR-expressing immature cells that subsequently become positively selected and mature into cells expressing TCRs that bind self-peptide-MHC-class-I complexes (CD8+ T cells) or self-peptide-MHC-class-II ligands (CD4+ T cells) (von Boehmer et al., 1989).
The cell and tissue identities in multicellular organisms are maintained by their particular epigenome (Bernstein et al., 2007). DNA methylation of the fifth position of cytosine (5mC) is a key epigenetic marker that plays important roles in mammalian development, differentiation and maintenance of cellular identity (Kim and Costello, 2017; Smith and Meissner, 2013). 5-hydroxymethylcytosine (5hmC) is an intermediate marker that is present during passive and active demethylation (Guo et al., 2011; Serandour et al., 2012), but 5hmC has also been suggested to play an important role in gene regulation (Kim et al., 2014; Yu et al., 2012). 5hmC is enriched in the gene bodies of highly expressed genes and at active enhancers (Ichiyama et al., 2015; Tsagaratou et al., 2014).
Here, we investigated the genome-wide changes in DNA methylation and DNA hydroxymethylation and their influences on stage-specific gene expression during T-cell precursor development. We identified stage-specific differentially methylated regions (ssDMRs) and differentially hydroxymethylated regions (ssDhMRs) and found that many of these regions overlap with the binding sites of stage-specific TFs. We also identified many stage-specific splicing variants. Our study may provide a framework for the understanding of the complexity of T-cell development and lineage commitment.
MATERIALS AND METHODS
Mice
C57BL/6J mice (gender- and age-matched, 6–8 weeks) were purchased from Daehan Biolink (Korea). All the animal experiments were performed according to the guidelines and regulations for rodent experiments provided by the Institutional Animal Care and Use Committee (IACUC) of KAIST. The protocols used in this study were approved by the KAIST IACUC (KA2010-21).
Antibodies
The antibodies used for flow cytometry were purchased from BD Biosciences, eBioscience, and BioLegend, unless otherwise indicated. The antibodies included CD4 (RM4-5), CD8α (53-6.7), CD11b (M1/70), CD11c (HL3), CD19 (1D3), CD25 (PC61), CD44 (IM7), CD45R (B220, RA3-6B2), NK1.1 (PK136), and TCRγ/δ (GL3).
Flow cytometry
Thymus cells were isolated and dispersed into single-cell suspensions by passage through a 70-μm cell strainer (SPL, Korea). The cells were blocked with anti-CD16/32 and then stained for surface molecules. DAPI (4,6-diamidino-2-phenylindole; Roche) was used for the dead cell exclusion. The data were acquired using an Aria II flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Sorting
Single-cell suspensions of thymus cells were prepared as described above (flow cytometry) and blocked with anti-CD16/32. The whole thymus cell suspensions were enriched via the depletion of CD4+ or CD8+ cells by staining with a biotin-conjugated anti-CD4 or CD8α antibody, followed by incubating with streptavidin-microbeads (Miltenyi) and magnetic separation. The enriched cells were stained for surface molecules, and the dead cells were excluded by staining with DAPI. Sorting was performed using an Aria II system (BD Biosciences) and an 85-μm nozzle.
Sorting of murine thymus T-cell sub-populations
To isolate cells during the early stage of T-cell differentiation, cells were obtained from the thymus of C57BL/6J mice and stained with antibodies. The obtained cells were separated using the following process: 1) DAPI-integrated dead cells were excluded via FSC-A versus DAPI plots; 2) the cells were gated for singlet cells from FSC-A versus FSC-H plots; and 3) among the cells that were negative for lineage (B220-CD11b-CD11c-CD19-NK1.1-) and TCRγ/δ expression, the double-negative (DN), double-positive (DP), and single-positive (SP) T-cell subsets were isolated based on the CD4 and CD8 expression level. The DN3 and DN4 T cells were further identified according to the CD25 and CD44 expression (Supplementary Fig. S1B). Throughout this process, the isolated cells demonstrated purity based on an immediate reanalysis of the collected cells at each stage (Supplementary Fig. S1C).
MBD-seq
MBD-seq was performed as previously described (Kim et al., 2014). Methylated DNA was precipitated using a Methyl-Miner methylated DNA enrichment kit (Invitrogen). The purified methylated DNA fragments were ligated to a pair of adaptors for sequencing using an Illumina NextSeq500 to generate 75 bp single-end reads.
hMeDIP-seq
hMeDIP-seq was performed as previously described (Kim et al., 2014). Fragmented DNA was end-repaired, A-tailed, and ligated to paired-end adapters. The pre-adapted DNA was subjected to immunoprecipitation using a hMeDIP kit (Diagenode). The immunoprecipitated DNA was amplified by 18 cycles of PCR using Illumina PCR primers and then size fractioned on a 2% agarose gel to obtain 200–300 bp fragments. The sequencing was performed using an Illumina NextSeq500 to generate 75 bp single-end reads.
RNA-seq
RNA-seq was performed as previously described (Baek et al., 2016). The RNA sequencing library was prepared using a TruSeq RNA Sample Prep Kit (Illumina, USA), and the sequencing was performed using Illumina NextSeq500 to generate 100-bp paired-end reads.
RNA-seq data analysis
FastQC (FastQC v0.11.3) (A Andrews, 2010) was used to filter the low-quality sequencing reads. Then, the reads were mapped to mouse genome build mm10 using TopHat (TopHat v2.0.11) (Trapnell et al., 2009) with the default parameters. DESeq (DESeq v3.1) (Anders and Huber, 2010) was used to select the differentially expressed genes among the five different developmental stages (fold-change>2 and P-value < 0.05). Hierarchical clustering was performed to characterize the overall expression patterns of the differentially expressed genes (DEGs) among the five T-cell stages using R statistical language (R version 3.3.1) (Team, 2013) with the Euclidean distance and complete linkage method. The developmental lineage-specific alternative splicing was analyzed using cufflinks and MISO tools. First, the cuffdiff program was used to quantify the expression of the transcripts across all stages. Then, we compared the alternative exon usage among the stepwise developmental stages using the mixture-of-isoforms statistical model, which assigns a “percentage spliced in” (PSI) value to each exon by estimating its abundance compared to the adjacent exons (Katz et al., 2010).
Methylome analysis (DMR, DhMR, ssDMR and ssDhMR)
The sequenced MBD-seq and hMeDIP-seq reads were mapped to the mouse reference genome (mm10) using Bowtie (Bowtie v1.1.1) (Langmead et al., 2009) (parameters: -m 1), indexed and sorted using SAMtools (v.0.1.19) (Li et al., 2009). To identify the differentially methylation regions (DMRs) and differentially hydroxymethylated regions (DhMRs), we used MEDIPS (MEDIPS v1.18.0) (Lienhard et al., 2014) (parameters: extend = 200, shift = 0, window size = 200, and unique = TRUE) and various preprocessing steps (saturation estimation, sequence pattern coverage, CpG enrichment bias and GC bias normalization). The DMRs and DhMRs were selected based on the following parameters: diff.method = edgeR, prob.method = poisson, MeDIP = T, CNV = F, type = RPKM, and p-value = 0.05. HOMER (HOMER v4.7.2) (Heinz et al., 2010) was used to annotate the stage-specific DMRs and DhMRs (ssDMR and ssDhMR). To identify the hypermethylated ssDMRs or ssDhMRs, we first generated the union of DMRs and DhMRs from the five developmental stages using the mergedPeak.pl command in HOMER. Then, we used the getDifferentialPeaks.pl script in HOMER to select hypermethylated regions with a statistical threshold of p < 0.05. All the sequence data were deposited to the Gene Expression Omnibus (GSE59117).
Enhancer region analysis
To identify the enhancer regions, we obtained the ChIP-seq data of active chromatin markers (H3K4me1, H3K4me3, H3K27ac, and Pol II) at the DP stage from the NCBI GEO database (GSE20898, GSE47995, and GSE63732). Each ChIP-seq data set was mapped to the reference genome (mm10), and the peaks were identified using HOMER. We identified the regions overlapping with the H3K4me1, H3K27ac and Pol II peaks and then filtered the H3K4me3-enriched regions because H3K4me3 peaks are enriched at promoter regions.
Motif identification
To identify the TF binding motifs at stage-specific DMR or DhMR regions, we used the findMotifsGenome.pl command in HOMER. This command identifies motifs enriched in specific regions compared with randomly selected background regions (enrichment threshold: P-value < 0.05). Then, we identified the statistically significant stage-specific TF binding motifs at each stage. To eliminate the false positive TF binding motifs, we filtered the stage-specific binding motifs of TFs that were not expressed in the RNA-seq data.
RESULTS
Global gene expression changes during T-cell development
To investigate the gene expression changes, we performed an RNA-seq analysis. Based on the read per kilobase per million mapped reads (RPKM) ≥ 1, approximately 17,500 genes were expressed among 20,861 reference transcripts (mm10). According to the pairwise comparisons of the gene expression changes between two stages along the T-cell developmental process, the most dramatic changes occurred between the DN4 and DP stages, involving 1,874 (11.4%) differentially expressed genes (DEGs; fold-change ≥ 2, P < 0.05; Fig. 1A). Many genes altered between the DN4 and DP stages were related to epigenetic modifications, such as histone modification and chromatin remodeling, suggesting that development from DN4 to DP requires the expression of genes associated with epigenetic modification before T-cell lineage commitment.
Fig 1. Patterns of gene expression changes during each stage of T-cell development.
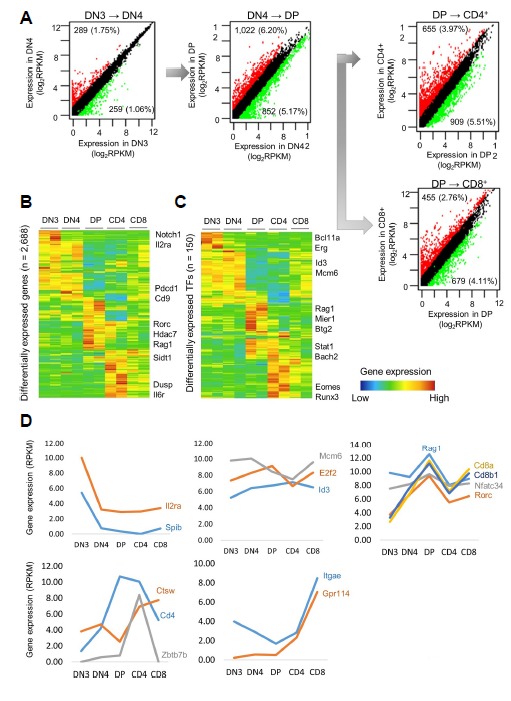
In total, 2,688 DEGs were selected based on a log2FC(RPKM) > 2 and p-value < 0.05. (A) Overall patterns of gene expression changes across the stages of the T-cell development process. The most dramatic changes occurred from DN4 to DP (11.37%), whereas fewer gene expression changes were observed from DN3 to DN4 (2.81%). Moderate gene expression changes occurred from DP to CD4 (9.46%) and DP to CD8 (6.87%). (B) Hierarchical clustering of gene expression patterns across developmental stages. (C) Hierarchical clustering of gene expression patterns of differentially expressed transcription factors across developmental stages. Major transcription factors during each stage. (D) Stage-specific gene expression at each stage (DN3 to CD4 and CD8).
To understand the patterns of the gene expression changes during each stage of development, we performed hierarchical clustering of 2,688 genes with a greater than twofold change in expression between any stages (FC ≥ 2, P < 0.05; Fig. 1B). Many gene expression changes reflected stage-specific identity. For example, the DN3-specific genes included several key genes in the Notch signaling pathway, which is important for selective T/B-cell commitment. Interestingly, the DN3-specific genes were expressed exclusively during DN3 and repressed during the subsequent stages. At DN4, during which cells proliferate explosively, the genes responsible for cell proliferation and the cell cycle, such as e2f2, Id3, and mcm6, were up-regulated. Interestingly, many genes were distinctly expressed during DP, suggesting that a major transition in the gene expression pattern occurs with TCR alpha/beta selection. Furthermore, the CD4+-specific genes included genes involved in protein recycling within the lysosome and the maturation of the MHC class II complex. The CD8+-specific genes included many cytotoxicity-associated genes, such as Gpr114, Ctsw, Gzmk, Prf1, and Fgfbp2 (Fig. 1D).
Subsequently, we focused on the changes in the TF expression levels. We obtained a list of 1,646 TFs from the GO term “DNA-dependent regulation of transcription” (GO:0006350) after removing genes with ambiguous annotation (Zhang et al., 2012). Among the 1,646 TFs, 150 genes were selected (FC ≥ 2, P < 0.05; Fig. 1C). From DN3 to DN4, the most strongly up-regulated TFs were LEF1 and Bcl11b (>10-fold increase). Pou6f1, SpiB, Ikzf3, and Ets1 exhibited increases greater than 8-fold, and moderate increases in Id3, Tcf12, and Hes1 were observed. In contrast, many TFs important for hematopoietic progenitors, including Gfi1b, Lmo2, Mef2c, Hoxa9, Sfpi1, Gata2, and interferon regulatory factors, were down-regulated from DN3 to DN4. Thus, a major regulatory shift occurred from DN4 to DP characterized by the repression of many progenitor cell TFs and the induction of several key factors, such as Rorc, Nfatc3, Cd8a, and Cd8b, that are important for the T-cell developmental program and TCR signaling. After positive selection occurred during the CD4 and CD8 stages, the differentially expressed TFs reflected the specific identity of each stage. For instance, many cytotoxicity-associated genes, such as Ctsw, Gzmk, Prf1, and Fdfp2, were exclusively up-regulated during the CD8 stage. The genes up-regulated during the CD4 stage included CCR4, Zbtb7b, and Ctsl1, which are involved in protein recycling within the lysosome and the maturation of the MHC class II complex (Fig. 1D).
We performed GO and pathway analyses of the lists of DEGs at each stage of T-cell development (Supplementary Table S1). Two broad categories of genes were up-regulated during the DN3 to DN4 transition. First, genes involved in cell division processes, such as mitosis and cell proliferation, were up-regulated. Second, genes associated with T-cell development and activation, hematopoietic cell lineage, and the T-cell receptor signaling pathway were up-regulated. In contrast, the DN3 to DN4 progression was accompanied by the down-regulation of genes involved in Notch signaling. During the DN4 to DP transition, cell adhesion-, T-cell activation-, and T-cell aggregation-associated genes were up-regulated. The cell adhesion-related genes are likely up-regulated because the transition from DN4 to DP represents a preparation step for the TCR-alpha rearrangement that occurs during the DP stage. In contrast, genes involved in rRNA processing and DNA replication were remarkably down-regulated. Expectedly, during the DP to CD4 transition, the expression of genes related to the role of CD4 cells was prominent. The expression of genes related to cytokines, the innate immune response, and the cytokine-cytokine receptor interaction pathway was increased. In contrast, cell cycle- and cell division-related genes were markedly down-regulated. Between the DP and CD8 stages, the expression of genes related to the role of CD8 cells, such as MHC class I and cell killing, was also increased, whereas the expression of genes related to V(D)J recombination, which are activated during the DP stage, was decreased (Supplementary Fig. S3).
Alternative splicing
Alternative splicing is a regulatory mechanism of gene expression that produces two or more distinct mRNA species from a single gene. We used the MISO method to identify 435 exons differentially spliced during early T-cell development (PSI > 10% and Bayes factor > 5) (Wang et al., 2008). Exon-skipping was the most frequent event (Fig. 2A), and, among the five stages, exon-skipping occurred most frequently during the DP stage. Interestingly, many alternative splicing events occurred in genes that play an important role in the development and activation of T cells. For example, the Ssbp2 gene exhibited an alternative splicing event in which exon 14 was skipped during the DN4 to DP transition (Fig. 2B). The expression of NM_024186, which is an iso-form of the Ssbp2 gene, was decreased during the DP stage, whereas the expression of the other isoform, i.e., NM_024272, was increased during the DP stage (Fig. 2C). To understand why the alternative splicing events occurred most frequently during the DP stage, we examined the expression of genes involved in alternative splicing and the regulation of alternative splicing events. First, we examined two genes, i.e., Celf2 and Map2k7, that are known to regulate alternative splicing during T-cell signaling (Fig. 2D). Then, we examined four genes (Clk3, Brdt, Mbnl2, and Esrp2) that are involved in alternative splicing and found that these four genes were specifically expressed during the DP stage. According to the Gene Ontology analysis, the DEGs at the DP stage are enriched with genes related to mRNA alternative splicing (Fig. 2E). Many genes associated with T-cell development and identity were related to stage-specific alternative splicing events. Thus, the increased expression of Celf2 in DP cells could contribute to the abundant alternative splicing events occurring during the transition from DN to DP (Mallory et al., 2015).
Fig 2. Alternative splicing events during T-cell development.
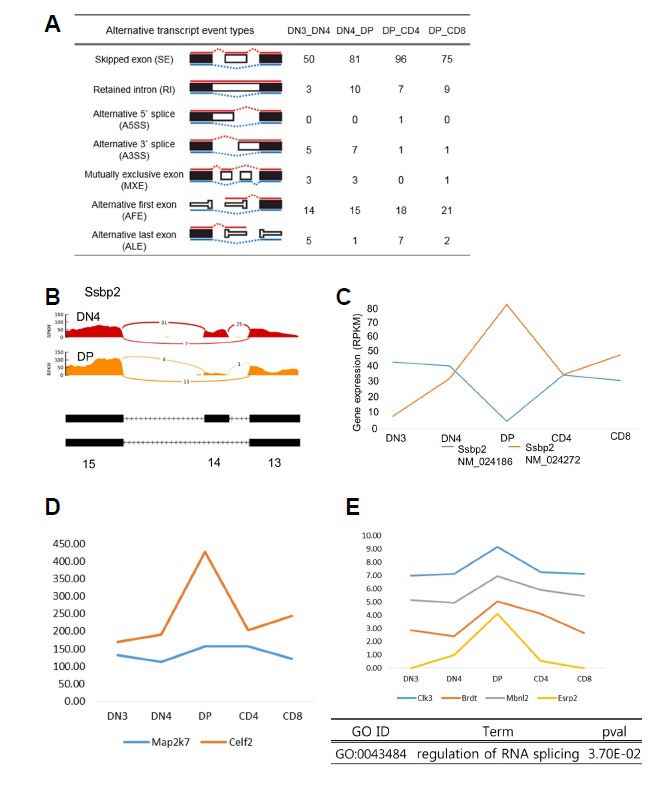
(A) Various alternative splicing events occurred during T-cell development. The skipped exon (SE) event was the most common. (B) Expression of exon 14 of the Ssbp2 gene almost disappeared upon conversion from DN4 to DP. (C) Comparison of the expression of the Ssbp2 gene isoforms. Isoform expression of the Ssbp2 gene is approximately 3 to 4 times higher during the DP stage than that during the other stages. (D) Expression patterns of Celf2 and Map2k7. Both genes are highly expressed during the DP stage. (E) Expression patterns of genes involved in alternative splicing. Most genes are highly expressed during the DP stage.
Also, we observed changes in DNA methylation and hydroxymethylation of alternative promoters of isoforms to observe epigenetic changes of the alternative first exons discovered during T-cell development. We found that several genes important for T-cell development showed DN3 stage-specific hypermethylation in the alternative promoters of the coding region. For example, Ogt, a gene critical for Notch mediated self-renewal and malignant transformation of β-selected T cell progenitors during thymus development (Swamy et al., 2016), showed DN3-specific hypermethylation in the isoform of the first exon (Supplementary Fig. S8). These results suggest that an alternative promoter within the coding region of some genes during T cell development is activated or inhibited by DNA methylation, resulting in the production of a protein lacking the N-terminal region.
Differentially methylated or hydroxymethylated regions during T-cell development
To understand how epigenetic changes, such as DNA methylation and hydroxymethylation, are associated with the gene expression changes during T-cell development, we first observed the expression patterns of the Dnmt and Tet family proteins, which mediate DNA methylation and hydroxymethylation (Figs. 3A and 3B). During each stage of T-cell development, significant changes in DNA methylation and hydroxymethylation were observed over the course of the T-cell lineage, and the most significant changes occurred between DN3 and DN4 (Fig. 3C). Most changes in DNA methylation and hydroxymethylation occurred at intragenic regions (Supplementary Figs. S4A and 4B). In total, 61,313 differentially methylated regions were identified from DN3 to CD4 or CD8. Among the DMRs, most methylation changes occurred during the DN3 to DN4 transition (31,054 DMRs; 50.64%). Of these DMRs, 26,678 DMRs were hypomethylated during the DN4 stage. Interestingly, the expression of Dnmt3b also gradually decreased from the DN3 stage to DN4 and DP.
Fig 3. Genome-wide analysis of the changes in methylation and hydroxymethylation during T-cell development.
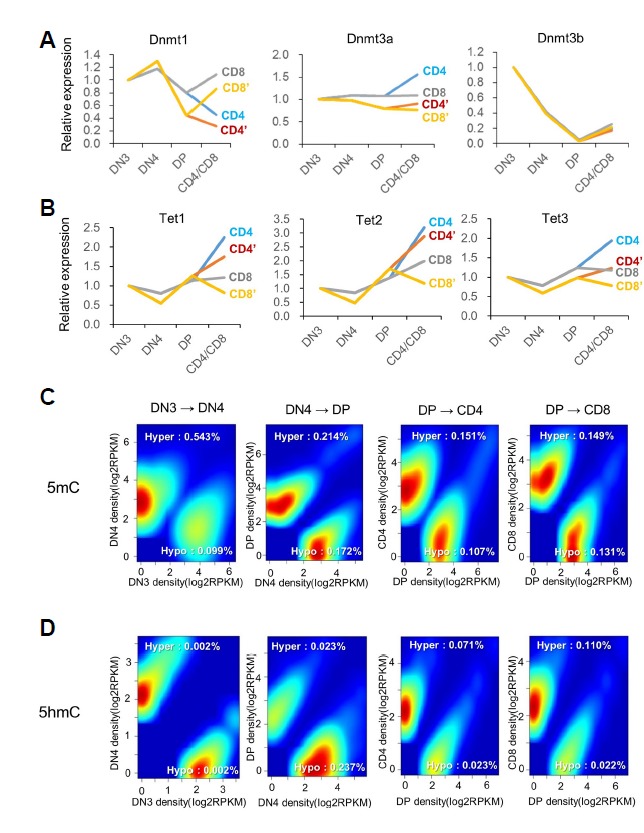
(A, B) Genome-wide identification of differentially methylated or hydroxymethylated regions. (C, D) Distribution of DMRs and DhMRs according to various genomic features.
We further investigated the relationships between gene expression and DNA methylation or hydroxymethylation. A negative correlation was observed between promoter methylation and gene expression during all stages. In contrast, a positive correlation was observed between gene-body hydroxymethylation and gene expression during most stages. Thus, increased promoter hydroxymethylation is likely associated with increased gene expression, while promoter hypermethylation is likely associated with decreased gene expression. The average density profile of 5hmC and 5mC around transcription start sites (TSSs) and gene-body regions clearly showed that the 5mC density is depleted from the TSSs of stage-specific DEGs, whereas 5hmC is enriched in the gene bodies of stage-specific DEGs (Figs. 4A and 4B).
Fig 4. Profiling of distinct, stage-specific differentially methylated and hydroxymethylated regions.
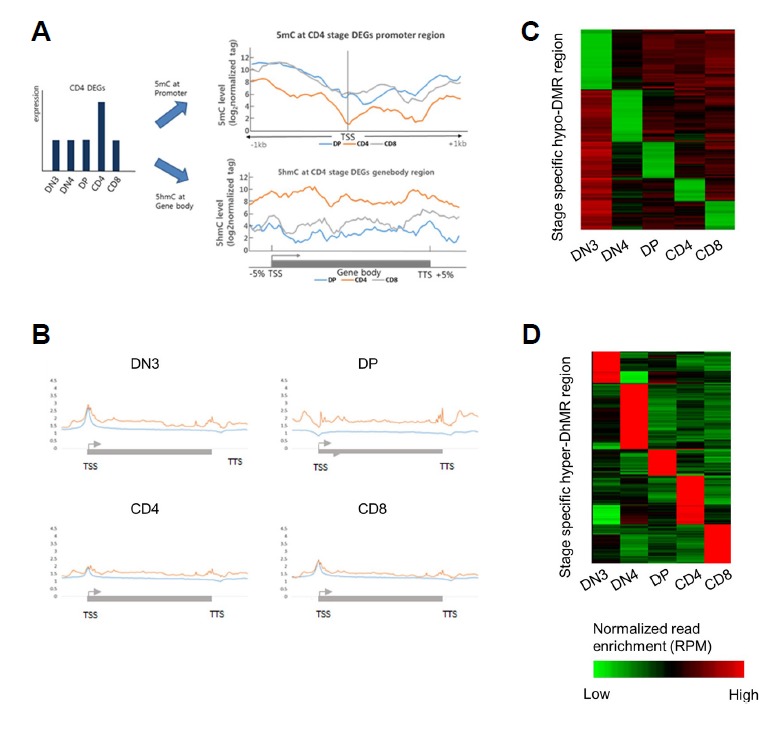
(A) Expression of specific DEGs at each stage and correlation between 5mC and 5hmC. The methylation level in the promoter region of stage-specific up-regulated genes was lower at the CD4 stage than that at the other stages, whereas the 5hmC level in the gene-body region was abundant during the other stages. (B) 5hmC distribution in the gene-body region of stage-specific DEGs at each stage. The red line represents the 5hmC distribution at each stage, and the blue line represents the 5hmC distribution at the other stages. (C) Snapshot of stage-specific DNA hypomethylation at each stage of early T-cell development. (D) Snapshot of stage-specific DNA hyperhydroxymethylation at each stage of early T-cell development. The abundance of methylation and hydroxymethylation was calculated from non-overlapping 200-bp bins spanning each stage.
Identification of stage-specific differentially methylated regions (ssDMR) and differentially hydroxymethylated regions (ssDhMR)
We selected stage-specific differentially methylated regions (ssDMRs) and hydroxymethylated regions (ssDhMRs) at each stage (Figs. 4C and 4D). Two examples (Myl10 and Ncaph) are shown to illustrate the relationships among gene expression, DNA methylation and hydroxymethylation. Myl10 is a DN3 stage-specific gene with a hypomethylated promoter region, and its gene-body region is highly hydroxymethylated. After differentiation into DN4, its promoter region is hypermethylated, whereas the gene-body hydroxymethylation is reduced. Thus, the DN3-specific expression of Myl10 is associated with both promoter hypomethylation and gene-body hydroxymethylation. The second example is the relationship between stage-specific hypomethylation and cis-regulatory regions. The stage-specific hypomethylation region at the gene body of the Ncaph gene overlaps the H3k4me1, H3K27ac, and Pol II ChIP-seq data obtained during the DP stage (Supplementary Figs. S5a and b). Additionally, the CD4-specific gene Cd4 is specifically expressed only in CD4+ cells, and its promoter region is hypomethylated only in CD4+ cells. Interestingly, no significant changes were observed in the hydroxymethylation of Cd4 gene-body regions, suggesting that promoter region methylation is mainly associated with Cd4 gene expression (Supplementary Fig. S6).
Stage-specific DMRs and DhMRs are predominantly located in regulatory elements
Many stage-specific, hypomethylated DMRs were located in the regulatory regions of stage-specific TFs (Fig. 5A). For example, many DN3-specific TFs, such as Gata3, Klf4, and Id1, were hypomethylated in their promoter regions. According to the Gene Ontology analysis of the stage-specific DMR nearest genes, many genes were involved in processes, such as ‘regulation of gene expression’, ‘T-cell receptor signaling pathway’, ‘T-cell selection’, and/or ‘T-cell differentiation’ (Fig. 5B). The sequence motif analysis of sequences from stage-specific, hypomethylated regions revealed that the enriched motifs corresponded to the dominant TFs during each stage. For example, the sequences from the DP-specific DMRs were enriched with motifs, such as Myb, Ets1, Maz, and Tcf12, all of which are DP-specific TFs (Fig. 5C). Thus, the stage-specific expression of many stage-specific TFs is regulated by promoter region methylation.
Fig 5. Analysis of stage-specific hypomethylated regions.
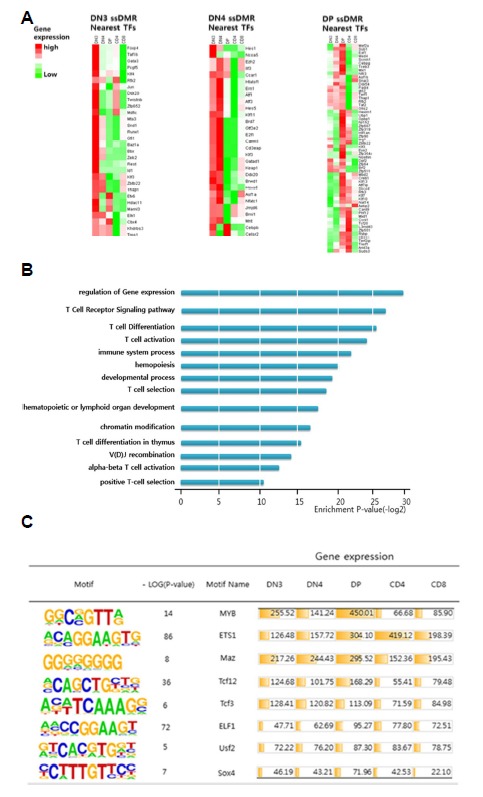
(A) Stage-specific hypomethylated regions near transcription factors exhibited predominant expression patterns at each stage. (B) Transcription factors near stage-specific hypomethylated regions belonging to GO terms associated with the development of T cells. (C) Transcription factors with binding motifs in stage-specific hypomethylated regions exhibited predominant expression patterns at each stage.
DISCUSSION
Genome-wide changes in DNA methylation and hydroxymethylation associated with T cells have been previously reported (Ko et al., 2015; Lee et al., 2001; Wilson et al., 2005; Zhao et al., 2016). Sellars et al. demonstrated that the dynamic regulation of DNA methylation plays an important role in enhancing the differential expression of CD4 and selecting the fate of helper and cytotoxic T cells (Sellars et al., 2015; Tsagaratou et al., 2014). Tsagaratou et al. (2014) showed that 5hmC was abundant in the gene body and cell type-specific enhancer regions during the DP, CD4, and CD8 T-cell stages, indicating that the 5hmC levels change dynamically during T-cell development and differentiation (Tsagaratou et al., 2014). However, the abovementioned studies focused on the process of determining cell fate. In contrast, our study evaluated the relationship between the gene expression changes and 5mC, 5hmC, alternative splicing and distal regulatory elements from cell fate determination to functional fate determination and from the early differentiation stage.
We observed a dynamic network of genes and TF expression that was previously reported in transcriptome analysis during the development of T-cell. During the developmental transition from DN3 to CD4 and CD8, we classified the patterns of gene expression into 24 distinct clusters (Supplementary Fig. S7). For example, the genes in the Notch signaling pathway were induced to regulate differentiation from precursor cells to T cells until beta-selection during the DN3 stage, and their expression was rapidly repressed following beta-selection. Then, the expression of genes mediating beta-selection, such as E2A or Id3, was increased. Many previous studies have suggested that the expression of diverse TFs is precisely controlled to regulate the transcription of genes required at different stages and that this regulation is mediated by chromatin remodeling, cis-regulatory elements, chromatin interaction, and DNA methylation (David-Fung et al., 2009; Huang et al., 2008; Naito et al., 2011; Samstein et al., 2012; Yui and Rothenberg, 2014).
We analyzed DNA methylation and DNA hydroxymethylation to observe epigenetic changes that regulate these gene expression changes. Similar to previously reported tissues and cells, we observed reverse correlation between promoter and methylation during development and positive correlation between gene body and hydroxymethylation. But alternative splicing events were more prevalent during the DP stage than during the other stages, and genes known to mediate alternative splicing were also highly expressed during this stage. We classified hundreds of alternative splicing events occurring during the early T-cell developmental stages into six categories and observed the relationship between these genes and the DNA methylation. During T-cell development, we have identified splicing variants of various genes, such as contrastive differential expression between isoforms or specific exons, and the presence of DMR in the gene body region of these genes and stage-specific hypermethylation of alternative promoters. We also compared DNA methylation of genes with or without splicing variants during the T-cell developmental stages to examine the potential role of DNA methylation in the alternative splicing (Supplementary Fig. S9). DNA methylation in the gene body of the genes without splicing variant was changed little, but DNA methylation of gene body of the genes with splicing variants showed dynamic methylation changes. Then, these genes with splicing variants in the coding region were enriched in the GO and pathway term associated with T-cell development.
We observed changes in stage-specific methylation in many cis-regulatory elements. Many DMRs and DhMRs were stage-specific and associated with the expression of corresponding genes. A recent study analyzing the methylation patterns of 17 normal tissues showed that many tissue-specific 5mC regions are located in distal regulatory regions (Hon et al., 2013; Lister et al., 2009). We also found that many stage-specific DMRs were located in intergenic regions rather than the promoter or gene body and that these DMRs overlapped with enhancer elements defined by H3K27ac peaks. Interestingly, these genes were enriched with GO terms related to T-cell development, such as T-cell differentiation and T-cell selection. Interestingly, a recent study reported that the stable CD4 expression in CD4+ T cells is regulated by the enhancer-dependent demethylation of the Cd4 locus, whereas CD4 silencing in CD8+ T cells is regulated by the methylation of the cis-acting silencer region of the proximal Cd4 locus (Feng and Rudensky, 2015; Komori et al., 2015; Sellars et al., 2015). Thus, dynamic DNA methylation/demethylation processes in cis-regulatory regions (both enhancers and promoters) provide new opportunities for depicting the signaling pathways involved in the selection of the thymus lineage (Hon et al., 2014; Huang et al., 2014; Stadler et al., 2011).
In conclusion, we systematically analyzed the changes in gene expression, DNA methylation/hydroxymethylation, and stage-specific splicing variant from the initial T-cell differentiation stage to the post-T-cell fate stage. Deletion of alternative promoters and exons by CRISPR technology will validate the functional roles of alternative promoter expression in thymocyte development, and the mechanisms mediating selective promoter activity suggested by our study. Recently, rapid developments in methods used in single-cell analyses have enabled the analysis of gene expression and epigenomic changes at the single-cell level (Kakaradov et al., 2017; Villani et al., 2017). Considering the recent technical advances, our study is limited because it is based on an analysis of cell populations. Integrated analyses of transcriptomes and epigenomes (i.e., DNA methylation, hydroxymethylation, ATAC-seq, and Hi-C) at the single-cell level could be highly valuable for elucidating the complex molecular networks underlying T-cell lineage commitment.
Supplementary data
ACKNOWLEDGMENTS
This study was supported by the Collaborative Genome Program for Fostering New Post-Genome Industry of the National Research Foundation (NRF) funded by the Ministry of Science and ICT (MSIT) (NRF-2017M3C9A5028693 to M.K., NRF-2014M3C9A3068554 and NRF-2017MBA9B5060884 to S.Y.K).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Andrews AS. FastQC: a quality control tool for high throughput sequence data. 2010. [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamas SP, Choi J, Yurovsky VV, White B. An alternative splice variant of human IL-4, IL-4 delta 2, inhibits IL-4-stimulated T cell proliferation. J Immunol. 1996;156:435–441. [PubMed] [Google Scholar]
- Baek SJ, Kim M, Bae DH, Kim JH, Kim HJ, Han ME, Oh SO, Kim YS, Kim SY. Integrated epigenomic analyses of enhancer as well as promoter regions in gastric cancer. Oncotarget. 2016;7:25620–25631. doi: 10.18632/oncotarget.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- David-Fung ES, Butler R, Buzi G, Yui MA, Diamond RA, Anderson MK, Rowen L, Rothenberg EV. Transcription factor expression dynamics of early T-lymphocyte specification and commitment. Dev Biol. 2009;325:444–467. doi: 10.1016/j.ydbio.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Rudensky AY. DNA methylation secures CD4(+) and CD8(+) T cell lineage borders. Nature Immunol. 2015;16:681–683. doi: 10.1038/ni.3207. [DOI] [PubMed] [Google Scholar]
- Germain RN. T-cell development and the CD4-CD8 lineage decision. Nature Reviews Immunol. 2002;2:309–322. doi: 10.1038/nri798. [DOI] [PubMed] [Google Scholar]
- Godfrey DI, Kennedy J, Suda T, Zlotnik A. A developmental pathway involving four phenotypically and functionally distinct subsets of CD3-CD4-CD8-triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J Immunol. 1993;150:4244–4252. [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. 2013;45:1198–1206. doi: 10.1038/ng.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, Yen CA, Ye Z, Mao SQ, Wang BA, et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell. 2014;56:286–297. doi: 10.1016/j.molcel.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Zhang P, Hirai H, Elf S, Yan X, Chen Z, Koschmieder S, Okuno Y, Dayaram T, Growney JD, et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat Genet. 2008;40:51–60. doi: 10.1038/ng.2007.7. [DOI] [PubMed] [Google Scholar]
- Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, Zepeda-Martinez JA, Pape UJ, Jacobsen SE, Peters B, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. P Nat Acad Sci USA. 2014;111:1361–1366. doi: 10.1073/pnas.1322921111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng Y, Zou Y, Zheng P, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity. 2015;42:613–626. doi: 10.1016/j.immuni.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JY, Tong A, Pan Q, Topp JD, Blencowe BJ, Lynch KW. Global analysis of alternative splicing during T-cell activation. RNA. 2007;13:563–572. doi: 10.1261/rna.457207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, Yu B, Wehrens EJ, Lopez J, Kim SH, et al. Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat Immunol. 2017;18:422–432. doi: 10.1038/ni.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med. 2017;49:e322. doi: 10.1038/emm.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Park YK, Kang TW, Lee SH, Rhee YH, Park JL, Kim HJ, Lee D, Lee D, Kim SY, et al. Dynamic changes in DNA methylation and hydroxymethylation when hES cells undergo differentiation toward a neuronal lineage. Hum Mol Genet. 2014;23:657–667. doi: 10.1093/hmg/ddt453. [DOI] [PubMed] [Google Scholar]
- Ko M, An J, Rao A. DNA methylation and hydroxymethylation in hematologic differentiation and transformation. Curr Opin Cell Biol. 2015;37:91–101. doi: 10.1016/j.ceb.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori HK, Hart T, LaMere SA, Chew PV, Salomon DR. Defining CD4 T cell memory by the epigenetic landscape of CpG DNA methylation. J Immunol. 2015;194:1565–1579. doi: 10.4049/jimmunol.1401162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing, S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 2014;30:284–286. doi: 10.1093/bioinformatics/btt650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory MJ, Allon SJ, Qiu J, Gazzara MR, Tapescu I, Martinez NM, Fu XD, Lynch KW. Induced transcription and stability of CELF2 mRNA drives widespread alternative splicing during T-cell signaling. P Nat Acad Sci USA. 2015;112:E2139–2148. doi: 10.1073/pnas.1423695112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NM, Agosto L, Qiu J, Mallory MJ, Gazzara MR, Barash Y, Fu XD, Lynch KW. Widespread JNK-dependent alternative splicing induces a positive feedback loop through CELF2-mediated regulation of MKK7 during T-cell activation. Genes Dev. 2015;29:2054–2066. doi: 10.1101/gad.267245.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingueneau M, Kreslavsky T, Gray D, Heng T, Cruse R, Ericson J, Bendall S, Spitzer MH, Nolan GP, Kobayashi K, et al. The transcriptional landscape of αβ T cell differentiation. Nat Immunol. 2013;14:619–632. doi: 10.1038/ni.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito T, Tanaka H, Naoe Y, Taniuchi I. Transcriptional control of T-cell development. Int Immunol. 2011;23:661–668. doi: 10.1093/intimm/dxr078. [DOI] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellars M, Huh JR, Day K, Issuree PD, Galan C, Gobeil S, Absher D, Green MR, Littman DR. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat Immunol. 2015;16:746–754. doi: 10.1038/ni.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serandour AA, Avner S, Oger F, Bizot M, Percevault F, Lucchetti-Miganeh C, Palierne G, Gheeraert C, Barloy-Hubler F, Peron CL, et al. Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic Acids Res. 2012;40:8255–8265. doi: 10.1093/nar/gks595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nature reviews Genetics. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM, Cantrell DA. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol. 2016;17:712–720. doi: 10.1038/ni.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC. R: A language and environment for statistical computing. 2013. [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagaratou A, Aijo T, Lio CW, Yue X, Huang Y, Jacobsen SE, Lahdesmaki H, Rao A. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. P Nat Acad Sci USA. 2014;111:E3306–3315. doi: 10.1073/pnas.1412327111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356 doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Boehmer H, Teh HS, Kisielow P. The thymus selects the useful, neglects the useless and destroys the harmful. Immunol Today. 1989;10:57–61. doi: 10.1016/0167-5699(89)90307-1. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CB, Makar KW, Shnyreva M, Fitzpatrick DR. DNA methylation and the expanding epigenetics of T cell lineage commitment. Semin Immunol. 2005;17:105–119. doi: 10.1016/j.smim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol. 2014;14:529–545. doi: 10.1038/nri3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell. 2012;149:467–482. doi: 10.1016/j.cell.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Wang J, Liao W, Li D, Li M, Wu H, Zhang Y, Gershwin ME, Lu Q. Increased 5-hydroxymethylcytosine in CD4(+) T cells in systemic lupus erythematosus. J Autoimmun. 2016;69:64–73. doi: 10.1016/j.jaut.2016.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.