

Abstract
This review describes target-enrichment approaches followed by next generation sequencing and their recent application to the research and diagnostic field of modern and past infectious human diseases caused by viruses, bacteria, parasites and fungi.
Keywords: hybrid capture, next generation (deep) sequencing, target enrichment, infectious disease, paleomicrobiology
Introduction
The development of next-generation sequencing (NGS) approaches has revolutionized human clinical research because of its ability to rapidly generate large volumes of sequencing data per run, with a concomitant decrease of sequencing costs (Shendure and Ji, 2008). Unbiased ultra-deep sequencing of complex samples is now accessible, although bioinformatics analyses may still be long and tedious. This issue is particularly problematic in the field of infectious disease diagnostic, where the rapid identification and functional characterization of a particular pathogen is critical for the clinical management of infected patients. So far, polymerase chain reaction (PCR) has been the gold standard method for the clinical diagnosis of infectious diseases (Edwards and Gibbs, 1994). This approach, which is based on the amplification of a generally short and conserved genomic region, can provide information on the presence/absence and abundance of a targeted microbial pathogen. PCR has numerous advantages, such as low cost, rapid processing and results acquisition, automation, sensitivity and specificity. However, and precisely because of its high specificity, PCR may not detect microorganisms whose sequences are too divergent from those targeted by the primers and probes designed. In addition, PCR will provide only partial information on the genetic diversity, genotype, functional potential, nutritional requirements as well as virulence or antibiotic-resistance. Such information, that could only be retrieved from whole genome sequencing (WGS), usually requires culture of the pathogen, which can be unsuccessful in the majority of cases (and particularly for viruses and other intracellular organisms which need host cells), can take several weeks for fastidious microorganisms or can be prevented by early administration of antimicrobial drugs. The power of NGS might thus be of particular interest in that cases for reconstructing full genomes of pathogens directly from nucleic acids extracted from clinical samples. However, due to the low pathogen/nucleic acid ratio in these complex biological samples, NGS may fail to detect/reconstruct genomes from pathogens present in low copy numbers in the sample. To overcome these limitations, capture methods, such as hybridization capture followed by NGS sequencing (also called hybrid-capture sequencing or target-enrichment sequencing) applied directly on human clinical samples have been developed (Mamanova et al., 2010). These approaches allow retrieving large genomic fragments to complete genomes with high sequencing coverage, which facilitate downstream investigations, such as phylogenetics, evolution, epidemiology, and drug resistance. In this review, we will briefly describe the different principles of hybridization capture coupled with NGS, its early developments on human genetic studies and applications in the recent years to the study of present and past human infectious diseases (Gasc et al., 2016) directly from biological samples.
Overview of the Experimental Procedure and Applications
Next-generation sequencing hybridization-based capture is an approach directly applied after nucleic acid extraction and library preparation (Figure 1). Fragmented shotgun libraries are denatured by heating and subjected to hybridization with DNA or RNA single-stranded oligonucleotides (called also ‘probes’ or ‘baits’) specific to the region of interest (Kozarewa et al., 2015). RNA baits are preferable, because RNA:DNA duplexes are better in term of hybridization efficiency and stability, compared to DNA:DNA hybrids (Lesnik and Freier, 1995). Non-specific unbound molecules are washed away, and the enriched DNA is eluted for NGS (Kozarewa et al., 2015). The hybridization between DNA libraries and baits can be carried out in solution or on a solid support. In “solid-phase,” DNA probes are bound to a solid support, such as a glass microarray slide (Albert et al., 2007; Okou et al., 2007), where in “solution-capture,” free DNA or RNA probes are biotinylated allowing them to isolate the targeted fragment-probe heteroduplexes using magnetic streptavidin beads (Gnirke et al., 2009). So far, there is no standardize protocol for target enrichment processes and several adjustments can be made on library fragment size, sample fragmentation, cleanup procedures, number of PCR amplification cycles, and/or the hybridization duration. Detailed protocols are described in the work of Mamanova et al. (2010) along with values on specificity, sensitivity and reproducibility of each tested procedure. The ultimate step of NGS hybridization-based capture is the sequencing of the enriched nucleic acids and bioinformatic analyses of the reads. The last process usually includes steps of trimming (for adapter sequences, low quality and duplicate reads), mapping of the remaining reads on reference genomes for pathogen detection and identification, and/or assembly into contigs for genome reconstruction (Figure 1). The information provided by the genome can further be explored to investigate the genetic diversity (strain genotyping, variant detection), epidemiology, evolutionary history, transmission networks, and/or antimicrobial resistance of the target pathogen(s).
FIGURE 1.
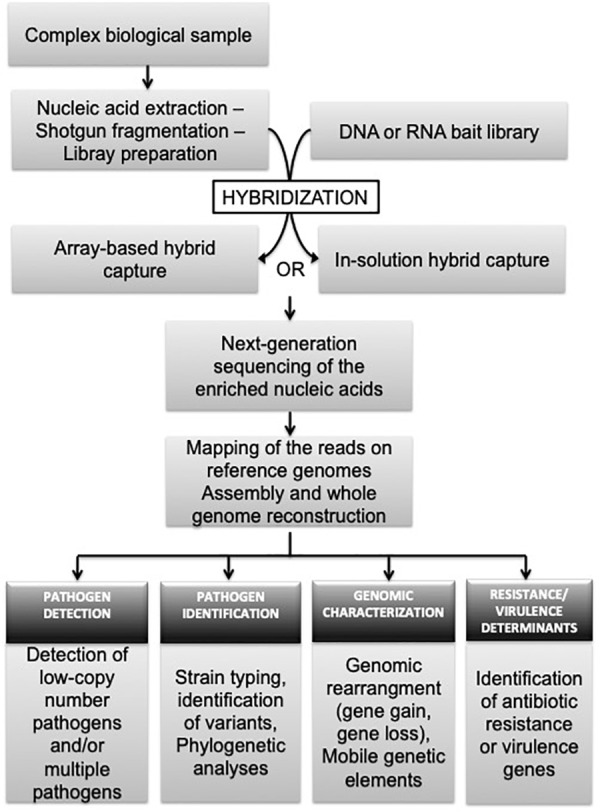
Overview of target-enrichment sequencing procedure and its application to research and diagnostic infectiology.
Early Developments of Hybrid-Capture Strategies: Human Genetic Studies on Modern and Ancient Samples
Target-enrichment strategy using hybrid capture was originally developed for human genomic studies for which it was used to capture and sequence the entire human exome. This genomic technique, also called exome sequencing (or whole exome sequencing) was first applied by using an array-based hybrid capture method in 2007 (Hodges et al., 2007). In this study, the authors developed six customized NimbleGen arrays to capture about 180,000 coding exons with overlapping 60–90-nt probes allowing an average enrichment of exon DNA sequences of 323 folds. Whole exome sequencing using capture arrays has proven its usefulness in identifying rare variants and mutations causing disease (Choi et al., 2009; Ng et al., 2009). The limitations of this technique include the need to design an array and a relatively large amount of DNA. To overcome some of the weaknesses of the previous method, Gnirke et al. (2009) have developed an in-solution hybrid capture method for human whole exome sequencing. To do so, biotinylated RNA baits of 170 bases in length were constructed, targeting 5,565 human protein-coding exons. In this study, authors have demonstrated the possibility to perform hybrid selection in solution. Following this, many targeted human exome in-solution enrichment methods for NGS have been developed, including those commercialized by Illumina 1 and Agilent Genomics 2 (Chen et al., 2015a,b). In-solution capture for exome sequencing turned out to be an effective approach applied to discover the causal mutation of rare Mendelian disorders (Shearer et al., 2010; Martignetti et al., 2013; Nectoux et al., 2015; Rousseau-Nepton et al., 2015), of complex disorders (Poultney et al., 2013; Guipponi et al., 2014; Griesi-Oliveira et al., 2015; Pérez-Serra et al., 2015), mitochondrial disorders (Calvo et al., 2012; Gai et al., 2013) and more recently of the screening of potential genetic mutation of patients suffering from cancer (Sikkema-Raddatz et al., 2013; Drilon et al., 2015; Xie et al., 2016; Rozenblum et al., 2017; Xu et al., 2017; Clark T.A. et al., 2018; Schrock et al., 2018).
The power of hybridization capture has been also successfully used to study human ancient DNA (aDNA) preserved in ancient human remains. Indeed, in ancient human samples, DNA is highly fragmented (thus a shotgun fragmentation step is usually not required) and dominated by a large contamination of environmental and bacterial DNA, which poses a limitation in shotgun aDNA sequencing experiment (Knapp and Hofreiter, 2010). Another characteristic of aDNA is cytosine deamination on the ends of DNA fragments. Library construction can be done directly on the double stranded DNAs (dsDNAs) or single stranded DNAs (ssDNAs) and may include cytosine deamination removal by the use of a damage treatment step with uracil DNA glycosylase and/or endonuclease VIII (Briggs et al., 2010). The first genetic marker analyzed in human paleogenetic studies was mitochondrial DNA (mtDNA) because of its higher copy number in the cell than nuclear DNA. Probe hybridization assays used biotinylated DNA or RNA probes targeting the two hypervariable segments of the mtDNA control region (CR) (Briggs et al., 2009; Krause et al., 2010; Maricic et al., 2010; Enk et al., 2013; Kihana et al., 2013; Templeton et al., 2013; Eduardoff et al., 2017; Loreille et al., 2018). Another uniparental marker, the Y-chromosome DNA (Y-DNA), was also used to study aDNA. As each cell possesses only one copy of the Y chromosome, the hybridization capture was carried out to enrich specific genomic regions of the Y chromosome both on solid support (Fu et al., 2013) and in solution (Cruz Dávalos et al., 2017). However, targeting mitochondrial DNA or Y chromosome involves discarding a large proportion of potentially informative sequences present in autosomal DNA. For this reason, Carpenter et al. (2013) reported a new capture-based method, called whole-genome in-solution capture (WISC), using modern DNA as bait covering the entire human genome. This method was applied to 12 ancient human DNA libraries and showed an enrichment of 6 to 159 folds of the sequence mapping to the human genome with enrichments of 2 to 13 folds for unique fragments (Carpenter et al., 2013). As for modern human genetic studies, commercial kits targeting mitochondrial DNA, custom loci, or entire nuclear genomes, such as those developed by Arbor Bioscience (myBaits®3) are now employed in the genetic sequencing of ancient DNA (Enk et al., 2014; Lindo et al., 2017).
Applications of Target-Enrichment Sequencing to Human Infectious Diseases
Parasites and Fungi
The first application of hybrid selection method for infectious diseases was in the field of human parasitology research(Melnikov et al., 2011; Table 1). To overcome the low proportion of Plasmodium falciparum sequences relative to that of their human host, authors have proposed to adapt in-solution NGS hybrid capture method to enrich this pathogen. This protocol has been tested in both mock mixtures composed of 99% human DNA and 1% Plasmodium but also in P. falciparum clinical samples. For this purpose, synthetic 140 bp oligos labeled with biotin were designed to capture exonic regions of the P. falciparum genome, whereas 250 bp oligos were constructed to target the entire genome. Processed and unprocessed samples were then sequenced with an Illumina technology. In the mockmetagenome, sequencing of the hybrid-selected samples yielded between 37 to 44-fold enrichment of the parasite DNA. In the human clinical sample, Illumina sequencing showed that at least 5.9% of reads mapped to Plasmodium, but no data was provided regarding the percentage of Plasmodium reads obtained without hybrid capture (Melnikov et al., 2011). However, this first study highlighted the good performance of NGS hybrid capture to sequence parasite genome from human clinical samples. In 2012, other studies confirmed the good performance of in-solution hybrid capture to enrich P. falciparum (Smith et al., 2012) sequences and P. vivax (Bright et al., 2012).
Table 1.
Example of studies that used target-enrichment sequencing for parasitic, fungal, bacterial, or viral diseases in modern and ancient samples.
| Targeted organism | Probes design | Infectious disease | Sample tested | Methods | Nucleic acids | Sequencing | Reference |
|---|---|---|---|---|---|---|---|
| Parasitic and fungal disease | |||||||
| Plasmodium falciparum 3D7 | Whole genome | Malaria | Mock sample whole blood | In-solution | DNA | Illumina GAIIx Illumina HiSeq | Melnikov et al., 2011 |
| P. falciparum 3D7 | Whole genome | Malaria | Mock sample Whole blood | In-solution | DNA | Illumina GAIIx | Smith et al., 2012 |
| Plasmodium vivax | Whole genome | Malaria | Whole blood | In-solution | DNA | Illumina HiSeq | Bright et al., 2012 |
| Candida albicans | 6094 ORFS | Systemic candidiasis | Animal model of infection | In-solution | RNA (cDNA) | Illumina HiSeq | Amorim-Vaz et al., 2015 |
| Bacterial disease | |||||||
| Chlamydia trachomatis | 74 References whole genome | Trachoma | Vaginal swabs Urine | In-solution | DNA | Illumina MiSeq | Christiansen et al., 2014 |
| Mycobacterium tuberculosis H37Rv | Whole genome | Multidrug-resistant tuberculosis | Sputum | In-solution | DNA | Illumina MiSeq | Brown et al., 2015 |
| Neisseria meningitidis | 77 Complete reference genome and 2898 drafts | Invasive meningococcal disease | CSF Whole blood | In-solution | DNA | Illumina MiSeq | Clark S.A. et al., 2018 |
| Viral disease | |||||||
| Varicella-zoster virus Epstein–Barr virus Kaposi’s Sarcoma-Associated Herpesvirus | Whole genome | Zoster-vaccine rash, wild-type zoster, encephalitis | Clinical and cultured samples | In-solution | DNA | Illumina GAIIx | Depledge et al., 2011 |
| Merkel cell polyomavirus (MCPyV) | 23 Overlapping PCR products that tiled across the MCPyV genome | Merkel cell carcinoma | Formalin-Fixed, Paraffin-Embedded Tissue | In-solution | DNA | Illumina GAIIx | Duncavage et al., 2011 |
| Human Herpesvirus 7 (HHV7) | Whole genome | Exanthem subitum | Cellular culture | In-solution | DNA | Illumina MiSeq | Donaldson et al., 2013 |
| Epstein–Barr Virus (EBV) | Whole genome | Nasopharyngeal carcinoma (NPC) | NPC tumor biopsy | In-solution | DNA | Illumina MiSeq | Kwok et al., 2014 |
| Zika virus (ZIKV) | Whole genome | ZIKV infection | Blood, urine, cerebrospinal fluid, and saliva | In-solution | RNA (cDNA) | Illumina HiSeq | Metsky et al., 2017 |
| Lassa virus | 2 References complete genome | Hemorrhagic fevers | Plasma or serum | In-solution | RNA (cDNA) | Illumina HiSeq | Matranga et al., 2014 |
| Varicella Zoster Virus (VZV) | Whole genome | Vaccine-associated rashes | Vesicular fluid | In-solution | DNA | Illumina (GAIIx, HiSeq, and MiSeq) | Depledge et al., 2014 |
| ViroCap 337 DNA and RNA viral species infecting human and animals | 1456 RefSeq + genome neighbor sequences (185,835 fasta total) | n.a. | Panel of human samples | In-solution | DNA and RNA (cDNA) | Illumina HiSeq | Wylie et al., 2015 |
| VirCapSeq-VERT 207 viral taxa infecting vertebrates | 342,438 Coding sequences (1,993,200 oligonucleotide probes) | n.a. | Mock samples Human clinical samples and animal samples | In-solution | DNA and RNA (cDNA) | Illumina HiSeq | Briese et al., 2015 |
| Norovirus | 662 References complete or partial genome | Acute gastroenteritis | Stool suspensions | In-solution | RNA (cDNA) | Illumina MiSeq | Brown et al., 2016 |
| Hepatitis C Virus (HCV) | 953 Reference complete genomes | HCV infections | Plasma Mock samples RNA transcripts | In-solution | RNA (cDNA) | Illumina MiSeq | Thomson et al., 2016 |
| Exogenous retroviruses Immunodeficiency virus type-1 (HIV-1) and the human T-cell leukemia virus type-1 (HTLV-1) | Whole genome | n.a. | Infected cells | In-solution | DNA | Illumina MiSeq | Miyazato et al., 2016 |
| Herpes simplex virus 1 and 2 (HSV-1/HSV-2) | Whole genome | Skin rash, herpetic lesions | Swab of the lesion | In-solution | DNA | Illumina MiSeq | Greninger et al., 2018 |
| ViroFind 535 DNA and RNA viral species that are known to infect humans | Whole genome | Progressive multifocal leukoencephalopathy (PML) | :Post mortem PML brain samples | In-solution | DNA and RNA (cDNA) | Illumina MiSeq | Chalkias et al., 2018 |
| Infectious diseases in paleomicrobiology | |||||||
| Yersinia pestis strain CO92 | Whole genome or pCD1 and pMT1 plasmids | Plague (Black Death) 1347–1351 | Teeth | Microarray | DNA | Illumina GAIIx | Bos et al., 2011 |
| Y. pestis strain CO92 | Portion of the Y. pestis pPCP1 plasmid | Plague (Black Death) 1347–1351 | Bones and Teeth | In-solution | DNA | Illumina GAIIx | Schuenemann et al., 2011 |
| Y. pestis strain CO92 | Core genome, plasmids and 155 other genes | Plague (Justinian) (6–8th centuries) | Teeth | In-solution | DNA | Illumina HiSeq | Wagner et al., 2014 |
| Mycobacterium tuberculosis | rpoB, gyrA, gyrB, katG, and mpt40 genes | Tuberculosis (1000 year old) | Skeletal samples | In-solution | DNA | Illumina HiSeq | Bos et al., 2014 |
| Mycobacterium leprae | 3 Genomic loci | Leprosy (11th to 14th century) | Bones and teeth | In-solution | DNA | Illumina MiSeq and HiSeq | Schuenemann et al., 2013 |
| Variola virus | Whole genome | Smallpox (1643–1665) | Mummified tissues of a child | In-solution | DNA | Illumina HiSeq | Duggan et al., 2016 |
| P. falciparum | Mitochondrial genomes of Plasmodium spp. | Malaria (1st–2nd century) | Teeth | In-solution | DNA | Illumina HiSeq | Marciniak et al., 2016 |
| Treponema pallidum | Whole genome | Syphilis (1681 to 1861) | Bones | Microarray | DNA | Illumina HiSeq | Schuenemann et al., 2018 |
n.a., not applicable.
Fungi are also a major cause of human diseases that can be particularly serious in immunocompromized patients or in patients hospitalized for serious diseases (Pfaller and Diekema, 2010). For example, systemic infections with Candida albicans in immunocompromized patients result in mortality rates of about 50% (Pfaller and Diekema, 2010). The prevention, diagnosis and therapy of fungi infections remain very difficult and comprehension of transcriptional regulation between fungal pathogens and host is an important step to identify potential novel targets for drug development (Pfaller and Diekema, 2010). Again, the limitations of host and pathogen transcriptome analysis lie in the low proportion of fungal RNA present in the total extracted RNA. The use of specific enrichment procedures before RNASeq analysis has then been proposed as an alternative method to overcome the problem of low fungus/host RNA ratio. For this purpose, Amorim-Vaz et al. (2015) have designed a set of 55,342 biotinylated 120 bp-RNA probes covering 6,094 C. albicans ORFs. cDNA libraries were established using SureSelect (Agilent) after extraction of RNA from mice kidney or Galleria mellonella larvae infected with C. albicans, and were subjected to capture with biotinylated probes before Illumina HiSeq sequencing. Results showed up to a 1670-fold enrichment of C. albicans reads in a given biological sample and a detection of more than 86% of its genes. Many genes that have been regulated in in vivo infection experiments have functions that have not yet been characterized and will require further research to understand their role during infection (Amorim-Vaz et al., 2015).
Bacteria
In bacteriological research and diagnostic, targeted capture strategies prior to sequencing could be a powerful tool in the management and therapeutics of patients with infectious disease. Indeed, the rapid identification of antimicrobial resistance is essential for a rapid and effective treatment. Regarding Mycobacterium tuberculosis, current methods of screening for antimicrobial resistance, which are based on the culture of the organism from sputum samples before sequencing, can take up to several weeks. To overcome these limitations, Brown et al. have proposed to use oligonucleotide enrichment technology to capture M. tuberculosis genome sequences directly from positive smear sputum samples (Brown et al., 2015; Table 1). Whole genome baits (120-mer RNA baits) were designed to span the entire positive strand of the H37Rv M. tuberculosis reference genome and synthesized by Agilent Technologies. The authors demonstrated the reliability of targeted sequencing to recover and sequence, in less than 96 h, nearly complete genomes directly from 81% (21/26) smear positive sputa but also its robustness to identify the genotype and resistance determinants of all samples that were previously tested positive samples. This study emphasizes the use of hybrid selection target enrichment that could allow personalized antimicrobial treatment in multidrug-resistant tuberculosis (Brown et al., 2015). Other studies have used biotinylated baits spanning entire genomes for high-resolution strain genotyping directly from clinical samples. Indeed, discrimination of Chlamydia trachomatis serovars from genital samples would facilitate the study of population structures and modes of transmission (Christiansen et al., 2014) while genomic data from uncultured Neisseria meningitidis not grown in the case of invasive meningococcal would allow increased surveillance of vaccine antigens and studies on possible vaccine deficiencies (Clark S.A. et al., 2018).
Viruses
In viral research and diagnostic laboratories, viral WGS is also essential for the detection of drug resistance and the development of novel treatments and vaccines (Houldcroft et al., 2017). In this domain, the first study that demonstrated the effectiveness of target capture technology for reconstructing full herpesvirus genomes from complex biological samples was proposed by Depledge et al. (2011) (Table 1). In this study, 120-mer RNA baits generating a 2× coverage for Varicella-Zoster Virus (VZV), a 5× coverage for Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated Herpesvirus (KSHV), were synthetized and hybridized with DNA extracted from a range of clinical samples including blood, saliva, vesicle fluid, cerebrospinal fluid, and tumor cell lines. Full-length herpes virus genomes were reconstructed at high read depth for the 13 samples tested and generated further studies on the structure and diversity of the viral population (Depledge et al., 2011). Following this study, the capture of whole genomic hybrids made it possible to study the genomic diversity of eight new complete EBV genomes isolated from biopsy specimens of primary nasopharyngeal carcinomas (Kwok et al., 2014), 37 Zika virus genomes (ZIKV) samples out of 66 attempts (Metsky et al., 2017), 453 complete genomes (with >90% genome coverage and >100-fold read depth) of different norovirus genotypes from 509 stool samples (Brown et al., 2016) and to achieve sufficient coverage for de novo genome assembly and detection of single nucleotide variants of Lassa virus (LASV) from ultra-low input samples (Matranga et al., 2014). This approach has been also used to characterize other clinically relevant viruses, such as hepatitis C virus (HCV) (Thomson et al., 2016), varicella zoster virus (Depledge et al., 2014), human herpesvirus 7 (HHV-7) (Donaldson et al., 2013, 7) and the herpes simplex virus 1 and 2 (HSV-1 and HSV-2) (Greninger et al., 2018). Hybrid capture associated with shotgun sequencing could also be performed using a combination of several viral species used as baits. Indeed, Wylie et al. (2015) developed ViroCap, a panel of probes designed to enrich nucleic acid from 34 families of DNA and RNA viruses (190 viral genera and 337 species) that infect vertebrate hosts, except human endogenous retrovirus. These probes were tested both on a pool of 14 clinical samples, which tested positive for a viral infection, and on eight samples from young children with fever, also positive for one or more viruses. Libraries were sequenced before capture (pre-capture) and following capture using ViroCap (post-capture). Combining results from both experiments, 32 viruses were detected (11 additional in the post-captured samples), including diverse DNA and RNA viruses (with genomes ranging from 5–161 kb) with genomic coverage >80% for 16 of the 32 genomes. Several complete genomes were reconstructed and belonged to Human bocavirus 1, Human parvovirus B19, human adenovirus B (type 3), human adenovirus C (type 1), KI polyomavirus, sapovirus, and human astrovirus 1. Finally, although ViroCap cannot detect viral sequences that are completely novel, its design, which includes neighbor genomes of reference sequences, allows variants with nucleotide sequence identity as low as 58% to be identified (Wylie et al., 2015). The same year, VirCapSeq-VERT, a virome capture sequencing platform targeting 207 viral taxa infecting vertebrates was described (Briese et al., 2015). VirCapSeq-VERT allowed reduction of background human DNA and a 100 to 10,000 fold enrichment in viral reads when compared with other enrichment procedures such as treatment with nucleases or RiboZero rRNA depletion (Briese et al., 2015). In 2018, a similar approach called ViroFind was designed to target 535 DNA and RNA viruses, which are known to infect humans or cause zoonoses. This in-solution target enrichment was applied to the brain biopsy samples of five patients with progressive multifocal leukoencephalopathy (PML) (Chalkias et al., 2018). It allowed the description of highly complex Polyoma virus JC populations as well as the detection of large genetic divergence among variants, with some of these mutations conferring viral fitness advantages (Chalkias et al., 2018). Lastly, other applications of target-enrichment sequencing have been described, such as the study of viral genome integrations within the human genome. This approach was powerful and efficient to identify Merkel Cell Polyomavirus (MCPyV) insertion sites on DNA extracted from formalin-fixed and paraffin-embedded tissue from Merkel cell carcinoma (Duncavage et al., 2011). It also allowed to analyze retroviral genomes integrated within host genomic DNA in case of human T-cell leukemia virus type-1 (HTLV-1) and human immunodeficiency virus type-1 (HIV-1) infections (Miyazato et al., 2016).
Paleomicrobiology
Paleomicrobiology is an emerging research field dedicated to the detection, identification and characterization of microorganisms (bacteria, viruses, and parasites) in ancient specimens. Elucidating past infectious diseases can lead to a better understanding of the temporal and geographical distributions of infected individuals, the introduction of microorganisms into human populations, the host-pathogen relationships but also the genetic evolution of the microorganisms (Drancourt and Raoult, 2005). The main limitations of paleomicrobiological studies concern the degradation of ancient DNA (aDNA) and the risk of contamination by modern DNA (Riviera-Perez et al., 2016). Target-enrichment prior to sequencing is therefore a particularly relevant tool in this context for genomes study. The first two studies using targeted enrichment in paleomicrobiology have investigated genetic changes and virulence factor of Yersinia pestis, the causal agent of the second plague pandemics (Black Death, 14–17th centuries) (Bos et al., 2011; Schuenemann et al., 2011; Table 1). To this end, an array-based enrichment using probe targeting either the full Y. pestis chromosome or pestis-specific virulence plasmids was applied directly after the DNA extraction from ancient bones (Schuenemann et al., 2011, 1) and/or teeth (Bos et al., 2011; Schuenemann et al., 2011). Using targeted DNA capture approach combined with high-throughput sequencing, the authors reconstructed 99% of the pPCP1 plasmid sequence (Schuenemann et al., 2011) and a draft genome of Y. pestis (Bos et al., 2011) with the molecular damages typically associated with aDNA. Comparisons with modern genomes did not identify any significant genetic variation that could explain the differences between the ancient and modern forms of the disease (Schuenemann et al., 2011). More recently, three other draft genomes of Y. pestis have been recovered from individuals who died during the first plague pandemics (the Plague of Justinian, 6–8th centuries) in two different rural sites in southern Germany (Wagner et al., 2014; Feldman et al., 2016). Genetic characterization showed that these 3 drafts derived from a single Justinianic strain which is unique and harbors novel substitutions and structural polymorphism (Wagner et al., 2014; Feldman et al., 2016). Finally, target enrichment sequencing also allowed the reconstruction of new Y. pestis strains from Bronze Age individuals (∼3,800 BP) (Spyrou et al., 2018) providing further datas into the early stages of Y. pestis genome evolution including on genomic characteristics supporting flea-borne transmission in rodents or humans (Spyrou et al., 2018). Finally, target-enrichment sequencing approaches in the paleomicrobiological research field have not been exclusively applied to the study of ancient plague pandemics, but have also allowed genomic investigation of ancient Mycobacterium tuberculosis (Bos et al., 2014), M. leprae (Schuenemann et al., 2013), Variola virus (Duggan et al., 2016), P. falciparum (Marciniak et al., 2016), and Treponema pallidum (Schuenemann et al., 2018) in human remains.
Conclusion
Target-enrichment sequencing is an efficient approach that allows large fragments and even entire sequences of the genome of targeted microorganisms to be reconstructed directly from modern and ancient complex biological samples containing a low pathogen/host nucleic acid ratio. The information provided by the genome can be used to explore the genetic diversity, epidemiology, evolutionary traits, transmission networks, host-pathogen interactions or antimicrobial resistance of the target pathogen or its variants. The main current limitations of democratizing target-enrichment sequencing in clinical diagnostic laboratories are its elevated cost, the high expertise required for library preparation and the necessary time to generate biotinylated probes from reference genomes, which hampers a rapid response to an emerging pathogen. Above all, it is not suitable for the detection and characterization of completely novel microorganisms, including viruses whose emergence may represent one of the main threats to human health in the near future.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This work was supported by the Agence Nationale de la Recherche (reference: ANR-13-JSV6-0004), by the IHU Méditerranée Infection, Marseille, France, by the French Government under the “Investissements d’Avenir” Program (reference: Méditerranée Infection 10-IAHU-03), by the Région Provence Alpes Côte d’Azur, and by the European funding FEDER PRIMI.
References
- Albert T. J., Molla M. N., Muzny D. M., Nazareth L., Wheeler D., Song X., et al. (2007). Direct selection of human genomic loci by microarray hybridization. Nat. Methods 4 903–905. 10.1038/nmeth1111 [DOI] [PubMed] [Google Scholar]
- Amorim-Vaz S., Tran V. D. T., Pradervand S., Pagni M., Coste A. T., Sanglard D. (2015). RNA enrichment method for quantitative transcriptional analysis of pathogens in vivo applied to the fungus Candida albicans. mBio 6:e00942-15. 10.1128/mBio.00942-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos K. I., Harkins K. M., Herbig A., Coscolla M., Weber N., Comas I., et al. (2014). Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 514 494–497. 10.1038/nature13591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos K. I., Schuenemann V. J., Golding G. B., Burbano H. A., Waglechner N., Coombes B. K., et al. (2011). A draft genome of Yersinia pestis from victims of the Black Death. Nature 478 506–510. 10.1038/nature10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese T., Kapoor A., Mishra N., Jain K., Kumar A., Jabado O. J., et al. (2015). Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 6:e01491-15. 10.1128/mBio.01491-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs A. W., Good J. M., Green R. E., Krause J., Maricic T., Stenzel U., et al. (2009). Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325 318–321. 10.1126/science.1174462 [DOI] [PubMed] [Google Scholar]
- Briggs A. W., Stenzel U., Meyer M., Krause J., Kircher M., Pääbo S. (2010). Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 38:e87. 10.1093/nar/gkp1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright A. T., Tewhey R., Abeles S., Chuquiyauri R., Llanos-Cuentas A., Ferreira M. U., et al. (2012). Whole genome sequencing analysis of Plasmodium vivax using whole genome capture. BMC Genomics 13:262. 10.1186/1471-2164-13-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C., Bryant J. M., Einer-Jensen K., Holdstock J., Houniet D. T., Chan J. Z. M., et al. (2015). Rapid whole-genome sequencing of Mycobacterium tuberculosis isolates directly from clinical samples. J. Clin. Microbiol. 53 2230–2237. 10.1128/JCM.00486-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. R., Roy S., Ruis C., Yara Romero E., Shah D., Williams R., et al. (2016). Norovirus whole-genome sequencing by sureselect target enrichment: a robust and sensitive method. J. Clin. Microbiol. 54 2530–2537. 10.1128/JCM.01052-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo S. E., Compton A. G., Hershman S. G., Lim S. C., Lieber D. S., Tucker E. J., et al. (2012). Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 4:118ra10. 10.1126/scitranslmed.3003310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter M. L., Buenrostro J. D., Valdiosera C., Schroeder H., Allentoft M. E., Sikora M., et al. (2013). Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am. J. Hum. Genet. 93 852–864. 10.1016/j.ajhg.2013.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkias S., Gorham J. M., Mazaika E., Parfenov M., Dang X., DePalma S., et al. (2018). ViroFind: A novel target-enrichment deep-sequencing platform reveals a complex JC virus population in the brain of PML patients. PLoS One 13:e0186945. 10.1371/journal.pone.0186945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Im H., Snyder M. (2015a). Whole-exome enrichment with the agilent sureselect human All exon platform. Cold Spring Harb. Protoc. 2015 626–633. 10.1101/pdb.prot083659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Im H., Snyder M. (2015b). Whole-exome enrichment with the illumina TruSeq exome enrichment platform. Cold Spring Harb. Protoc 2015:db.rot084863. 10.1101/pdb.prot084863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M., Scholl U. I., Ji W., Liu T., Tikhonova I. R., Zumbo P., et al. (2009). Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 106 19096–19101. 10.1073/pnas.0910672106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen M. T., Brown A. C., Kundu S., Tutill H. J., Williams R., Brown J. R., et al. (2014). Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples. BMC Infect. Dis. 14:591. 10.1186/s12879-014-0591-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S. A., Doyle R., Lucidarme J., Borrow R., Breuer J. (2018). Targeted DNA enrichment and whole genome sequencing of Neisseria meningitidis directly from clinical specimens. Int. J. Med. Microbiol. 308 256–262. 10.1016/j.ijmm.2017.11.004 [DOI] [PubMed] [Google Scholar]
- Clark T. A., Chung J. H., Kennedy M., Hughes J. D., Chennagiri N., Lieber D. S., et al. (2018). Analytical validation of a hybrid capture-based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J. Mol. Diagn. 20 686–702. 10.1016/j.jmoldx.2018.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz Dávalos D. I., Nieves-Colón M. A., Sockell A., Poznik D. G., Schroeder H., Stone A. C., et al. (2017). In-solution Y-chromosome capture-enrichment on ancient DNA libraries. BMC Genomics 19:608. 10.1186/s12864-018-4945-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depledge D. P., Kundu S., Jensen N. J., Gray E. R., Jones M., Steinberg S., et al. (2014). Deep sequencing of viral genomes provides insight into the evolution and pathogenesis of Varicella zoster virus and its vaccine in humans. Mol. Biol. Evol. 31 397–409. 10.1093/molbev/mst210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depledge D. P., Palser A. L., Watson S. J., Lai I. Y.-C., Gray E. R., Grant P., et al. (2011). Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS One 6:e27805. 10.1371/journal.pone.0027805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson C. D., Clark D. A., Kidd I. M., Breuer J., Depledge D. D. (2013). Genome sequence of human herpesvirus 7 strain UCL-1. Genome Announc. 1:e00830-13. 10.1128/genomeA.00830-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drancourt M., Raoult D. (2005). Palaeomicrobiology: current issues and perspectives. Nat. Rev. Microbiol. 3 23–35. 10.1038/nrmicro1063 [DOI] [PubMed] [Google Scholar]
- Drilon A., Wang L., Arcila M. E., Balasubramanian S., Greenbowe J. R., Ross J. S., et al. (2015). Broad, hybrid capture-based next-generation sequencing identifies actionable genomic alterations in “driver-negative” lung adenocarcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 21 3631–3639. 10.1158/1078-0432.CCR-14-2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan A. T., Perdomo M. F., Piombino-Mascali D., Marciniak S., Poinar D., Emery M. V., et al. (2016). 17th century variola virus reveals the recent history of smallpox. Curr. Biol. 26 3407–3412. 10.1016/j.cub.2016.10.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncavage E. J., Magrini V., Becker N., Armstrong J. R., Demeter R. T., Wylie T., et al. (2011). Hybrid capture and next-generation sequencing identify viral integration sites from formalin-fixed, paraffin-embedded tissue. J. Mol. Diagn. 13 325–333. 10.1016/j.jmoldx.2011.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eduardoff M., Xavier C., Strobl C., Casas-Vargas A., Parson W. (2017). Optimized mtDNA control region primer extension capture analysis for forensically relevant samples and highly compromised mtDNA of different age and origin. Genes 8:E237. 10.3390/genes8100237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M. C., Gibbs R. A. (1994). Multiplex PCR: advantages, development, and applications. PCR Methods Appl. 3 S65–S75. 10.1101/gr.3.4.S65 [DOI] [PubMed] [Google Scholar]
- Enk J., Rouillard J.-M., Poinar H. (2013). Quantitative PCR as a predictor of aligned ancient DNA read counts following targeted enrichment. BioTechniques 55 300–309. 10.2144/000114114 [DOI] [PubMed] [Google Scholar]
- Enk J. M., Devault A. M., Kuch M., Murgha Y. E., Rouillard J.-M., Poinar H. N. (2014). Ancient whole genome enrichment using baits built from modern DNA. Mol. Biol. Evol. 31 1292–1294. 10.1093/molbev/msu074 [DOI] [PubMed] [Google Scholar]
- Feldman M., Harbeck M., Keller M., Spyrou M. A., Rott A., Trautmann B., et al. (2016). A high-coverage Yersinia pestis genome from a sixth-century justinianic plague victim. Mol. Biol. Evol. 33 2911–2923. 10.1093/molbev/msw170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q., Meyer M., Gao X., Stenzel U., Burbano H. A., Kelso J., et al. (2013). DNA analysis of an early modern human from Tianyuan Cave. China. Proc. Natl. Acad. Sci. U.S.A. 110 2223–2227. 10.1073/pnas.1221359110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai X., Ghezzi D., Johnson M. A., Biagosch C. A., Shamseldin H. E., Haack T. B., et al. (2013). Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 93 482–495. 10.1016/j.ajhg.2013.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasc C., Peyretaillade E., Peyret P. (2016). Sequence capture by hybridization to explore modern and ancient genomic diversity in model and nonmodel organisms. Nucleic Acids Res. 44 4504–4518. 10.1093/nar/gkw309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirke A., Melnikov A., Maguire J., Rogov P., LeProust E. M., Brockman W., et al. (2009). Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27 182–189. 10.1038/nbt.1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greninger A. L., Roychoudhury P., Xie H., Casto A., Cent A., Pepper G., et al. (2018). Ultrasensitive capture of human herpes simplex virus genomes directly from clinical samples reveals extraordinarily limited evolution in cell culture. mSphere 3:e00283-18. 10.1128/mSphereDirect.00283-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesi-Oliveira K., Acab A., Gupta A. R., Sunaga D. Y., Chailangkarn T., Nicol X., et al. (2015). Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry 20 1350–1365. 10.1038/mp.2014.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guipponi M., Santoni F. A., Setola V., Gehrig C., Rotharmel M., Cuenca M., et al. (2014). Exome sequencing in 53 sporadic cases of schizophrenia identifies 18 putative candidate genes. PLoS One 9:e112745. 10.1371/journal.pone.0112745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges E., Xuan Z., Balija V., Kramer M., Molla M. N., Smith S. W., et al. (2007). Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 39 1522–1527. 10.1038/ng.2007.42 [DOI] [PubMed] [Google Scholar]
- Houldcroft C. J., Beale M. A., Breuer J. (2017). Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 15 183–192. 10.1038/nrmicro.2016.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihana M., Mizuno F., Sawafuji R., Wang L., Ueda S. (2013). Emulsion PCR-coupled target enrichment: an effective fishing method for high-throughput sequencing of poorly preserved ancient DNA. Gene 528 347–351. 10.1016/j.gene.2013.07.040 [DOI] [PubMed] [Google Scholar]
- Knapp M., Hofreiter M. (2010). Next generation sequencing of ancient DNA: requirements, strategies and perspectives. Genes 1 227–243. 10.3390/genes1020227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarewa I., Armisen J., Gardner A. F., Slatko B. E., Hendrickson C. L. (2015). Overview of target enrichment strategies. Curr. Protoc. Mol. Biol. 112:7.21.1–7.21.23. 10.1002/0471142727.mb0721s112 [DOI] [PubMed] [Google Scholar]
- Krause J., Fu Q., Good J. M., Viola B., Shunkov M. V., Derevianko A. P., et al. (2010). The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464 894–897. 10.1038/nature08976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok H., Wu C. W., Palser A. L., Kellam P., Sham P. C., Kwong D. L. W., et al. (2014). Genomic diversity of Epstein-barr virus genomes isolated from primary nasopharyngeal carcinoma biopsy samples. J. Virol. 88 10662–10672. 10.1128/JVI.01665-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnik E. A., Freier S. M. (1995). Relative thermodynamic stability of DNA, RNA, and DNA:RNA hybrid duplexes: relationship with base composition and structure. Biochemistry 34 10807–10815. 10.1021/bi00034a013 [DOI] [PubMed] [Google Scholar]
- Lindo J., Achilli A., Perego U. A., Archer D., Valdiosera C., Petzelt B., et al. (2017). Ancient individuals from the North American Northwest Coast reveal 10,000 years of regional genetic continuity. Proc. Natl. Acad. Sci. U.S.A. 114 4093–4098. 10.1073/pnas.1620410114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loreille O., Ratnayake S., Bazinet A. L., Stockwell T. B., Sommer D. D., Rohland N., et al. (2018). Biological sexing of a 4000-year-old egyptian mummy head to assess the potential of nuclear DNA recovery from the most damaged and limited forensic specimens. Genes 9:E135. 10.3390/genes9030135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamanova L., Coffey A. J., Scott C. E., Kozarewa I., Turner E. H., Kumar A., et al. (2010). Target-enrichment strategies for next-generation sequencing. Nat. Methods 7 111–118. 10.1038/nmeth.1419 [DOI] [PubMed] [Google Scholar]
- Marciniak S., Prowse T. L., Herring D. A., Klunk J., Kuch M., Duggan A. T., et al. (2016). Plasmodium falciparum malaria in 1st-2nd century CE southern Italy. Curr. Biol. 26 R1220–R1222. 10.1016/j.cub.2016.10.016 [DOI] [PubMed] [Google Scholar]
- Maricic T., Whitten M., Pääbo S. (2010). Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS One 5:e14004. 10.1371/journal.pone.0014004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignetti J. A., Tian L., Li D., Ramirez M. C. M., Camacho-Vanegas O., Camacho S. C., et al. (2013). Mutations in PDGFRB cause autosomal-dominant infantile Myofibromatosis. Am. J. Hum. Genet. 92 1001–1007. 10.1016/j.ajhg.2013.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matranga C. B., Andersen K. G., Winnicki S., Busby M., Gladden A. D., Tewhey R., et al. (2014). Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 15:519. 10.1186/PREACCEPT-1698056557139770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A., Galinsky K., Rogov P., Fennell T., Van Tyne D., Russ C., et al. (2011). Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 12:R73. 10.1186/gb-2011-12-8-r73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsky H. C., Matranga C. B., Wohl S., Schaffner S. F., Freije C. A., Winnicki S. M., et al. (2017). Zika virus evolution and spread in the Americas. Nature 546 411–415. 10.1038/nature22402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazato P., Katsuya H., Fukuda A., Uchiyama Y., Matsuo M., Tokunaga M., et al. (2016). Application of targeted enrichment to next-generation sequencing of retroviruses integrated into the host human genome. Sci. Rep. 6:28324. 10.1038/srep28324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nectoux J., de Cid R., Baulande S., Leturcq F., Urtizberea J. A., Penisson-Besnier I., et al. (2015). Detection of TRIM32 deletions in LGMD patients analyzed by a combined strategy of CGH array and massively parallel sequencing. Eur. J. Hum. Genet. 23 929–934. 10.1038/ejhg.2014.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S. B., Turner E. H., Robertson P. D., Flygare S. D., Bigham A. W., Lee C., et al. (2009). Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461 272–276. 10.1038/nature08250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okou D. T., Steinberg K. M., Middle C., Cutler D. J., Albert T. J., Zwick M. E. (2007). Microarray-based genomic selection for high-throughput resequencing. Nat. Methods 4 907–909. 10.1038/nmeth1109 [DOI] [PubMed] [Google Scholar]
- Pérez-Serra A., Toro R., Campuzano O., Sarquella-Brugada G., Berne P., Iglesias A., et al. (2015). A novel mutation in lamin a/c causing familial dilated cardiomyopathy associated with sudden cardiac death. J. Card. Fail. 21 217–225. 10.1016/j.cardfail.2014.12.003 [DOI] [PubMed] [Google Scholar]
- Pfaller M. A., Diekema D. J. (2010). Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 36 1–53. 10.3109/10408410903241444 [DOI] [PubMed] [Google Scholar]
- Poultney C. S., Goldberg A. P., Drapeau E., Kou Y., Harony-Nicolas H., Kajiwara Y., et al. (2013). Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am. J. Hum. Genet. 93 607–619. 10.1016/j.ajhg.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviera-Perez J. I., Santiago-Rodriguez T. M., Toranzos G. A. (2016). Paleomicrobiology: a snapshot of ancient microbes and approaches to forensic microbiology. Microbiol. Spectr. 4:EMF-0006-2015. 10.1128/microbiolspec.EMF-0006-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau-Nepton I., Okubo M., Grabs R., Mitchell J., Polychronakos C., Rodd C. (2015). A founder AGL mutation causing glycogen storage disease type IIIa in Inuit identified through whole-exome sequencing: a case series. CMAJ Can. Med. Assoc. J. 187 E68–E73. 10.1503/cmaj.140840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblum A. B., Ilouze M., Dudnik E., Dvir A., Soussan-Gutman L., Geva S., et al. (2017). Clinical impact of hybrid capture-based next-generation sequencing on changes in treatment decisions in lung cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 12 258–268. 10.1016/j.jtho.2016.10.021 [DOI] [PubMed] [Google Scholar]
- Schrock A. B., Pavlick D., Klempner S. J., Chung J. H., Forcier B., Welsh A., et al. (2018). Hybrid capture–based genomic profiling of circulating tumor dna from patients with advanced cancers of the gastrointestinal tract or anus. Clin. Cancer Res. 24 1881–1890. 10.1158/1078-0432.CCR-17-3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuenemann V. J., Bos K., DeWitte S., Schmedes S., Jamieson J., Mittnik A., et al. (2011). Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the Black Death. Proc. Natl. Acad. Sci. U.S.A. 108 E746–E752. 10.1073/pnas.1105107108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuenemann V. J., Lankapalli A. K., Barquera R., Nelson E. A., Hernández D. I., Alonzo V. A., et al. (2018). Historic Treponema pallidum genomes from Colonial Mexico retrieved from archaeological remains. PLoS Negl. Trop. Dis. 12:e0006447. 10.1371/journal.pntd.0006447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuenemann V. J., Singh P., Mendum T. A., Krause-Kyora B., Jäger G., Bos K. I., et al. (2013). Genome-wide comparison of medieval and modern Mycobacterium leprae. Science 341 179–183. 10.1126/science.1238286 [DOI] [PubMed] [Google Scholar]
- Shearer A. E., DeLuca A. P., Hildebrand M. S., Taylor K. R., Gurrola J., Scherer S., et al. (2010). Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl. Acad. Sci. U.S.A. 107 21104–21109. 10.1073/pnas.1012989107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J., Ji H. (2008). Next-generation DNA sequencing. Nat. Biotechnol. 26 1135–1145. 10.1038/nbt1486 [DOI] [PubMed] [Google Scholar]
- Sikkema-Raddatz B., Johansson L. F., de Boer E. N., Almomani R., Boven L. G., van den Berg M. P., et al. (2013). Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum. Mutat. 34 1035–1042. 10.1002/humu.22332 [DOI] [PubMed] [Google Scholar]
- Smith M., Campino S., Gu Y., Clark T. G., Otto T. D., Maslen G., et al. (2012). An In-solution hybridisation method for the isolation of pathogen DNA from human DNA-rich clinical samples for analysis by NGS. Open Genom. J. 5 18–29. 10.2174/1875693X01205010018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyrou M. A., Tukhbatova R. I., Wang C.-C., Valtueña A. A., Lankapalli A. K., Kondrashin V. V., et al. (2018). Analysis of 3800-year-old Yersinia pestis genomes suggests Bronze Age origin for bubonic plague. Nat. Commun. 9:2234. 10.1038/s41467-018-04550-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton J. E. L., Brotherton P. M., Llamas B., Soubrier J., Haak W., Cooper A., et al. (2013). DNA capture and next-generation sequencing can recover whole mitochondrial genomes from highly degraded samples for human identification. Investig. Genet. 4:26. 10.1186/2041-2223-4-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson E., Ip C. L. C., Badhan A., Christiansen M. T., Adamson W., Ansari M. A., et al. (2016). Comparison of next generation sequencing technologies for the comprehensive assessment of full-length hepatitis C viral genomes. J. Clin. Microbiol. 54 2470–2484. 10.1128/JCM.00330-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner D. M., Klunk J., Harbeck M., Devault A., Waglechner N., Sahl J. W., et al. (2014). Yersinia pestis and the plague of Justinian 541-543 AD: a genomic analysis. Lancet Infect. Dis. 14 319–326. 10.1016/S1473-3099(13)70323-2 [DOI] [PubMed] [Google Scholar]
- Wylie T. N., Wylie K. M., Herter B. N., Storch G. A. (2015). Enhanced virome sequencing using targeted sequence capture. Genome Res. 25 1910–1920. 10.1101/gr.191049.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Lu X., Wu X., Lin X., Zhang C., Huang X., et al. (2016). Capture-based next-generation sequencing reveals multiple actionable mutations in cancer patients failed in traditional testing. Mol. Genet. Genomic Med. 4 262–272. 10.1002/mgg3.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M.-D., Liu S.-L., Feng Y.-Z., Liu Q., Shen M., Zhi Q., et al. (2017). Genomic characteristics of pancreatic squamous cell carcinoma, an investigation by using high throughput sequencing after in-solution hybrid capture. Oncotarget 8 14620–14635. 10.18632/oncotarget.14678 [DOI] [PMC free article] [PubMed] [Google Scholar]