

Abstract
Botanical medicines have been utilized for centuries, but it remains challenging to identify bioactive constituents from complex botanical extracts. Bioassay-guided fractionation is often biased towards abundant or easily-isolatable compounds. To comprehensively evaluate active botanical mixtures, methods that allow for the prioritization of active compounds are needed. To this end, a method integrating bioassay-guided fractionation, biochemometric selectivity ratio analysis, and molecular networking was devised and applied to Angelica keiskei Koidzumi to comprehensively evaluate its antimicrobial activity against Staphylococcus aureus. This approach enabled the identification of putative active constituents early in the fractionation process, and provided structural information for these compounds. A subset of chalcone analogs were prioritized for isolation, yielding 4-hydroxyderricin (1, MIC ≤ 4.6 µM, IC50 = 2.0 µM), xanthoangelol (2, MIC ≤ 4.0 µM, IC50 = 2.3) and xanthoangelol K (4, IC50 = 168 µM). This approach allowed for the identification of a low abundance compound (xanthoangelol K) that has not been previously reported to possess antimicrobial activity, and facilitated a more comprehensive understanding of the compounds responsible for A. keiskei’s antimicrobial activity.
Keywords: Biochemometrics, chalcones, selectivity ratio, molecular networking, mass spectrometry, Angelica keiskei, Apiaceae
Introduction
The complexity of botanicals makes them a rich source for medicinally useful compounds, but leads to many analytical challenges. The traditional workflow for natural product discovery is bioassay-guided fractionation [1–2], in which bioactive extracts and subsequent fractions are chromatographically separated and retested for bioactivity until active compounds have been isolated. Because botanical extracts contain thousands of individual constituents, it is often difficult to assign activity to individual components, thus, the most abundant or easily isolatable compounds are often presumed to be responsible for bioactivity [3–4]. New methods are needed that will enable isolation efforts to be focused on those components most likely to be responsible for the desired biological activity.
Compounds from nature have been utilized to treat microbial infections throughout history [5], and some sources estimate that up to two-thirds of antibacterial agents on today’s market are derived from natural products [6]. The virtually limitless chemical diversity of natural products, particularly botanicals, results from their complex biosynthetic pathways, and many plant secondary metabolites, including flavonoids, alkaloids, and coumarins, have shown antimicrobial activity [1, 5, 7–10]. Angelica keiskei Koidzumi (Apiaceae), or ashitaba, is a member of the Angelica genus native to the southernmost islands of Japan that is popularly utilized as a food and a medicinal herb, purportedly to extend life expectancy, increase vitality, and to treat a broad range of diseases and infections [11]. Most of these activities result from the action of unique prenylated chalcones, as well as coumarins and flavanones (reviewed in [12]). Two compounds from A. keiskei, 4-hydroxyderricin (1) and xanthoangelol (2) have been shown to possess activity against methicillin-resistant Staphyloccoccus aureus (MRSA) [13]. Additionally, A. keiskei chalcones xanthoangelol F and isobavachalcone are active against other Gram-positive organisms, though they have not been tested against pathogenic bacteria such as MRSA [14]. With this study, we sought to employ antimicrobial extracts of A. keiskei as a test case for the development of new methods to prioritize bioactive compounds early in the isolation process for a complex botanical.
In combination with chromatographic techniques, mass spectrometry can be utilized to analyze hundreds of secondary metabolites simultaneously [3, 15–16]. Using a process called biochemometrics, quantitative chemical information and biological activity data can be incorporated into a statistical model. With this statistical modeling approach, it is possible to discovery chemical patterns related to bioactivity [3]. Partial least squares (PLS) analysis can be used in combination with chromatographic and mass spectrometric data to correlate metabolite profiles with biological data [17]. A recent study from our laboratory showed that selectivity ratio analysis was useful for the identification of trace bioactive constituents in fungal extracts without being confounded by highly abundant compounds [3]. The selectivity ratio compares the correlation and covariance to the residual variance, and provides a quantitative measurement of the ability of a given variable to differentiate between active and inactive groups [18].
Biochemometric analysis is helpful for distinguishing between active and inactive chemical constituents, but it is also useful to obtain structural information for the purpose of prioritizing new compounds for isolation. To address this, we have utilized the Global Natural Product Social Molecular Networking (GNPS) database [19] to build molecular networks from mass spectral fragmentation data. These fragmentation data provide useful chemical information, and structurally similar molecules should possess similar mass spectral fragmentation patterns. By comparing cosine similarity scores of individual compounds’ fragmentation patterns, GNPS can produce visual networks comprised of chemically related compounds and enables the identification of known compounds, molecular families, and structural analogs. By combining GNPS networking with biochemometric analysis, we propose that it would be possible to identify the structural classes of putative active molecules. The goal of this project is to utilize this integrated approach to prioritize isolation efforts on biologically relevant compounds from A. keiskei and to gain a more comprehensive understanding of which constituents contribute to the antimicrobial activity of this botanical against MRSA.
Results and Discussion
The first goal of this study was to utilize biochemometric analysis to identify putative bioactive constituents contributing to the antimicrobial activity of A. keiskei. Bioactivity screening demonstrated complete inhibition of methicillin-resistant S. aureus (MRSA, strain USA300 LAC strain AH1263) [20] by the A. keiskei extract at 10 µg/mL. This extract was then fractionated in several stages (see fractionation schemes, Supplementary Fig. S1 and S2), with the fractions displaying the most pronounced antimicrobial activity against MRSA prioritized for further isolation (Table 1).
Table 1.
Antimicrobial Activity of Angelica keiskei Koidzumi (AK) crude extract (CR) and second-stage fractions AK-3-1 through AK-4-4a
| Sample | Methicillin-resistant S. aureus growth inhibition (%) | |
|---|---|---|
| 50 µg/mL | 5 µg/mL | |
| Chloramphenicolb | 100 ± 0 | 46.7 ± 1.8 |
| AK-CR | 99.22 ± 0.39 | 6.4 ± 6.0 |
| AK-3-1 | 0 ± 0b | 21 ± 16 |
| AK-3-2 | 99.35 ± 0.65 | 26.0 ± 1.3 |
| AK-3-3 | 99.09 ± 0.91 | 11.14 ± 0.79 |
| AK-3-4 | 100 ± 0 | 0 ± 0 |
| AK-3-5 | 90.7 ± 3.3 | 99.61 ± 0.23 |
| AK-3-6 | 0 ± 0b | 26 ± 15 |
| AK-3-7 | 0 ± 0 | 0 ± 0 |
| AK-3-8 | 0 ± 0 | 0 ± 0 |
| AK-4-1 | 97.4 ± 2.4 | 19.76 ± 0.26 |
| AK-4-2 | 98.8 ± 1.2 | 98.95 ± 0.47 |
| AK-4-3 | 99.74 ± 0.26 | 3.2 ± 1.2 |
| AK-4-4 | 0 ± 0 | 0.66 ± 0.66 |
Growth inhibition of methicillin-resistant S. aureus strain (MRSA USA300 LAC strain AH1263) [20] relative to vehicle control measured turbidimetrically by OD600. Data presented are the result of triplicate analyses ± SEM. b Chloramphenicol (Sigma-Aldrich, 98% purity) served as the positive control.
Higher concentration samples of AK-3-1 and AK-3-6 show lower activity than their low-concentration counterparts, likely due to low solubility in aqueous media at high concentrations.
Bioactivity and mass spectral data from the second stage of fractionation (AK-3-1 through AK-3-8 and AK-4-1 through AK-4-4) (Table 1) were utilized to produce a biochemometric model predicting which constituents were responsible for antimicrobial activity. The internally cross-validated model generated five components that accounted for 83.61% of the independent (mass spectral), and 99.93% of the dependent (growth inhibition) variation (component 1: 32.58% independent, 53.16% dependent; component 2: 24.85% independent, 30.29% dependent; component 3: 11.54%, 13.98%; component 4: 7.86%, 1.93%; component 5: 6.79%, 0.57%).
To interpret the model and tentatively identify the chemical entities responsible for the MRSA growth inhibition, a selectivity ratio plot was generated (Fig. 1A). This plot revealed several marker ions that were strongly correlated with bioactivity, but could not provide structural information about these components. To generate such structural information, molecular networks were generated using MS/MS data from second-stage and third-stage chromatographic fractions (fractions resulting from two or three rounds of chromatographic separation, Supplementary Fig. S1). The resulting molecular networks were filtered using the biochemometric selectivity scores to identify molecular families of putative active compounds and assign tentative structures to candidate molecules (Supplementary Fig. S3). Interestingly, one second-stage molecular network and one third-stage molecular network identified the chalcones 4-hydroxyderricin (1) and xanthoangelol (2), which are the only known anti-MRSA compounds from A. keiskei [13]. Other known A. keiskei chalcones were also identified (Fig. 2). The same networks also contained masses of seven of the top ten contributors to bioactivity (marker ions A–G, Table 2) based on the biochemometric model (Fig. 1B–1C), suggesting that chalcones are responsible for A. keiskei’s antimicrobial efficacy against MRSA. The combination of biochemometrics and molecular networking enabled identification of a subset of these chalcones for prioritization and subsequent analysis, making it possible to predict the identity of biologically active extract components prior to isolating them.
Fig. 1.
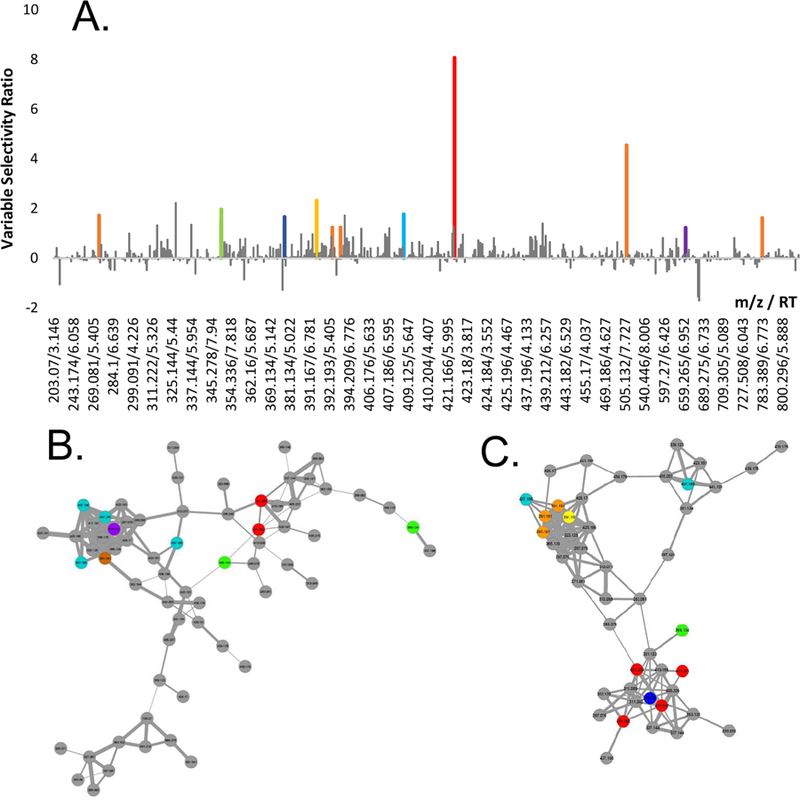
Selectivity plot (A) and selected molecular networks of second-stage (B) and third-stage (C) fractions of A. keiskei root extract. Bars have been color coded in A and points have been color coded in B and C only if they were both correlated with bioactivity and appeared in molecular networks of interest. Predicted active compounds in A appeared almost exclusively in these networks, indicating that a particular class of compounds is responsible for A. keiskei’s antimicrobial activity.
Fig. 2.
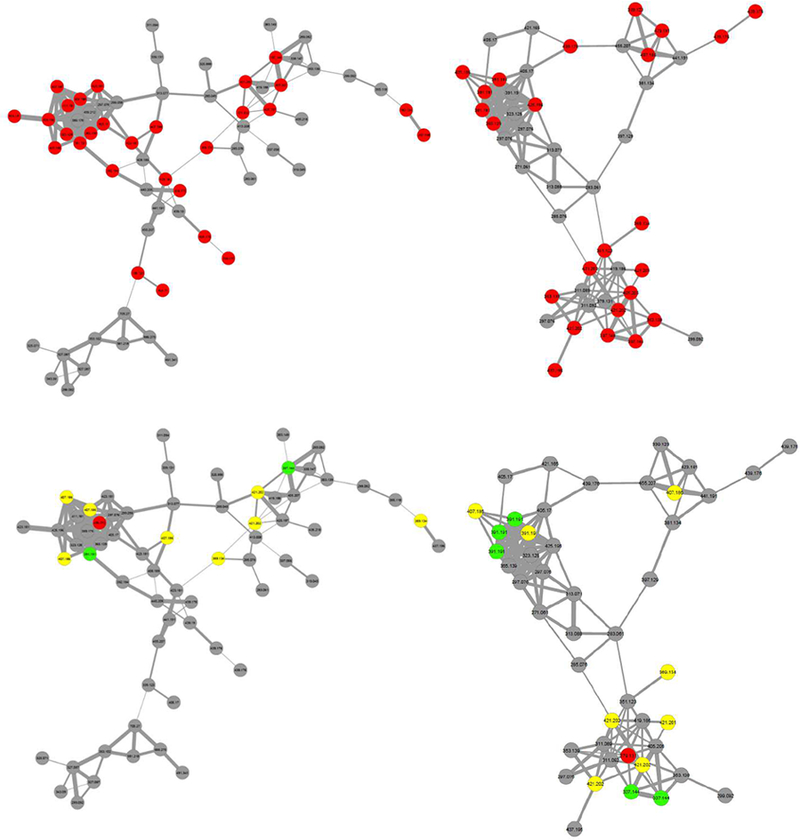
Molecular networks comprised of compounds detected in A. keiskei built from fractions following one (left) and two (right) stages of fractionation. In top networks, compounds marked in red match accurate masses of known A. keiskei chalcones. In bottom networks, green compounds match accurate masses of known antimicrobials 1 and 2, yellow compounds match known chalcones that have not been shown to possess anti-MRSA activity, and red compounds were correlated with bioactivity based on biochemometric selectivity ratio analysis but do not match known masses from the literature.
Table 2.
Tentative Identification of Putative Bioactive Chalcones from A. keiskei.
| Marker ion | Ion/retention time (molecular formula, δ (ppm)) | Adducts and fragments (molecular formula, δ (ppm)) | Tentative identification(s) |
|---|---|---|---|
| A | 421.202 [M-H]- / 6.23 (C26H29O5-, 1.189) |
4,2’,4’-trihydroxy-3’-[(2E,
5E)-7-methoxy-3,7-dimethyl- 2,5-octadienyl]chalcone a Xanthoangelol G a |
|
| B | 391.191 [M-H]- / 6.77 (C25H27O4-, 0.168) |
505.184 [M-H + TFA]-
(C25H27O4 + C2HF3O2, 0.399) 271.134 [M-H – C8H8O]- (C17H19O3-, 2.141) 783.389 [2M-H]- (2C25H28O4 – H, 0.886) |
Xanthoangelol b |
| C | 391.191 [M-H]- / 5.59 (C25H27O4-, 0.168) |
Xanthoangelol I a | |
| D | 351.123 [M-H]- / 5.52 (C21H19O5-, 0.708) |
Xanthoangelol K b | |
| E | 407.186 [M-H]- / 6.58 (C25H27O5- ,0.371) |
Xanthoangelol B a (2E)-1-[3,5-dihydroxy-2- methyl-2-(4-methyl-3- [penten-1-yl)-3,4-dihydroxy- 2H-chromen-8-yl]-3-(4- hydroxyphenyl-2-propen-1- one) a (2E)-1-[4-hydroxy-2-(2- hydroxy-6-methyl-5-hypten- 2-yl)-2,3-dihydro-1- benzofuran-5-yl]-3-(4- hydroxyphenyl)-2-propen-1- one a |
|
| F | 379.155 [M-H]- / 5.97 (C23H23O5-, 1.19) |
Potentially new chalcone derivative c |
|
| G | 439.211 [M-H]- / 5.17 (C26H31O6-, 2.422) |
Potentially new chalcone derivative c |
previously reported from Angelica keiskei Koidzumi, identified using accurate mass data [12].
isolated and confirmed by NMR
accurate masses do not match accurate masses of known A. keiskei chalcones, yet these masses appeared in chalcone molecular networks, indicating that they may be new chalcone derivatives.
Fifteen of the features in networks of interest matched the reported accurate masses of known chalcones [12] that have not yet been associated with antimicrobial activity (Fig. 2). Of these, five were predicted as potentially contributing to bioactivity by the biochemometric model, including the top contributor at m/z 421.202. Two additional compounds in these networks were identified among the top ten contributors by the biochemometric model that did not match accurate masses of bioactive chalcones from A. keiskei (Fig. 2). Because these compounds clustered with known chalcones based on similarities in mass spectral fragmentation patterns (Fig. 2), it was predicted that other chalcone antimicrobials might be present.
Biochemometric and molecular networking analysis identified marker ions associated with activity (Table 2). Purification of active A. keiskei fractions was conducted to assess the predictive accuracy of this approach, and four compounds were isolated (Fig. 3). The two known anti-MRSA compounds from A. keiskei, 1 and 2, were isolated using a combination of normal- and reversed-phase chromatography. Compound 1 was isolated at 98% purity following two stages of normal-phase flash chromatography and one stage of reversed-phase flash chromatography. Compound 2 was obtained at 95% purity following three stages of fractionation using both normal-phase flash chromatography and reversed-phase preparative-scale HPLC. The structures of compounds 1 and 2 were confirmed with 1H and 13C NMR by comparing to literature data [21] (Supplementary Fig. S4 – S7).
Fig. 3.
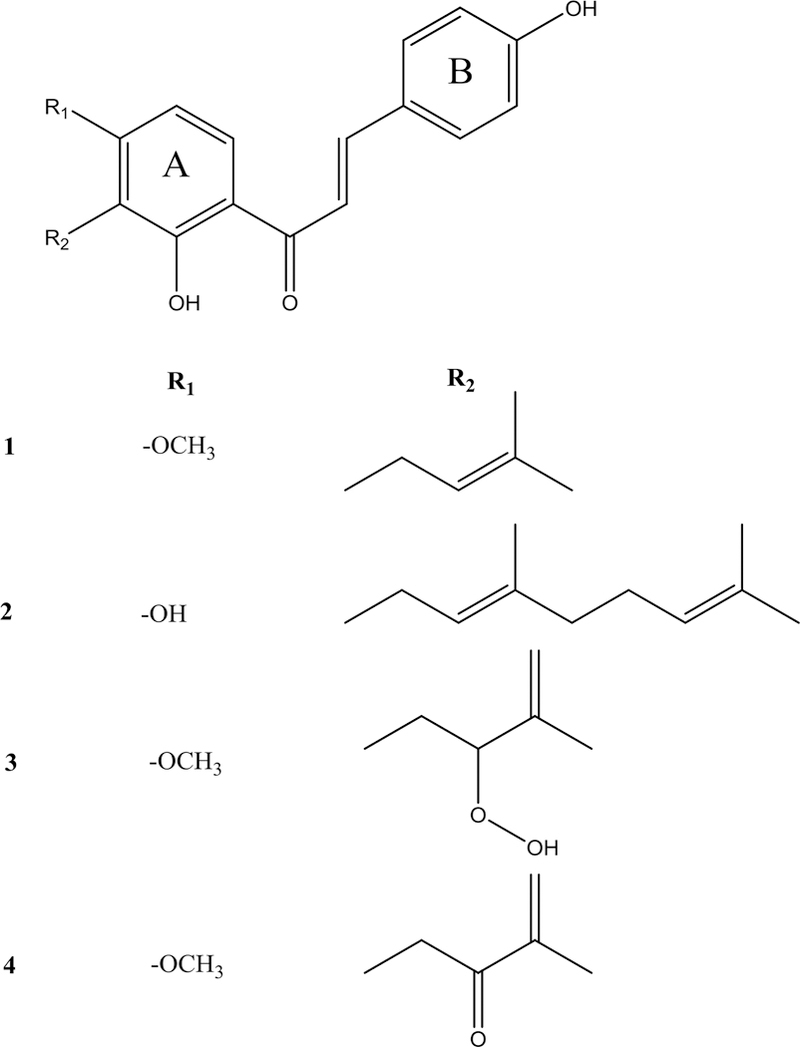
Structures of compounds 1–4, which were isolated from ashitaba (Angelica keiskei) and assessed for antimicrobial activity.
Two additional chalcones, 3 and 4, were isolated following a scale-up extraction and isolation. Compound 3 was isolated with 96% purity following two rounds of normal-phase flash chromatography, and 4 at 99% purity required an additional round of reversed-phase preparative HPLC. 1H and 13C NMR were utilized to confirm the identities of these compounds by comparing to published data [22–23] (Supplementary Fig. S8–S12). For 4, HMBC data were collected to confirm the presence of a ketone peak that did not appear in the 13C NMR spectra (Fig. S12), likely due to keto-enol tautomerization.
By integrating biochemometrics and molecular networking into the traditional bioassay-guided fractionation workflow, it was possible to prioritize minor constituents in A. keiskei for isolation (see workflow, Supplementary Fig. S3). Using biochemometrics to filter molecular networks and focus on specific structural classes, a subset of chalcone derivatives were identified that were most likely to possess antimicrobial activity and were prioritized for isolation. With this method, known, abundant antimicrobial compounds 1 and 2 were isolated, similar to previous bioassay-guided fractionation approaches alone [13]. Compounds 1 and 2 demonstrated MICs against MRSA (USA300 LAC strain AH1263) [20] of 4.6 µM and 4.0 µM, respectively (Table 3, Fig. S13). The biochemometrics/GNPS approach also enabled isolation of an additional low abundance antimicrobial compound (4), marker ion D (Table 2) that has not previously been reported to possess antimicrobial activity. In selectivity ratio plots, 4 was listed as the fourth top contributor to the observed biological activity of A. keiskei despite its low relative abundance (Fig. 1A, Table 2). In the base-peak chromatogram of the A. keiskei root extract, the peak area associated with 4 only accounted for 0.8% of the total fraction (Fig. 4). Compound 4 did inhibit growth of MRSA (IC50 at 168 µM, Table 3, Fig. S13) although did not reach MIC at the highest concentration tested (284 µM). Finally, as additional confirmation, we also isolated 3, which appeared in the chalcone molecular network (Fig. 2) but was not predicted to be antimicrobial. As predicted by biochemometrics, 3 did not possess antimicrobial activity, despite structural similarity to active compounds. Collectively, the agreement between predicted and observed biological activity of 1–4 demonstrates that the biochemometrics process as employed can be effective for identifying a subset of molecules for isolation based on their likely biological activity.
Table 3.
MIC and IC50 data for compounds 1–4 against methicillin-resistant S. aureus (MRSA USA300 LAC strain AH1263) [20] relative to vehicle control measured turbidimetrically by OD600. Presented data were calculated using four-parameter logistic curves of triplicate data.
| Compound | MICa | IC50 |
|---|---|---|
| 1 | 4.6 µM | 2.0 µM |
| 2 | 4.0 µM | 2.3 µM |
| 3 | -- | -- |
| 4 | -- | 168 µM |
The MIC value expressed is likely higher than the actual MIC value, which lies somewhere between the lowest tested concentration that inhibited bacterial growth and the highest tested concentration that did not completely inhibit bacterial growth [27].
Fig. 4.
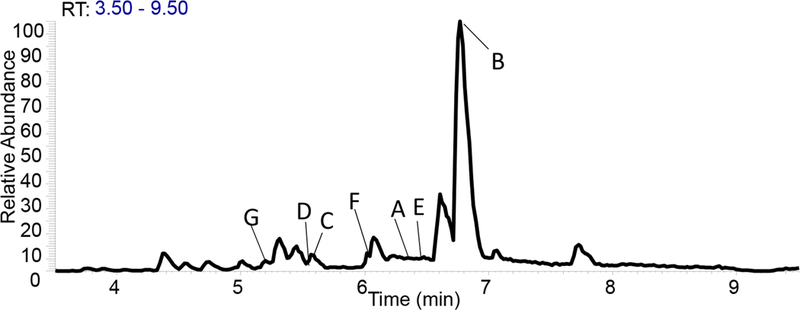
Base-peak chromatogram of ethyl acetate A. keiskei root extract with peaks of interest identified by biochemometric selectivity ratio analysis. This analysis was successful in enabling prioritization of trace peaks of interest for isolation.
The results described here are consistent with previous studies which suggest that prenyl- and geranyl- moieties on the A-ring of chalcones (present in 1 and 2) are associated with antimicrobial activity [14]. Compound 3 has a markedly different side chain from 1 and 2, with a flexible peroxide group, which is likely responsible for its decreased antimicrobial activity. Compound 4, though it does not contain an prenyl side chain, could possess weak activity due to the similarity of its side chain in rigidity and size to the prenyl substituent seen in 1.
Several additional features identified as possibly contributing to biological activity were identified in GNPS as chemically related to isolated chalcones 1–4 (Fig. 1 and 2). Based on these networks and accurate mass data of these compounds, we tentatively identified these compounds (Table 2). Unfortunately, material was too limited to isolate these compounds or assess biological activity. From a drug discovery standpoint, however, this approach is useful in dereplication, as it allowed us to identify these compounds as chalcones early in the fractionation process. Since chalcones are well documented antimicrobials [24], we did not complete an additional scale up to pursue their isolation.
In this example, marker ion A (Table 2) at m/z [M-H]- 421.202 eluting at 6.2 minutes was identified as the constituent most correlated with bioactivity and accounted for 0.4% of the total extract based on peak area. Unfortunately, even with a scale up extraction and chromatographic efforts tailored to this specific compound, isolation efforts for this compound were unsuccessful. This failure to isolate the active constituent demonstrates one of the inherent limitations of the biochemometric approach for identifying bioactive compounds. While it is possible based on mass spectrometric data to identify minor compounds that may have important biological activity, it may be infeasible (due to limited quantity) to isolate such minor compounds for confirmation of structure and activity.
One limitation of this study is that biochemometric analysis did not predict biological activity for the most abundant isotopes of 1 and 2, despite the confirmed antimicrobial activity of these compounds. Based on relative peak area, 2 accounted for 37.8% of the relative abundance in the EtOAc extract, and 1 accounted for 12.5%. The high abundance and antimicrobial potency of these compounds likely led to a mismatch in biological and chemical data. While the relative peak area of these compounds varied in every fraction under study, the biological activity was saturated at 100% in multiple fractions. Consequently, the linearity between the relative abundance of these compounds and their corresponding bioactivity was likely skewed, leading to false negative results. Although the [M-H]- peak for the most abundant (12C) isotope of 2 was not identified as active (Fig. 1), several of the 13C isotopes as well as the TFA adduct, and an in-source fragment of this compound were predicted to be active (marker ion B, Table 2). The adducts and isotopes of 2 were only evident in fractions where 2 was extremely abundant, and consequently, they were identified by the selectivity score as marker ions related to bioactivity. The identification of an active isotope of a compound that is not itself predicted to be active is clearly an artifact of an error in the data analysis process, given that all isotopes co-occur in the sample, and the adducts are formed in the ionization process and likely not present in the sample at all.
An important goal for the comprehensive characterization of a botanical medicine should be to isolate minor constituents within the extract. However, it is not feasible to isolate all minor constituents in a complex mixture, so putative bioactive constituents must be prioritized. A major strength of the biochemometric selectivity ratio analysis is its ability to identify low-abundance constituents contributing to activity without being confounded by compounds of high abundance. However, this strength comes with an important weakness in that bioactive compounds of high abundance may be overlooked. This weakness can easily be overcome, however, if this statistical analysis is incorporated into the traditional bioassay-guided fractionation workflow, which favors the isolation of abundant active compounds. It is also possible that this limitation could be addressed by diluting samples to reduce the level of high abundance compounds, although this approach would come at the expense of sacrificing response of those present at low abundance.
In combination with bioassay-guided fractionation, biochemometrics and molecular networking can be utilized to identify structural families of putative active constituents present at very low levels, allowing for the prioritization of isolation of both high and low abundance components that contribute to activity, or alternately, enabling the dereplication of known bioactive compounds and their structural analogs. The latter application is important because it prevents time being wasted on reisolating known active compounds. Had we been searching for bioactive compounds with novel structures only, we may have chosen not to pursue further isolation with A. keiski once we identified chalcones as the major class of active constituents within this plant. However, for the purpose of this study, a botanical containing known antimicrobial constituents served as a useful test case. The approach employed here not only facilitated the identification of a trace antimicrobial constituent from A. keiskei, but also yielded new and more complete information about which constituents are responsible for the antimicrobial activity of this botanical. Additionally, it provided insight into which structural characteristics of chalcones are associated with their antimicrobial effects.
Materials and Methods
General experimental procedures
NMR spectra were obtained using a JEOL ECA-500 MHz spectrometer. UPLC-MS analysis was completed in both negative and positive modes using an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) connected to an Acquity UPLC system (Waters Corporation). When collecting UPLC-MS data, 3 μL of 1 mg/mL samples suspended in MeOH were injected into the column. Using a flow rate of 0.3 mL/min, samples eluted from the column (BEH C18 1.7 μm, 2.1 × 50 mm, Waters Corporation) using the following gradient with solvent A consisting of water with 0.1% formic acid and solvent B consisting of acetonitrile with 0.1% formic acid: 90:10 (A:B) from 0–0.5 min, increasing to 0:100 (A:B) from 0.5–8.0 min. The gradient was held at 100% B for 0.5 min, before returning to starting conditions over 0.5 min and held from 9.0–10.0 min. Mass analysis was completed in both positive and negative ionization modes over a scan range of 150–2000 with the following settings: capillary voltage at −21.00 V, capillary temperature at 275.00 °C, tube lens offset at −95.00 V, spray voltage at 3.50 kV, sheath gas flow at 30.00, and auxiliary gas flow at 15.00. The top 4 most intense ions were fragmented with CID set to 35.0.
Flash chromatographic separations were completed using a CombiFlash RF system (Teledyne-Isco) and examined using a PDA detector and an evaporative light scattering detector (ELSD). Preparative and analytical HPLC separations were conducted with a Varian HPLC system (Agilent Technologies) using Galaxie Chromatography Workstation software (version 1.9.3.2, Agilent Technologies). All chemicals were acquired through Sigma-Aldrich and were spectroscopic or microbiological grade.
Plant Material
Fresh roots of Angelica keiskei Koidzumi were collected on November 14, 2015 from Strictly Medicinal Seeds in Williams, Oregon (Sample # 12421, N 42°12’17.211”, W 123°19’34.60). Scale up material was completed using plant material from the same source collected on December 29, 2016 (Sample #12444, N 42°12’17.211”, W 123°19’34.60). The identity of this plant material was confirmed by Richard A. Cech at Strictly Medicinal Seeds, and a voucher specimen was deposited at the herbarium of the University of North Carolina at Chapel Hill (NCU627665).
Extraction
Fresh A. keiskei roots were dried in a single-wall transite oven (Blue M Electric Company) at 40 °C for 24 hours. The resulting dry mass (138.90 g) was ground using a Wiley Mill Standard Model No. 3 (Arthur Thomas Company) and submerged in MeOH at 160 g/L for 24 hours three times. The resulting MeOH extract was concentrated in vacuo and then subjected to liquid-liquid extraction. First, the extract was defatted by partitioning between 10% aqueous MeOH and hexane (1:1). The dried aqueous MeOH layer was partitioned further between 4:5:1 EtOAc/MeOH/H2O. To remove hydrosoluble tannins, the EtOAc layer was washed with a 1% NaCl solution. The resulting EtOAc extract was dried under nitrogen, yielding 3,650.32 mg dried extract, before further experimentation. Scale up material (964 g) was dried, extracted, and partitioned using the same methods listed above, ultimately yielding 18.10 g of dried EtOAc extract for subsequent chromatographic separation.
Chromatographic Separation and Isolation.
The isolation scheme is provided as Supporting information (Fig. S1 and S2). The first-stage separations of the EtOAc extract (3,100 mg) were conducted using normal-stage flash chromatography (40 g silica gel column) at a 40 mL/min flow rate with a 35 min hexane/CHCl3/MeOH gradient. The last two fractions (AK-3 and AK-4) were subjected to a second stage of normal-phase flash chromatography. Fraction 3 (AK-3, 1355 mg) was separated again with a 40 g silica gel column at a flow rate of 40 mL/min and fraction 4 (AK-4, 536 mg) was separated on a 12 g silica column with a flow rate of 30 mL/min. Each run lasted 45 minutes, and was completed using a hexane/EtOAc/MeOH gradient. The most active fraction from the separation of AK-3 (fraction 2, AK-3-2, 1000 mg) was subjected to a final round of reversed-phase flash chromatography using a 130g C18 reversed phase RediSep Rf column with an 85 mL/min flow rate. A 25-minute gradient of CH3CN/H2O was used, starting at 40:60 and increasing to 85:15. It was increased to 100:0 for 5 minutes, upon which starting conditions were re-established. Compound 1 eluted at 18 min (234.45 mg, 98% purity, 7.6% yield). Fraction AK-4-2 (364 mg) was also subjected to a final round of reversed-phase preparative HPLC injected onto a Luna preparatory column (5 µm PFP, 250 × 21.20mm; Phenomenex). The 35 minute run began at 40:60 CH3CN:H2O and was increased to 100:0 over thirty minutes. Compound 2 was collected from 28–35 minutes (284.59 mg, 95% purity, 9.1% yield).
Compounds 3 and 4 were isolated following scale up extraction. First, 17.5 g of EtOAc extract were separated on a 120g silica column with an 85mL/min flow rate using the same hexane/CHCl3/MeOH gradient as used for the first fractionation of original extract. The second fraction (S-AK-2, 5.3 g) was separated again using normal-phase flash chromatography on a 120 g silica column at 85 mL/min flow rate with a 55-minute gradient of hexane/EtOAc/MeOH. Compound 3 eluted at 31 minutes (150 mg, 96% purity, 0.85% yield). Fraction 4 (S-AK-2-4, 172 mg) was subjected to a final 45-minute round of reversed-phase preparative HPLC on a Gemini-NX preparatory column (5 µm C18, 250 × 21.20 mm; Phenomenex) at a flow rate of 21.4 mL/min with a gradient of 55:45 CH3CN:H2O. Compound 4 (1.5 mg, 99% purity, 0.0086% yield) eluted at 19 minutes.
4-hydroxyderricin (1):
yellow crystalline solid; HRESIMS m/z 337.1438 [M-H]- (calculated for C21H21O4-, 337.1440); 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) chemical shifts matched literature values [25] and are provided in Supporting Information (Fig. S4 and S5).
Xanthoangelol (2):
yellow crystalline solid; HRESIMS m/z 391.1907 [M-H]- (calculated for C25H27O4-, 391.1909); 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) chemical shifts matched literature values [25] and are provided in Supporting Information (Fig. S6 and S7).
Xanthoangelol E (3):
yellow, amorphous powder; HRESIMS m/z 369.1340 [M-H]- (calculated for C21H21O6-, 369.1338); 1H NMR (500 MHz, DMSO) and 13C NMR (125 MHz, DMSO) chemical shifts matched literature values [22] and are provided in Supporting Information (Fig. S8 and S9).
Xanthoangelol K (4):
yellow amorphous powder; HRESIMS m/z 351.1231 [M-H]- (calculated for C21H19O5-, 351.1232); 1H NMR (500 MHz, CDCl3),13C NMR (125 MHz, CDCl3), and HMBC (400 MHz, CDCl3) chemical shifts matched literature values [23] and are provided in Supporting Information (Fig. S10-S12).
Antimicrobial Assay
Antimicrobial activity was monitored by assessing growth inhibition of a laboratory strain of Staphylococcus aureus (SA1199) [26] and a clinically relevant strain of methicillin-resistant S. aureus (MRSA USA300 LAC strain AH1263) [20], obtained from Dr. Alexander Horswill at the University of Colorado Anschutz Medical Campus. Cultures were grown from a single colony isolate of each strain in Müeller-Hinton broth (MHB) and diluted to 1.0 × 105 CFU/mL based on absorbance at 600 nm (OD600).
Samples were screened in triplicate at final concentrations of 10 and 100 μg/mL or 5 and 50 μg/mL. Samples were dissolved in 1:1 EtOH/DMSO (v/v) and diluted with MHB to prepare final concentrations in broth with less than 2% EtOH/DMSO. The known antibiotic chloramphenicol (98% purity, Sigma-Aldrich) was used as a positive control at the same concentrations as tested extracts. The vehicle was 2% EtOH/DMSO in MHB. Each well was inoculated with bacteria and incubated for 24 hours at 37 °C. OD600 was evaluated after incubation and used to calculate the percent growth inhibition. All fractions were subjected to analysis and active fractions were chosen for further fractionation.
Minimal inhibitory concentrations (MICs) were calculated for pure compounds based on the Clinical Laboratory Standards Institute (CLSI) standard protocols [27]. Isolated compounds or chloramphenicol (positive control, 98% purity, Sigma-Aldrich) were added to 96-well plates in triplicate at concentrations ranging from 0–100 µg/mL in MHB. Broth containing 2% 1:1 EtOH/DMSO was used as the vehicle control. The concentration of EtOH/DMSO was set at a fixed value of 2% for all wells. After a 24-hour incubation at 37 °C, OD600 values were measured using a Synergy H1 microplate reader (Biotek). The MIC was defined as the concentration at which no statistically significant difference between the blank wells (containing sample and broth but no bacteria) and the treated sample was observed.
Biochemometric Analysis
LC-MS data were collected in both negative mode and positive mode and individually analyzed, aligned, and filtered utilizing MZmine 2.21.2 (http://mzmine.sourceforge.net/) [28]. Raw mass spectral data files from second-stage fractions were uploaded for peak picking into MZmine based upon m/z values within each spectrum above a set baseline for all batch samples. Chromatograms were constructed for all m/z values lasting longer than 0.1 minute, following which they were deconvoluted using algorithms that were applied to chromatograms to recognize individual peaks. The peak detection parameters were set as follows: noise level (absolute value) at 1.25 × 106 (positive mode) and at 2 × 106 (negative mode), minimum peak duration at 0.5 seconds, m/z variation tolerance at 0.05, and m/z intensity variation tolerance at 20%. Peaks were aligned if their masses were within 5 ppm and their retention times were a maximum of 0.15 minutes from one another. Peak list filtering and retention time alignment were completed to produce an aligned peak list. The resulting data matrix, consisting of m/z, retention time, and peak area, was imported into Excel (Microsoft) and merged with bioactivity data from samples at tested at 5 μg/mL to form the final data set for biochemometric analysis.
Biochemometric analysis was completed using Sirius version 10.0 statistical software (Pattern Recognition Systems) [29]. Before analysis, data were adjusted using a fourth root transformation to normalize noise across treatments [30]. An internally cross-validated PLS model was then produced using 100 iterations and a significance level of 0.05. Statistical algorithms internal to the Sirius software utilized model predictions to produce selectivity ratios identifying putative antimicrobial constituents.
Molecular Networking Analysis
Mass spectral data were converted to mzXML format using FileZilla version 3.14.1, part of the ProteoWizard platform (http://proteowizard.sourceforget.net/#) [31]. Following file conversion, mass spectral and fragmentation data were uploaded to the GNPS data analysis portal in 3 groups, where fractions active at 5 μg/mL were included in Group 1, fractions active at 50 μg/mL were included in Group 2, and inactive fractions were included in Group 3. These data were then combined into consensus spectra using the MS-clustering algorithm [32] within the Global Natural Product Social Molecular Networking (GNPS) database [19].
Molecular networks were produced using the online GNPS workflow. First, MS/MS peaks within 17 Da of the precursor m/z were removed, and only the top six fragment peaks were compared for analysis. Using MS-Cluster, consensus spectra were produced with a parent mass tolerance of 0.5 Da and an MS/MS fragment ion tolerance of 0.3 Da. Consensus spectra containing fewer than 10 spectra were discarded. Molecular networks were subsequently produced, and compounds were connected if they had a cosine score (similarity score) above 0.65 and more than 6 matched fragment peaks. If more than 10 compounds shared a cosine score above this threshold with a given compound, only the top 10 most similar compounds were connected. Parameters for third-stage fractions were the same, except that the minimum cluster size was adjusted to 100. Fragmentation patterns were compared to databases within GNPS, including the GNPS Library, the GNPS-NIH-Natural Products Library, GNPS Prestwick Phytochemical Library, and the RESPECT Library to tentatively ID components matching MS/MS patterns already contained within the system. Networks were viewed in GNPS using the network visualizer in addition to being imported to Cytoscape [33] for visualization. To simplify investigation of networks, nodes containing accurate masses identified by biochemometric analysis as putative active compounds were prioritized for structural characterization.
Supplementary Material
Acknowledgements
This research was supported by the National Center for Complementary and Integrative Health of the National Institutes of Health under award numbers 5 T32 AT008938, and 1R01 AT006860. The authors would also like to thank Dr. Laura Sanchez for her help with molecular networking, and Dr. Alexander Horswill for his provision of microbial strains utilized for this project.
Footnotes
Conflict of Interest Disclosure
The authors declare that there are no conflicts of interest.
References
- [1].Sharma SB, Gupta R. Drug development from natural resource: a systematic approach. Mini Rev Med Chem 2015; 15 (1): 52–57. [DOI] [PubMed] [Google Scholar]
- [2].Kinghorn AD, Fong HHS, Farnsworth NR, Mehta RG, Moon RC, Moriarty RM, Pezzuto JM. Cancer Chemopreventative Agents Discovered by Activity-Guided Fractionation: A Review. Curr Org Chem 1998; 2 (6): 597–612. [Google Scholar]
- [3].Kellogg JJ, Todd DA, Egan JM, Raja HA, Oberlies NH, Kvalheim OM, Cech NB. Biochemometrics for Natural Products Research: Comparison of Data Analysis Approaches and Application to Identification of Bioactive Compounds. J Nat Prod 2016; 79 (2): 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Inui T, Wang Y, Pro SM, Franzblau SG, Pauli GF. Unbiased evaluation of bioactive secondary metabolites in complex matrices. Fitoterapia 2012; 83 (7): 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Roemer T, Xu D, Singh SB, Parish CA, Harris G, Wang J, Davies JE, Bills GF. Confronting the challenges of natural product-based antifungal discovery. Chem Biol 2011; 18 (2): 148–164. [DOI] [PubMed] [Google Scholar]
- [6].Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod 2016; 79: 629–661. [DOI] [PubMed] [Google Scholar]
- [7].Cowan MM. Plant products as antimicrobial agents. Clin Microbiol Rev 1999; 12(4): 564–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kingston DG. Modern natural products drug discovery and its relevance to biodiversity conservation. J Nat Prod 2011; 74 (3): 496–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gurjar MS, Ali S, Akhtar M, Sing KS. Efficacy of plant extracts in plant disease management. Agricultural Sciences 2012; 3: 425–433. [Google Scholar]
- [10].Savoia D Plant-derived antimicrobial compounds: alternatives to antibiotics. Future Microbiol 2012; 7 (8): 979–990. [DOI] [PubMed] [Google Scholar]
- [11].Toyama K, Chokki S. Ashitaba-Hachijojima Island’s Reiso in the Early Modern Period. Kobe Health Welfare University Anal 2014; 15(1): 37–44. [Google Scholar]
- [12].Caesar LK, Cech NB. A Review of the Medicinal Uses and Pharmacology of Ashitaba. Planta Med 2016; 82 (14): 1236–1245. [DOI] [PubMed] [Google Scholar]
- [13].Inamori Y, Baba K, Tsujibo H, Taniguchi M, Nakata K, Kozawa M. Antibacterial activity of two chalcones, xanthoangelol and 4-hydroxyderricin, isolated from the root of Angelica keiskei KOIDZUMI. Chem Pharm Bull (Tokyo) 1991; 39(6): 1604–1605. [DOI] [PubMed] [Google Scholar]
- [14].Sugamoto K, Matsusita Y, Matsui K, Kurogi C, Matsui T. Synthesis and antibacterial activity of chalcones bearing prenyl or geranyl groups from Angelica keiskei. Tetrahedron 2011; 67(29): 5346–5359. [Google Scholar]
- [15].Wu C, Du C, Gubbens J, Choi YH, van Wezel GP. Metabolomics-Driven Discovery of a Prenylated Isatin Antibiotic Produced by Streptomyces species MBT28. J Nat Prod 2015; 78 (10): 2355–2363. [DOI] [PubMed] [Google Scholar]
- [16].Sidebottom AM, Johnson AR, Karty JA, Trader DJ, Carlson EE. Integrated Metabolomics Approach Facilitates Discovery of an Unpredicted Natural Product Suite from Streptomyces coelicolor M145. ACS Chem Biol 2013; 8 (9): 2009–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cox DG, Oh J, Keasling A, Colson KL, Hamann, MT. The utility of metabolomics in natural product and biomarker characterization. Biochim Biophys Acta 2014; 1840: 3460–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rajalahti T, Arneberg R, Berven FS, Myhr K-M, Ulvik RJ, Kvalheim OM. Biomarker discovery in mass spectral profiles by means of selectivity ratio plot. Chemom Intell Lab Syst 2009; 95: 35–48. [Google Scholar]
- [19].Yang JY, Sanchez LM, Rath CM, Liu X, Boudreau PD, Bruns N, Glukhov E, Wodtke A, de Felicio R, Fenner A, Wong WR, Linington RG, Zhang L, Debonsi HM, Gerwick WH, Dorrestein PC. Molecular networking as a dereplication strategy. J Nat Prod 2013; 76 (9): 1686–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boles BR, Thoendel M, Roth AJ, Horswill AR. Identification of Genes Involved in Polysaccharide-Independent Staphylococcus aureus Biofilm Formation. PLoS One 2010; e10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim JH, Son YK, Kim GH, Hwang KH. Xanthoangelol and 4-hydroxyderricin are the major active principles of the inhibitory activities against monoamine oxidases on Angelica keiskei K. Biomol Ther (Seoul) 2013; 21(3): 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Baba K, Nakata K, Taniguchi M, Kido T, Kozawa M. Chalcones from Angelica keiskei. Phytochemistry 1990; 29(12): 3907–3910. [Google Scholar]
- [23].Li J, Gao L, Meng F, Tang C, Zhang R, Li J, Luo C, Li J, Zhao W. PTP1B inhibitors from stems of Angelica keiskei (Ashitaba). Bioorg and Med Chem Lett 2015; 25(10): 2028–2032. [DOI] [PubMed] [Google Scholar]
- [24].Nowakowska Z A review of anti-infective and anti-inflammatory chalcones. Eur J Med Chem 2007; 42(2): 125–127. [DOI] [PubMed] [Google Scholar]
- [25].Kawabata K, Sawada K, Ikeda K, Fukuda I, Kawasaki K, Yamamoto N, Ashida H. Prenylated chalcones 4-hydroxyderricin and xanthoangelol stimulate glucose uptake in skeletal muscle cells by inducing GLUT4 translocation. Mol Nutr Food Res 2011; 55: 467–475. [DOI] [PubMed] [Google Scholar]
- [26].Kaatz GW, Seo SM. Inducible NorA-Mediated Multidrug Resistance in Staphylococcus aureus. Antimicrob Agents Chemother 1995; 39(12): 2650–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].CLSI. National Committee for Clinical Laboratory Standards. In: Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically—Tenth Edition: Approved Standard M7-A10 Wayne, PA; 2015. [Google Scholar]
- [28].Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZMine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinf 2010; 11: 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kvalheim OM, Chan H, Benzie IFF, Szeto T, Tzang AH, Mok DI, Chau F. Chromatographic profiling and multivariate analysis for screening and quantifying the contributions from individual components to the bioactive signature in natural products. Chemom Intell Lab Syst 2011; 107: 98–105. [Google Scholar]
- [30].Kvalheim OM, Brackstad F, Liang Y- Z. Preprocessing of analytical profiles in the presence of homoscedastic or heteroscedastic noise. Anal Chem 1994; 66: 43–51. [Google Scholar]
- [31].ProteoWizard. http://proteowizard.sourceforge.net/#, Ed., ProteoWizard Software Foundation: 2013 [Google Scholar]
- [32].Frank AM, Savitski MM, Nielsen ML, Zubarev RA, Pevzner PA. De novo peptide sequencing and identification with precision mass spectrometry. J Proteome Res 2007; 6: 114–123. [PubMed:17203955] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ahannon P, Markiel A, Ozier O, Balinga NS, Want JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated modules of biomolecular interaction networks. Genome Res 2003; 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.