

Abstract
Under both physiologic and clinical conditions GABAA receptors are exposed to multiple agonists, including the transmitter GABA, endogenous or exogenous neuroactive steroids, and various GABAergic anesthetic and sedative drugs. The functional output of the receptor reflects the interplay among all active agents. We have investigated the activation of the concatemeric α1β2γ2L GABAA receptor by combinations of agonists. Simulations of receptor activity using the coagonist concerted transition model demonstrate that the response amplitude in the presence of agonist combinations is highly dependent on whether the paired agonists interact with the same or distinct sites. The experimental data for receptor activation by agonist combinations were in agreement with the established views of the overlap of binding sites for several pairs of orthosteric (GABA, β-alanine, and piperidine-4-sulfonic acid) and/or allosteric agents (propofol, pentobarbital, and several neuroactive steroids). Conversely, the degree of potentiation when two GABAergic agents are coapplied can be used to determine whether the compounds act by binding to the same or distinct sites. We show that common interaction sites mediate the actions of 5α- and 5β-reduced neuroactive steroids, and natural and enantiomeric steroids. Furthermore, the results indicate that the anesthetics propofol and pentobarbital interact with partially shared binding sites. We propose that the findings may be used to predict the efficacy of drug mixtures in combination therapy and thus have potential clinical relevance.
Introduction
The GABAA receptor is a transmitter-gated ion channel and a key component in regulating the excitatory-inhibitory balance in the brain. The binding of the transmitter GABA to the two orthosteric binding sites in the extracellular domain of the receptor leads to opening of an anion-selective ion channel, thereby contributing to cellular inhibition (Bouzat, 2012; Chua and Chebib, 2017). Besides GABA, numerous endogenous and exogenous compounds, including many neurosteroids and intravenous anesthetics, can activate the receptor (Sieghart, 2015; Olsen, 2018). Coapplication of two (or more) GABAergic agents typically results in potentiation of the current response. Direct activation of the GABAA receptor and potentiation of transmitter-activated receptors underlie the clinical actions of GABAergic anesthetics.
The degree or magnitude of potentiation, and by extension the clinical efficacy of an anesthetic drug, depends on multiple factors. One such factor is whether the two GABAergic agents in a combination interact with the same site(s). Coapplication of two agonists acting at the same sites can result in potentiation because of “concentration additivity,” i.e., an increase in the effective concentration of the ligand. However, the exact nature of modulation depends on the efficacies and concentrations of each compound. For example, coapplication of a low-efficacy orthosteric agonist, such as piperidine-4-sulfonic acid (P4S), enhances the peak response to GABA when the concentrations of both agonists are low. At higher concentrations, P4S displaces GABA from the orthosteric binding sites and the response amplitude becomes limited by the gating efficacy of P4S. Coapplication of multiple allosteric agents that act through the same sites—for example, different species of structurally related neuroactive steroids—can be expected to perform analogously.
Agonist combinations where the individual agents interact with distinct sites produce potentiation via “energetic additivity.” In this instance, one agonist acts to independently reduce the free energy difference to be overcome by the other. This process is exemplified by coapplication of an allosteric agonist with an orthosteric agonist, e.g., coapplication of propofol with GABA. The peak response to the combination of GABA + propofol can be accurately predicted based on energetic additivity of the effects of each individual agent (Ruesch et al., 2012; Akk et al., 2018; Shin et al., 2018). Coapplication of two allosteric agonists that interact with distinct sites would be mechanistically similar, resulting in additivity of free energies provided by each agonist toward stabilization of the open state (Shin et al., 2017).
Here, we have analyzed activation of the concatemeric α1β2γ2L GABAA receptor by combinations of orthosteric and/or allosteric agents, using the coagonist concerted transition model (Monod et al., 1965; Forman, 2012; Akk et al., 2018). In this model (Fig. 1), the receptor can exist in two states, resting and active, which have different affinities for the agonist; when the receptor transitions from one state to the other, the properties of all sites change. Receptor activation by a given agonist can be fully characterized by four parameters: 1) basal activity of the receptor in the absence of agonist, 2) affinity of the resting receptor to the agonist, 3) affinity of the active receptor to the agonist, and 4) the number of binding sites for the agonist. The effect of coapplication of a second agonist interacting with distinct sites can be considered to modify basal activity with no specific effect on receptor interaction with the principal agonist. Coapplication of a second agonist interacting with the same sites as the principal agonist can be considered as a simple competitive interaction.
Fig. 1.

The state diagram of the activation model. The receptor is exposed to a single agonist X (A), or to the combination of agonists X and Y that interact with distinct sites (B) or the same sites (C). Note that the front plane in (B) is the scheme in (A), and that (C) is a subset of states shown in (B) (missing states indicated by gray color). The inactive states (R) are depicted on the bottom plane and active states (A) on the top plane. The equilibrium between the states is determined by the constants given next to the arrows. The KX value is the equilibrium dissociation constant of the inactive receptor and cXKX is the equilibrium dissociation constant of the active receptor. Parameter L (= A/R) describes the equilibrium between the inactive and active states. Note that in (B) two inactive states (YRX and Y2RX) are hidden and in (C) YRX is hidden.
The overall goal of the study was to compare the magnitude of potentiation for combinations of GABAergic compounds that act through the same or distinct sites. We show that the functional response to an agonist combination is a computable value and that it depends on the extent of overlap between the sites for the individual agents. Conversely, we propose that the response amplitude in the presence of an agonist combination can be used to determine whether the paired compounds interact with the same or distinct sites.
Materials and Methods
Receptor Expression.
The GABAA receptors were expressed in Xenopus oocytes. Harvesting of oocytes was conducted under the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. The animal protocol was approved by the Animal Studies Committee of Washington University in St. Louis (Approval No. 20170071).
The receptors comprised concatemeric β2-α1-γ2L (βαγ) and β2-α1 (βα) constructs. The design and properties of the receptors have been described previously (Bracamontes and Steinbach, 2009; Bracamontes et al., 2011; Akk et al., 2018). Receptors formed of βαγ and βα constructs without further mutations are referred to as wild-type concatemeric receptors. Constructs containing the α1(L263S) or β2(Y143W+M286W) mutations were generated using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The coding region was fully sequenced prior to use. The cDNAs in the pcDNA3 vector were linearized with Xba I (NEB Laboratories, Ipswich, MA) and the complementary RNAs were generated using mMessage mMachine (Ambion, Austin, TX). The oocytes were injected with a total of 12 ng complementary RNA in a 1:1 ratio for the concatemeric constructs and incubated in ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES; pH 7.4) with supplements (2.5 mM Na pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamycin) at 16°C for 1–3 days prior to conducting electrophysiological recordings.
Electrophysiology.
The recordings were done using standard two-electrode voltage clamp. The oocytes were clamped at −60 mV. The chamber (RC-1Z; Warner Instruments, Hamden, CT) was perfused with ND96 at 5–8 ml/min. Solutions were gravity applied from 30-ml glass syringes with glass luer slips via Teflon tubing. A typical experiment consisted of recording of a 10–20-second baseline, followed by drug application for 20–60 seconds and bath (ND96) application until full recovery. Solutions were switched manually. The concentration-response relationships were determined by exposing each oocyte to a full range of agonist concentrations (six-to-nine concentration points). Due to the low gating efficacy of neuroactive steroids, the concentration-response relationships for alfaxalone, allopregnanolone [(3α5αP) 5α-pregnan-3α-ol-20-one], pregnanolone [(3α5βP) 5β-pregnan-3α-ol-20-one], and the enantiomer of 3α5βP (ent-3α5βP) were conducted in the presence of a low concentration of GABA. The properties of etiocholanolone and alfaxalone were also investigated on a receptor containing the gain-of-function α1(L263S) mutation (Chang and Weiss, 1999).
The current responses were amplified with an Axoclamp 900A (Molecular Devices, Sunnyvale, CA) or OC-725C amplifier (Warner Instruments), digitized with a Digidata 1320 or 1200 series digitizer (Molecular Devices), and stored using pClamp (Molecular Devices). The current traces were analyzed using Clampfit (Molecular Devices) to determine the peak amplitude.
Data Analysis.
The current amplitudes were converted to units of open probability by matching the relative peak responses against a scale ranging from 0 to 1 of the open probability of the receptor (Popen) (Forman and Stewart, 2012; Eaton et al., 2016). Wild-type concatemeric receptors in the absence of agonist exhibit minuscule constitutive activity [i.e., open probability of a constitutively active receptor (Popen,const) = 0.00011] (Akk et al., 2018); therefore, the holding current in the absence of agonists was considered to have a value of Popen = 0. The current level corresponding to Popen = 1 was estimated by exposing the receptors to the combination of saturating GABA plus 100 μM pentobarbital (Ziemba and Forman, 2016).
In receptors containing the gain-of-function mutations, the current level corresponding to Popen = 0 was estimated by exposing the oocytes to 100–500 μM of the channel blocker picrotoxin. In these receptors, no increase in peak amplitude was observed during coapplication of pentobarbital with saturating GABA. Accordingly, the mutant receptors were considered to have a Popen value indistinguishable from 1 in the presence of saturating GABA alone. The open probability of the constitutively active mutant receptors (Popen,const) was calculated as Ipicrotoxin/(Ipicrotoxin − IGABA), where Ipicrotoxin is the current amplitude during the application of picrotoxin and IGABA is the current amplitude in the presence of saturating GABA.
We note that this approach for estimating the Popen values can lead to potential errors. One source of error is incomplete blockade of constitutive activity in the presence of picrotoxin that may result in overestimation of the holding current associated with zero activity. Desensitization, particularly in the presence of saturating GABA and a potentiator, may result in underestimated peak amplitude. This, however, is not a major concern because the majority of experiments were conducted at low concentrations of agonists, where desensitization is reduced.
The current response data in units of open probability were analyzed in the framework of the coagonist concerted transition model (Fig. 1A). The experimental concentration-response curves were fit to eq. 1 describing the state function of the receptor:
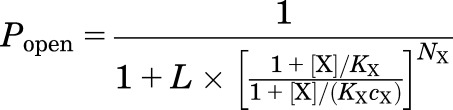 |
(1) |
where X is an agonist; KX is the equilibrium dissociation constant for X in the closed receptor; cX is the ratio of the equilibrium dissociation constant for X in the open receptor to KX; and NX is the number of binding sites for X. The number of binding sites was constrained to two for GABA, P4S, and β-alanine (Krogsgaard-Larsen et al., 1980; Amin and Weiss, 1993; Jones et al., 1998), six for propofol (Shin et al., 2018), two for pentobarbital (Ziemba and Forman, 2016), and two for all steroids (Hosie et al., 2006; Bracamontes et al., 2011). The parameter L is a measure of background activity. For wild-type concatemeric receptors in the absence of additional agonists, L, calculated as (1 − Popen,const)/Popen,const, was held at 8000 (Akk et al., 2018). To analyze the steroid concentration-response data recorded in the wild-type concatemeric receptor in the presence of a low concentration of GABA, L was constrained to (1 − Popen,GABA)/Popen,GABA. For receptors containing the α1(L263S) or β2(Y143W+M286W) mutations L was estimated experimentally as (1 − Popen,const)/Popen,const. Curve fitting was carried out using Origin version 7.5 (OriginLab, Northhampton, MA) on averaged data obtained from at least five cells.
Experimental and Predicted Responses to Agonist Combinations.
In experiments involving measurements of responses to agonist combinations, the cells were first exposed to each agonist separately, followed by the application of the combination. Additionally, each cell was exposed to 3 mM GABA + 100 μM pentobarbital (wild-type concatemeric receptors), 10 μM GABA [receptors containing the α1(L263S) mutation], or 300 μM GABA [receptors containing the β2(Y143W+M286W) mutations], which generated a response with a Popen value that was considered to be indistinguishable from 1 (Chang and Weiss, 1999; Ziemba and Forman, 2016; Shin et al., 2018). Activation of the wild-type concatemeric receptor by steroid combinations was recorded in the presence of a low concentration of GABA. In this case, the cells were initially exposed to GABA and combinations of GABA plus a single steroid. This was followed by application of GABA plus both steroids.
The predicted peak responses to agonist combinations were calculated using three models. First, a prediction was made assuming energetic additivity, i.e., that each agonist in the combination interacts with a distinct set of binding sites. The activation scheme for two agonists interacting with distinct sites is given in Fig. 1B. To calculate the predicted peak responses, we employed eq. 1 using KX and cX for the primary agonist and constrained L to the value calculated from the direct activating effect of the potentiator as (1 − Popen,potentiator)/Popen,potentiator. There are no objective criteria to designate one agonist in the pair as primary and the other as potentiator. In combinations that involved GABA as one of the agonists, we named GABA as primary. In other cases, we arbitrarily assigned one agonist as primary.
Second, predictions were made assuming that the paired agonists compete for common binding sites (Fig. 1C). The predicted peak responses in this model were calculated using eq. 2:
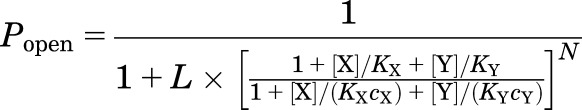 |
(2) |
where X and Y are the two agonists; N is the number of shared sites; KX and KY are the equilibrium dissociation constants for X and Y in the closed receptor, respectively; and cX and cY are the ratios of the equilibrium dissociation constants for X and Y in the open receptor to KX and KY, respectively. We note that this approach could only be employed when the number of binding sites was the same for each agonist in the pair. For example, this approach was not used when analyzing interactions of GABA (NGABA = 2) with propofol (Npropofol = 6).
We also explored a situation where one agonist interacts with a subset of binding sites available to the other compound. In this case, the interaction is a mixture of competition and energetic additivity. To test this scenario, the response predictions were made using eq. 3:
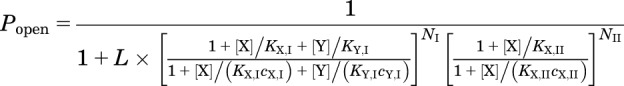 |
(3) |
In this model, agonist X binds to class I and II sites, whereas agonist Y binds only to class I sites. The terms in eq. 3 are as defined previously.
In cases where the predictions could be made using the distinct (eq. 1) and same site (eq. 2) models, the observed values of Popen were compared with predicted Popen values by calculating the log likelihood ratio (LLR) as follows (Burnham et al., 2011):
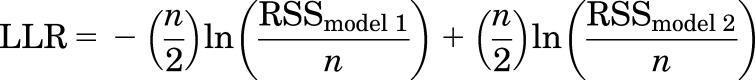 |
(4) |
where n is the number of cells for the condition; RSS is the residual sum of squares; and models 1 and 2 describe models assuming distinct and same sites, respectively, for the paired agonists. The likelihood ratio (LR = eLLR) is reported in Tables 2 and 3 as a gauge of how much one model is more likely than the other to describe the data. Additionally, we report in Tables 2 and 3 the values of the parameter Δ calculated as
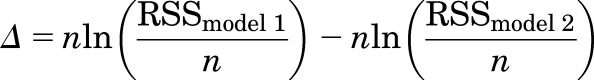 |
(5) |
where model 1 denotes the model with lower likelihood and model 2 denotes the model with higher likelihood. The value of Δ thus calculated is interpretable with regard to empirical support for a model. Models with a value of Δ up to 2 are considered to have substantial support, models with Δ = 4–7 have considerably less support, and those with Δ > 10 have essentially no empirical support (Burnham and Anderson, 2004).
TABLE 2.
Effects of agonist combinations on open probability
The columns give the paired agonists (agonists 1 and 2) and adjusted concentrations, open probabilities for each agonist separately (Popen,agonist 1 and Popen,agonist 2) or combined (Popen,agonist 1+agonist 2), and the predicted Popen value assuming the paired agonists act through the same or distinct sites. The column labeled LR shows the likelihood ratio (LR = eLLR) that quantifies by how many times the model with distinct sites is more likely than the model with the same sites to describe the data (and in parentheses the inverse of that likelihood ratio). The value of parameter Δ (see Materials and Methods) indicates the empirical support for the lower ranked model. Values of Δ of 4–7 are considered to indicate little empirical support and values of >10 indicate essentially no support (Burnham and Anderson, 2004). The data show mean ± S.D. from five to nine cells at each condition. The nominal concentrations of agonists are provided in the text. The predicted Popen values were not calculated for GABA + propofol and alfaxalone + propofol (N/A) using a model assuming the same sites because the number of binding sites is different for the agents in both pairs.
| Adjusted Concentration |
Popen,agonist 1 | Popen,agonist 2 | Popen,agonist 1+agonist 2 | Predicted Popen |
LR | Δ | ||
|---|---|---|---|---|---|---|---|---|
| Agonist 1 |
Agonist 2 |
Same Site |
Distinct Site |
|||||
| μM | μM | |||||||
| GABA (2.2) | propofol (9.4) | 0.013 ± 0.007 | 0.011 ± 0.004 | 0.47 ± 0.19 | N/A | 0.48 ± 0.24 | N/A | N/A |
| GABA (2.8) | pentobarbital (67) | 0.021 ± 0.016 | 0.013 ± 0.013 | 0.63 ± 0.11 | 0.052 ± 0.043 | 0.53 ± 0.25 | 1209 (8.3 × 10−4) | 14 |
| GABA (6.8) | alfaxalone (0.97) | 0.083 ± 0.022 | 0.0008 ± 0.0005 | 0.36 ± 0.05 | 0.054 ± 0.011 | 0.35 ± 0.21 | 39 (0.026) | 7 |
| GABA (4.0) | P4S (45) | 0.034 ± 0.004 | 0.049 ± 0.009 | 0.059 ± 0.010 | 0.085 ± 0.009 | 0.93 ± 0.02 | <10−6 (>106) | 35 |
| GABA (3.0) | β-alanine (206) | 0.021 ± 0.002 | 0.024 ± 0.004 | 0.054 ± 0.011 | 0.074 ± 0.010 | 0.80 ± 0.04 | <10−6 (>106) | 36 |
| alfaxalone (0.29)a | 3α5αP (0.07)a | 0.032 ± 0.16 | 0.034 ± 0.014 | 0.053 ± 0.022 | 0.053 ± 0.024 | 0.09 ± 0.04 | 2.5 × 10−5 (39,883) | 21 |
| 3α5αP (0.17)a | 3α5βP (0.26)a | 0.052 ± 0.011 | 0.044 ± 0.010 | 0.074 ± 0.013 | 0.071 ± 0.014 | 0.18 ± 0.06 | <10−6 (>106) | 37 |
| 3α5βP (0.28)a | ent-3α5βP (0.80)a | 0.10 ± 0.02 | 0.12 ± 0.03 | 0.17 ± 0.03 | 0.16 ± 0.03 | 0.36 ± 0.08 | <10−6 (>106) | 30 |
| alfaxalone (0.15)b | etiocholanolone (7.5)b | 0.18 ± 0.02 | 0.15 ± 0.01 | 0.19 ± 0.01 | 0.19 ± 0.01 | 0.23 ± 0.03 | 4.7 × 10−4 (2134) | 15 |
| alfaxalone (0.20)a | propofol (1.2)a | 0.18 ± 0.09 | 0.23 ± 0.12 | 0.31 ± 0.12 | N/A | 0.37 ± 0.17 | N/A | N/A |
| alfaxalone (0.63) | propofol (31) | 0.0006 ± 0.0001 | 0.10 ± 0.03 | 0.38 ± 0.03 | N/A | 0.33 ± 0.10 | N/A | N/A |
| alfaxalone (0.14)a | pentobarbital (5.3)a | 0.12 ± 0.04 | 0.19 ± 0.07 | 0.30 ± 0.13 | 0.23 ± 0.07 | 0.28 ± 0.08 | 16.4 (0.061) | 6 |
| alfaxalone (0.63) | pentobarbital (172) | 0.0006 ± 0.0001 | 0.057 ± 0.025 | 0.35 ± 0.09 | 0.042 ± 0.018 | 0.21 ± 0.10 | 228 (0.0044) | 11 |
LR, likelihood ratio.
The experiments with alfaxalone + 3α5αP, 3α5αP + 3α5βP, and 3α5βP + ent-3α5βP, and sets of experiments with alfaxalone + propofol, and alfaxalone + pentobarbital were conducted in the presence of a low concentration of GABA.
The experiments with alfaxalone + etiocholanolone were conducted on receptors containing the gain-of-function α1(L263S) mutation in both α subunits.
TABLE 3.
Effects of combinations of propofol and pentobarbital on open probability
The columns give the type of receptor, the total number of propofol sites, the number of sites that can bind either propofol or pentobarbital, the open probabilities in the presence of propofol, pentobarbital, or propofol + pentobarbital, the open probability calculated for the specified number of shared sites, and the open probability calculated assuming that propofol and pentobarbital bind to distinct sites. The number of cells was seven for wild-type concatemers and six for the mutant receptor. The model with shared sites was more likely—by >105- and 104-fold, respectively—than the model with distinct sites to describe the data for the wild-type concatemeric and mutant receptors. The Δ values (see Materials and Methods) for the lower ranked model were 27 and 19 for the wild-type and mutant concatemeric receptors, respectively.
| Receptor | NPRO | NPRO or PEB | Popen,PRO | Popen,PEB | Popen,PRO+PEB | Predicted Popen |
|
|---|---|---|---|---|---|---|---|
| Shared Site | Distinct Site | ||||||
| βαγ + βα | 6 | 2 | 0.023 ± 0.015 | 0.023 ± 0.019 | 0.29 ± 0.17 | 0.26 ± 0.18 | 0.67 ± 0.25 |
| β(Y143W+M286W)αγ + β(Y143W+M286W)α | 2 | 2 | 0.27 ± 0.06 | 0.27 ± 0.04 | 0.36 ± 0.06 | 0.37 ± 0.06 | 0.49 ± 0.10 |
PEB, pentobarbital; PRO, propofol.
In cases where the paired agonists were expected to act through a different number of distinct binding sites (GABA + propofol; alfaxalone + propofol) and only a prediction for Popen for the model with distinct sites could be made, the predicted and observed Popen values were compared using the paired t test. The results are reported subsequently.
Materials and Chemicals.
The inorganic salts used in ND96, GABA, β-alanine, P4S, pentobarbital, and picrotoxin were purchased from Sigma-Aldrich (St. Louis, MO). Propofol was purchased from MP Biomedicals (Solon, OH). The steroids (alfaxalone, 3α5αP, 3α5βP, and etiocholanolone) were bought from Sigma-Aldrich or Tocris/Bio-Techne (Minneapolis, MN). The enantiomer of 3α5βP was synthesized as described previously (Nilsson et al., 1998).
The stock solution of GABA was made in ND96 at 500 mM, stored in aliquots at −20°C, and diluted as needed on the day of experiment. The stock solutions of β-alanine and P4S were made on the day of experiment in ND96 at 100 and 5 mM, respectively, and further diluted immediately before experiment. Stock solutions of propofol [200 mM in dimethyl sulfoxide (DMSO)] and pentobarbital (5 mM in bath solution) were stored at room temperature. The steroids were dissolved in DMSO at 10–50 mM and stored at room temperature. The agonist solutions were pH adjusted when needed.
The highest final DMSO concentration in working solutions was 0.1%. We have previously found that DMSO at up to 0.5% is without effect on holding current or peak amplitude of the response to an EC50 concentration of GABA from oocytes expressing the closely related α1β3γ2L receptors (Germann et al., 2016).
Results
GABAA Receptor Activation by Orthosteric and Allosteric Agonists.
We commenced by examining activation of the concatemeric GABAA receptor by several orthosteric (P4S and β-alanine) and allosteric (pentobarbital and the steroids alfaxalone, 3α5αP, 3α5βP, ent-3α5βP, and etiocholanolone) activators. Receptor function was recorded at six-to-nine concentration points from at least five cells for each agonist.
The wild-type GABAA receptor is only weakly activated by neuroactive steroids. To obtain robust current responses, the properties of steroids were studied in the presence of ∼EC10 GABA and/or in receptors containing the gain-of-function α1(L263S) mutation. Sample current traces are given in Fig. 2.
Fig. 2.

Sample current traces in the presence of orthosteric or allosteric agonists, or combinations of agonists. (A) The wild-type concatemeric receptors were activated by 30 μM GABA, 100 μM propofol, 100 μM P4S, 3 mM β-alanine, or 1 mM pentobarbital. The concentrations were selected to generate approximately half-maximal responses for the given agonist. The amplitudes of the current responses are given in units of open probability for easier comparison of gating efficacy between the agonists. (B) The wild-type concatemeric receptors were activated by a low concentration of GABA (4–8 μM) in the absence (left trace in each pair) and presence of a steroid (1 μM alfaxalone, 0.3 μM 3α5αP, 0.3 μM 3α5βP, or 1 μM ent-3α5βP). The calibration bars apply to all traces in (B). (C) The concatemeric βα(L263S)γ + βα(L263S) receptors were activated by 1 μM alfaxalone (ALF) or 5 μM etiocholanolone (Etio). Note that these recordings were conducted in the absence of added GABA.
The activation properties of each agent were determined by fitting eq. 1 to the concentration-response data. The concentration-response curves are shown in Fig. 3, and the fitting results are summarized in Table 1.
Fig. 3.

Activation properties of GABAergic agonists. Estimated open probability of the concatemeric β2α1γ2L + β2α1 GABAA receptor is given as a function of concentration of GABA, propofol (PRO), P4S, β-alanine (β-Ala), pentobarbital (PEB), or the steroids alfaxalone (ALF), 3α5αP, 3α5βP, ent-3α5βP, and etiocholanolone (Etio). The data points and error bars show mean ± S.D. from five to eight cells. The curves were generated by fitting eq. 1 to the Popen data (see Materials and Methods). The fitted values of K and c are provided in Table 1. The data for GABA (dashed line) and propofol (dotted line) are from prior reports (Akk et al., 2018; Shin et al., 2018). The effects of alfaxalone, 3α5αP, 3α5βP, and ent-3α5βP on the wild-type concatemeric receptor were obtained in the presence of a low concentration of GABA that generated a background response with a Popen value of ∼0.1 in wild-type concatemeric receptors. Introduction of the α1(L263S) mutation increases the constitutive open probability and mimics the presence of GABA. A receptor containing the α1(L263S) mutation in both concatemeric constructs was used to determine the activation properties of alfaxalone and etiocholanolone.
TABLE 1.
Properties of GABAergic agonists
A summary of the activation properties of the GABAergic agonists employed in the study. Wild type is the ternary GABAA receptor consisting of βαγ + βα concatemeric constructs and α1(L263S) is the receptor consisting of βα(L263S)γ and βα(L263S) concatemeric constructs. The parameter KX is the equilibrium dissociation constant of the closed receptor for a given agonist. The parameter cX gives the ratio of the dissociation constants of the open receptor to that of the closed receptor. The parameter NX is the number of binding sites for the agonist. Gating energy was calculated as NXRT × ln(cX). The maximal predicted open probability (Popen,max) was calculated as 1/(1 + LcXN) with L held at 8000 for the wild-type receptor (Akk et al., 2018) and 8.1 for the mutant receptor (Shin et al., 2018).
| Receptor | Agonist |
KX |
cX | NX | Gating Energy |
Popen,max |
|---|---|---|---|---|---|---|
| μM | kcal/mol | |||||
| Wild type | GABAa | 72 ± 15 | 0.003 ± 0.000 | 2 | −6.74 | 0.92 |
| Wild type | P4S | 38 ± 4 | 0.027 ± 0.000 | 2 | −4.26 | 0.15 |
| Wild type | β-Alanine | 6664 ± 2947 | 0.002 ± 0.001 | 2 | −7.17 | 0.96 |
| Wild type | Propofola | 21 ± 3 | 0.222 ± 0.003 | 6 | −5.33 | 0.51 |
| Wild type | Pentobarbital | 1912 ± 1690 | 0.004 ± 0.002 | 2 | −6.52 | 0.89 |
| Wild type | Alfaxaloneb | 2.3 ± 0.3 | 0.159 ± 0.009 | 2 | −2.17 | 0.005 |
| Wild type | 3α5αPb | 0.27 ± 0.07 | 0.233 ± 0.018 | 2 | −1.72 | 0.002 |
| Wild type | 3α5βPb | 0.45 ± 0.06 | 0.265 ± 0.010 | 2 | −1.57 | 0.002 |
| Wild type | ent-3α5βPb | 2.4 ± 0.4 | 0.166 ± 0.012 | 2 | −2.12 | 0.005 |
| α1(L263S) | Alfaxalone | 3.0 ± 0.3 | 0.130 ± 0.006 | 2 | −2.41 | 0.88 |
| α1(L263S) | Etiocholanolone | 11.1 ± 1.5 | 0.685 ± 0.009 | 2 | −0.45 | 0.21 |
The data for GABA and propofol are from prior reports (Akk et al., 2018; Shin et al., 2018).
The wild-type concatemeric receptor is only weakly activated by neuroactive steroids. Accordingly, the properties of the steroids alfaxalone, 3α5αP, 3α5βP, and ent-3α5βP were determined in the presence of a low concentration (∼EC10) of GABA.
Coapplication of an Allosteric Agonist with the Transmitter GABA.
Coapplication of an allosteric agonist, such as propofol or pentobarbital, enhances the peak current response to GABA. In the coagonist model, description of receptor activity in the presence of an agonist combination does not require that there is specific interaction between the agonists; the potentiating effect is explained by each active compound independently and additively contributing free energy to stabilization of the open state (Ruesch et al., 2012; Ziemba and Forman, 2016; Shin et al., 2018). Potentiation can also be viewed as the change in receptor activation by the primary agonist due to reduction in L (increase in background activity) resulting from the direct activating effect of the potentiator.
To illustrate receptor potentiation by a combination of GABA and an allosteric agonist, we coapplied propofol or pentobarbital with GABA. The experimental peak responses to the combination were compared with the predicted peak responses, which were calculated assuming independent and additive energetic contributions by each agonist (eq. 1). Each cell was exposed to low concentrations of GABA, propofol (or pentobarbital), and the combination of GABA with propofol (or pentobarbital). The cells were also exposed to the combination of saturating (3 mM) GABA + 100 μM pentobarbital to generate a response with the estimated value of Popen = 1 (Ziemba and Forman, 2016), which was used as the reference response to which the responses to single agonists and agonist combinations from that cell were compared.
The application of 1.5 μM GABA generated a response with a value of Popen = 0.013 ± 0.007 (n = 9 cells). In the same set of cells, the application of 15 μM propofol generated a response with a value of Popen = 0.011 ± 0.004. The combination of GABA with propofol produced a response that had a value of Popen = 0.47 ± 0.19.
To predict the peak response to GABA + propofol, we first calculated the modified L (see Materials and Methods) from the direct activating response to propofol [modified L = (1 – 0.011)/0.011 = 89.9]. We then calculated, using eq. 1, the response to GABA employing the modified L, and the KGABA and cGABA values given in Table 1. This approach produced a predicted value of Popen = 0.48 ± 0.24 (mean ± S.D. for predictions made for each of the nine cells individually) for the combination. The experimental (0.47 ± 0.19) and predicted (0.48 ± 0.24) Popen values are not different (P = 0.78; paired t test).
Conversely, we calculated the value for modified L for the response to GABA [modified L = (1 – 0.013)/0.013 = 75.9] and then determined the response to propofol employing the modified L of 75.9, and the Kpropofol and cpropofol values given in Table 1. It is not crucial whether receptor activation by GABA is estimated on the background of propofol-elicited activity or activation by propofol is estimated on the background of GABA-elicited activity. In the coagonist model, either compound can be considered to enhance the background activity upon which the response to the other agonist is measured. As expected, both approaches produced identical results (predicted Popen = 0.48). From these experiments, we infer that the actions of GABA and propofol can be described through energetic additivity, and that the two agonists act on the GABAA receptor through distinct binding sites. Both inferences are in agreement with prior reports (O’Shea et al., 2000; Ruesch et al., 2012; Shin et al., 2018). We did not test the model in which GABA (NGABA = 2) and propofol (Npropofol = 6) share some of the binding sites.
In this analysis, the nominal concentrations of agonists were adjusted for each oocyte to account for day-to-day variability and to reflect the actual, observed peak amplitudes. This was done by matching the experimental peak amplitude with the concentration-response data given in Fig. 3 and Table 1. Thus, the prediction of the response to an agonist combination is based on the responses to the individual agonists at their observed Popen values rather than at their nominal concentrations. In this experiment, the mean adjusted concentrations were 2.2 ± 0.8 μM for GABA (nominal concentration = 1.5 μM) and 9.4 ± 2.1 μM for propofol (nominal concentration = 15 μM). The reasons for variability are not fully clear to us but may include errors in preparation of solutions, differences in levels of endogenous modulators, and/or slow rundown or hysteresis in the concentration-response measurements. The mean adjusted concentrations for each agonist are provided in Table 2.
Coapplication of pentobarbital (Popen = 0.013 ± 0.013; n = 6) with GABA (Popen = 0.021 ± 0.016) generated a response with the mean peak Popen value of 0.63 ± 0.11. The predicted Popen value for the combination, assuming distinct binding sites, was 0.53 ± 0.25. Both GABA and pentobarbital were postulated to bind to two sites; therefore, the predicted Popen value could also be calculated using a model in which GABA and pentobarbital interact with the same sites (eq. 2; 0.052 ± 0.043). The comparative ability of the two models to describe the observed response to the combined application was assessed by computing the likelihood ratio (see Materials and Methods). As shown in Table 2, the distinct site model was estimated to be 1209-fold more likely.
Coapplication of 1 μM alfaxalone, which by itself generated a response with the mean Popen value of 0.0007 ± 0.0005 (n = 6), with GABA (Popen = 0.087 ± 0.021) produced a response with the mean peak Popen value of 0.43 ± 0.19. The predicted Popen value for the pair assuming independent sites for GABA and alfaxalone was 0.33 ± 0.19, and assuming that the same sites mediate the actions of the two agonists, 0.060 ± 0.017. In this case, the likelihood ratio was 39-fold for the ability of the distinct site model over the same site model to describe the observations. We infer that GABA and pentobarbital, and GABA and alfaxalone act independently and energetically additively to stabilize the open channel. These findings are in agreement with prior reports (Ziemba and Forman, 2016; Shin et al., 2017). The data are summarized in Fig. 4 and Table 2.
Fig. 4.

Coapplication of GABA with an allosteric or orthosteric agonist. The experimental and predicted Popen values are given for combinations of GABA with propofol (PRO), pentobarbital (PEB), alfaxalone (ALF), P4S, or β-alanine (β-Ala). The open circles show experimental data from each cell separately. The open triangles and squares show predictions based on models assuming distinct or same sites for the paired compounds. The filled symbols and error bars shown mean ± S.D. for each condition. The data and results of statistical analyses are summarized in Table 2. Prediction with the same site model was not done for the GABA + PRO combination because of a difference in the number of postulated binding sites.
Coapplication of an Orthosteric Agonist with the Transmitter GABA.
The data given previously showed that the combination of an allosteric agonist with GABA results in potentiation of the current response. Receptor behavior is fundamentally different when two agonists acting at the orthosteric sites are coapplied. In particular, the effect of coapplication depends on whether the paired compounds have similar or different gating efficacies, and on the concentration of each agent.
Using eq. 2, we modeled the effect of coapplication of GABA with the low-efficacy orthosteric agonist P4S. The simulations were done at four concentrations of GABA, selected to elicit responses with Popen values of 0.05, 0.1, 0.2, or 0.5. The underlying assumption was that GABA and P4S act at the same sites (Krogsgaard-Larsen et al., 1980). The simulations (Fig. 5A) show that at low concentrations of GABA, coapplication with P4S generates a larger response than when either agonist is applied alone. As the concentration of P4S is increased and P4S out competes GABA at the transmitter binding site, the Popen value of the response to the combination approaches that of saturating P4S. At higher transmitter concentrations, when the response to GABA is greater than the response to saturating P4S, the latter acts as a competitive inhibitor at all concentrations (Fig. 5A).
Fig. 5.

Predicted responses to coapplication of two orthosteric agonists. The effects of coapplication of GABA with low-efficacy agonist P4S (A) or high-efficacy agonist β-alanine (B) on receptor open probability. The concentration-response data for P4S and β-alanine in the absence of GABA are shown as open circles. The concentrations of GABA were held at 5.0, 7.6, 12.2, or 29.8 μM, to elicit responses with Popen values of 0.05, 0.1, 0.2, and 0.5, respectively (filled circles, bottom to top in the graphs). The simulations were done using eq. 2 and the parameters provided in Table 1. The approach assumes that two shared sites mediate the actions of GABA + P4S and GABA + β-alanine.
GABA and β-alanine have similar maximal Popen values (Fig. 3; Table 1). Coapplication of β-alanine is predicted to lead to potentiation of GABA-activated receptors (Fig. 5B). At saturating concentrations of β-alanine, the response to GABA + β-alanine reaches the maximal open probability for β-alanine.
We experimentally tested receptor activation in the presence of the agonist pairs of GABA + P4S and GABA + β-alanine. Coapplication of P4S (Popen = 0.049 ± 0.009; n = 5) with GABA (Popen = 0.034 ± 0.004) resulted in a response with the mean peak Popen value of 0.059 ± 0.010. Coapplication of β-alanine (Popen = 0.024 ± 0.004; n = 5 cells) with GABA (Popen = 0.021 ± 0.002) generated a response with the mean peak Popen value of 0.054 ± 0.011. The predictions for the combinations were done using two models. In the model in which each agonist interacts with distinct sites (eq. 1), the predicted Popen values for GABA + P4S and GABA + β-alanine were 0.93 ± 0.02 and 0.80 ± 0.04, respectively. In the model assuming that the agonists bind to the same set of sites (eq. 2) the predicted Popen values were 0.085 ± 0.009 for GABA + P4S and 0.074 ± 0.010 for GABA + β-alanine. Thus, the assumption of shared binding sites resulted in predicted Popen values close to the experimental values. The likelihood ratios indicated that the same site model was >106 times more likely to describe the data. Summaries of the data are provided in Fig. 4 and Table 2.
Coapplication of Two Allosteric Agonists that Interact with the Same Binding Sites.
Receptor behavior is similar in principle when two allosteric agonists that interact with the same sites are coapplied. We tested receptor activation by several combinations of structurally related steroids, for which it was assumed that the same sites mediate their effects on the GABAA receptor (Hosie et al., 2006; Miller et al., 2017). The experiments were conducted in the presence of a low concentration of GABA or on receptors containing the gain-of-function α1(L263S) mutation. Both approaches increase background activity, enabling studies of weak activators such as neuroactive steroids (Akk et al., 2018).
The steroid pair of alfaxalone + 3α5αP was tested on the wild-type concatemeric receptor in the presence of 2 μM GABA. Application of GABA elicited a response with the mean Popen value of 0.012 ± 0.005 (n = 6). Coapplication of 0.5 μM alfaxalone with GABA generated a response with the mean Popen value of 0.032 ± 0.016. When GABA was combined with 0.2 μM 3α5αP the mean Popen value was 0.034 ± 0.014. Coapplication of both steroids with GABA generated a response with the mean Popen value of 0.053 ± 0.022. The predicted Popen value of the response to GABA + alfaxalone + 3α5αP using a model with two shared binding sites (eq. 2) for the steroids is 0.053 ± 0.024. Using a model with distinct sites for alfaxalone and 3α5αP, the predicted Popen value was 0.09 ± 0.04. The likelihood ratio indicated that the same site model was about 40,000-fold more likely. We infer that alfaxalone and 3α5αP interact with the same sites.
We next examined potentiation of GABA-activated receptors by the combination of 3α5αP + 3α5βP. The peak responses in the presence of GABA and 0.3 μM 3α5αP or 3α5βP had mean Popen values of 0.052 ± 0.011 (n = 6) and 0.044 ± 0.010, respectively. The mean Popen value in the presence of GABA + 3α5αP + 3α5βP was 0.074 ± 0.013. The predicted Popen value assuming shared sites was 0.071 ± 0.014. For comparison, the predicted Popen value assuming unique sites for 3α5αP and 3α5βP was 0.18 ± 0.06. The likelihood ratio indicated that the same site model was >106 times more likely. Our conclusion that 3α5αP and 3α5βP interact with the same sites is in agreement with prior data (Hosie et al., 2006; Miller et al., 2017).
We also examined potentiation of GABA-activated receptors by the combination of the natural steroid 3α5βP and its enantiomer (ent-3α5βP). Coapplication of 0.3 μM 3α5βP with GABA increased the Popen value from 0.025 ± 0.005 (n = 6) to 0.10 ± 0.02. In the presence of GABA + ent-3α5βP, the Popen value was 0.12 ± 0.03, and in the presence of GABA + 3α5βP + ent-3α5βP it was 0.17 ± 0.03. The predicted Popen value was 0.16 ± 0.03 using the model in which 3α5βP and ent-3α5βP bind to the same sites and it was 0.36 ± 0.08 with the model having unique sites for the two steroids. The likelihood ratio indicated that the same site model was >106 times more likely. We infer that 3α5βP and ent-3α5βP act on the GABAA receptor through the same sites.
Finally, we tested direct activation of the βα(L263S)γ + βα(L263S) receptor by the steroid combination alfaxalone + etiocholanolone. Previous studies of single-channel kinetics had suggested that etiocholanolone did not interact with all sites occupied by a more efficacious steroid, such as 3α5βP or alfaxalone (Li et al., 2007). This experiment was conducted in the absence of GABA. The gain-of-function mutation increases unliganded activity and enables studies of weak agonists (Popen,const = 0.11) (Akk et al., 2018).
Exposure of the mutant receptor to alfaxalone or etiocholanolone produced responses with the mean peak Popen value of 0.18 ± 0.02 (n = 6) or 0.15 ± 0.01, respectively. Coapplication of alfaxalone and etiocholanolone generated a response with the mean Popen value of 0.19 ± 0.01. The mean Popen value predicted from the model with two shared sites for alfaxalone and etiocholanolone was 0.19 ± 0.01. Using a model with distinct binding sites for alfaxalone and etiocholanolone, the predicted Popen value was 0.23 ± 0.03. The likelihood ratio indicated that the same site model was about 2000-fold more likely. The data are summarized in Fig. 6 and Table 2.
Fig. 6.

Coapplication of allosteric agonists. The experimental and predicted Popen values are given for the steroid combinations of alfaxalone (ALF) + 3α5αP, 3α5αP + 3α5βP, 3α5βP + ent-3α5βP, and alfaxalone + etiocholanolone (Etio), and the combinations of alfaxalone with propofol (PRO) or pentobarbital (PEB). The open circles show experimental data from each cell separately. The open triangles and squares show predictions based on models assuming distinct or same sites for the paired compounds. The filled symbols and error bars show mean ± S.D. for each condition. The experimental conditions, data, and results of statistical analyses are summarized in Table 2. Prediction with the same site model was not done for the ALF + PRO combination because of a difference in the number of postulated binding sites.
In conclusion, the actions of all tested steroid combinations (alfaxalone + 3α5αP, 3α5αP + 3α5βP, 3α5βP + ent-3α5βP, and alfaxalone + etiocholanolone) were best accounted for by a model where the paired steroids interacted with the same binding sites.
Coapplication of Two Allosteric Agonists that Interact with Distinct Binding Sites.
In the presence of two allosteric agonists with distinct—i.e., unshared binding sites—receptor behavior is similar to the situation where an allosteric agonist is coapplied with GABA. In this situation, either agonist can be considered to independently increase background activity and thereby promote activation by the other agonist.
We first examined receptor activation in the presence of alfaxalone and propofol, which are expected to act on the GABAA receptor through distinct sites (Nourmahnad et al., 2016), with Nalfaxalone = 2 and Npropofol = 6. The experiments were conducted both in the absence and presence of GABA. In the absence of GABA, the mean Popen values were 0.0006 ± 0.0001 (n = 6) for alfaxalone and 0.10 ± 0.03 for propofol. Coapplication of alfaxalone with propofol generated a peak response with a Popen value of 0.38 ± 0.03. Assuming independent actions of the two agonists, the predicted average open probability is 0.33 ± 0.10 (P = 0.15; paired t test).
The mean Popen value in the presence of 3 μM GABA + 0.2 μM alfaxalone was 0.18 ± 0.09 (n = 6). Coapplication of 2 μM propofol with GABA + alfaxalone increased the mean Popen value to 0.31 ± 0.12. Assuming independent actions of GABA, alfaxalone, and propofol (eq. 1), the predicted Popen value for the triple drug combination is 0.37 ± 0.17 (P = 0.09; paired t test). Thus, the data obtained for the alfaxalone + propofol combination support the previous finding of distinct sites for alfaxalone and propofol (Nourmahnad et al., 2016).
Analogously, we probed the effect of the combination of alfaxalone + pentobarbital. Exposure to alfaxalone elicited a peak response with the mean Popen value of 0.0006 ± 0.0001 (n = 6). Exposure to pentobarbital generated a mean Popen value of 0.057 ± 0.025. Coapplication of alfaxalone with pentobarbital produced responses with the mean Popen value of 0.35 ± 0.09. The predicted Popen value is 0.21 ± 0.10 assuming different sites for alfaxalone and pentobarbital, and 0.042 ± 0.018 assuming that the same sites mediate the actions. In this case, the likelihood ratio is 16 for the distinct sites over the same sites model.
The application of 3 μM GABA + 0.2 μM alfaxalone elicited a response with the mean Popen value of 0.12 ± 0.04 (n = 7). Coapplication of 25 μM pentobarbital with GABA + alfaxalone generated a response with the mean Popen value of 0.30 ± 0.13. Using the model with distinct binding sites for GABA, alfaxalone, and pentobarbital, the predicted Popen value for the triple drug combination was 0.28 ± 0.08. The mean Popen value predicted using a model in which alfaxalone and pentobarbital interact with the same sites is 0.23 ± 0.07. The likelihood ratio is 228 for the distinct sites over the same sites model. Summaries of the data are provided in Fig. 6 and Table 2.
Coapplication of Two Allosteric Agonists that Interact with Partially Shared Binding Sites.
In the models described previously, the two paired agonists were assumed to share all or none of the binding sites. As shown through modeling and experimental data, the two situations are associated with different levels of potentiation during coapplication. An extension of these models is one in which one of the compounds interacts with a subset of the binding sites available to the other. In this mechanism, the effect is a mix of the agonists binding to distinct sites (energetic additivity) and competition (concentration additivity) at the shared sites.
We hypothesized that such a model describes the interaction between propofol and pentobarbital on the GABAA receptor. Photolabeling studies have shown that propofol and a barbiturate analog bind with high affinity to overlapping sites at the α-β and γ-β interfaces near the β(M227) residue (Chiara et al., 2013). In addition, propofol binds to the β-α interface with high affinity (Jayakar et al., 2014; Nourmahnad et al., 2016). Thus, photolabeling experiments suggest that the αβγ GABAA receptor contains two common sites for propofol and pentobarbital in addition to distinct sites to which propofol binds.
To test this hypothesis, we exposed cells expressing wild-type concatemeric receptors to propofol, pentobarbital, or the combination of the two. In seven cells, the mean Popen value in the presence of propofol was 0.023 ± 0.015. The mean Popen value in the presence of pentobarbital was 0.023 ± 0.019. Coapplication of the two drugs generated a peak response with the mean Popen value of 0.29 ± 0.17.
The experimental data were compared with predicted Popen values calculated using two approaches. First, as the null hypothesis, we assumed that propofol and pentobarbital interact with distinct sets of sites and that the actions of the drugs are governed by energetic additivity. Such a model (eq. 1) predicts a mean Popen value of 0.67 ± 0.25 for propofol + pentobarbital. In the second approach (eq. 3), we constrained the total number of propofol binding sites to six (Shin et al., 2018) and varied the number of sites shared with pentobarbital from one to three. In this case, the activation curve for pentobarbital was fit with Npentobarbital = 1, 2, or 3 to obtain the appropriate values for K and c for pentobarbital. The predicted mean Popen values for propofol + pentobarbital were 0.44 ± 0.26, 0.26 ± 0.18, and 0.18 ± 0.12 for one, two, and three shared sites, respectively. We infer that the combination of a total of six sites for propofol, of which two alternatively can bind pentobarbital adequately, describes activation of the wild-type concatemeric receptor. The likelihood ratio indicated that this model of partially shared sites was >105 times more likely than the model with no shared sites.
Introduction of the β2(Y143W) and β2(M286W) mutations has been shown to reduce the number of functional binding sites for propofol such that concatemeric receptors containing the two mutations in each of the β subunits (a quadruple-mutant receptor) effectively retain only two propofol binding sites (Shin et al., 2018). These mutations are located in the β subunit or at the β-α interface where they are expected to disrupt the actions of propofol (Eaton et al., 2015; Franks, 2015; Shin et al., 2018). A change in the number of propofol binding sites can be expected to alter receptor activation by the propofol + pentobarbital combination in a predictable manner.
We tested activation of the β(Y143W+M286W)αγ + β(Y143W+M286W)α receptor by 5 μM propofol, 15 μM pentobarbital, and the combination of the two drugs. The mean Popen values were 0.27 ± 0.06 (n = 6 cells) in the presence of propofol and 0.27 ± 0.04 in the presence of pentobarbital. When propofol was coapplied with pentobarbital, the mean Popen was 0.36 ± 0.06. An activation model (eq. 2) with two common binding sites for propofol and pentobarbital predicted a Popen value of 0.37 ± 0.06. In contrast, a model where propofol and pentobarbital interact with distinct sites predicted a Popen value of 0.49 ± 0.10 for the drug combination. The likelihood ratio indicated that the model in which two sites could bind either propofol or pentobarbital was >104 times more likely than the model in which the sites were distinct. The data are summarized in Table 3.
The Predicted Isobolograms for Agonist Combinations.
The data shown previously demonstrate that receptor activity in the presence of various agonist combinations can be markedly different when the compounds bind to the same versus distinct binding sites. Specifically, the potentiating effect resulting from the addition of a second GABAergic drug is greater when the two drugs bind to distinct sites. This can have clinical implications when combination therapies are considered.
We have simulated isobolograms for situations where the transmitter is combined with another orthosteric agonist or an allosteric drug, and when two allosteric drugs are combined. The results (Fig. 7) demonstrate that the combination of two orthosteric agonists results in strict concentration additivity illustrated by the linear isobole of additivity. In contrast, the combination of GABA with the allosteric drug propofol generates a curvilinear isobole. This effect is mediated by energetic additivity that manifests as apparent synergy (Shin et al., 2017). Coapplication of propofol and pentobarbital also produces a curvilinear isobole; however, the curvature is less pronounced due to partial overlap of binding sites.
Fig. 7.

Predicted isobolograms for agonist combinations. The combination of GABA with β-alanine (A) or P4S (B) produces linear isoboles of additivity, indicative of concentration additivity. The combination of GABA with the allosteric agonist propofol (C) produces a highly curvilinear isobole. The combination of pentobarbital and propofol (D) also generates a curvilinear isobole but with reduced curvature because of partial overlap between the binding sites for pentobarbital and propofol. The dashed lines in (C and D) show hypothetical linear isoboles that would be observed if the paired compounds interacted with the same sites on the receptor. The isobolograms were calculated for the target Popen value of 0.15 in all panels.
Discussion
The goal of this study was to compare activation of the GABAA receptor by various combinations of orthosteric and allosteric agonists. We were motivated by the fact that native GABAA receptors under physiologic and clinical conditions can be exposed to multiple GABAergic drugs whose net action and interactions are not well understood. We examined receptor activity in the presence of combinations of orthosteric agonists (GABA + β-alanine and GABA + P4S), an orthosteric agonist + an allosteric agonist (GABA + propofol and GABA + steroid), and combinations of allosteric agents (steroid + steroid and propofol + pentobarbital). The experimental data were analyzed and compared with predictions made using variations of the coagonist activation model. The major finding, in agreement with simulations based on the model, is that the degree of potentiation resulting from combining GABAergic compounds depends on whether the individual agonists bind to the same or distinct sites. Conversely, we propose that receptor activity in the presence of agonist combinations can be used to determine whether the paired compounds interact with the same or distinct sites.
The simulated and experimental data indicate that the degree of potentiation is greater when agonists interacting with distinct sites are combined than when agonists interacting with the same sites are combined. We have used the terms shared or common sites when a site can be occupied by either agent in the pair, and unique or distinct sites when only one agent in the pair can bind to a given site. Combination of agonists interacting with distinct sites underlies classic potentiation that is observed when, for example, propofol or a neuroactive steroid is combined with GABA. Receptor activity in the presence of such combinations can be predicted by summing energetic contributions of the individual agents. Classic potentiation manifests as synergy in isobolographic analysis of the effects of drug combinations (Shin et al., 2017). In contrast, the amplitude of the response to a combination of agonists interacting with the same sites depends on the relative efficacies and concentrations of the individual agents but is rarely larger than the sum of responses to either drug alone. Importantly, the response amplitude to agonist combination in all cases is a computable value.
This approach can be used to assess whether two (or more) agents in a combination act through the same or distinct sites, since the responses predicted by models with different degrees of overlap of binding sites are in most cases well separated. By comparing the experimental and predicted responses we show that the same sites mediate the actions of a 5α-reduced steroid (3α5αP) and a 5β-reduced steroid (3α5βP), which is in agreement with previous structural and mutational data (Hosie et al., 2006; Miller et al., 2017). In addition, we show that a natural steroid (3α5βP) and its enantiomer (ent-3α5βP), and the weak steroid etiocholanolone (Li et al., 2007) and the strong steroid alfaxalone (Cao et al., 2018) act through the same binding sites. Finally, we propose that propofol and pentobarbital interact with partially shared sites. The electrophysiological data are best described by the receptor containing six sites for propofol (Shin et al., 2018), two of which can alternatively bind pentobarbital. Elimination of the four unique propofol sites through mutagenesis produced a receptor whose activity in the presence of propofol + pentobarbital was best described by the drugs competing for two common sites. These findings provide functional confirmation to previous photolabeling data indicating shared binding sites for these anesthetics (Jayakar et al., 2014).
The concerted transition model (Fig. 1) postulates that all binding sites for a given agonist possess identical properties, i.e., K and c values. Within experimental error this has largely been proven true, when tested with mutations introduced to individual binding sites for GABA (Baumann et al., 2003), the steroid 3α5αP (Bracamontes et al., 2011), or propofol (Shin et al., 2018), although not for etomidate (Maldifassi et al., 2016). Anesthetic agents, including barbiturates, have been shown to interact with varying affinities at different intersubunit interfaces in photolabeling studies (Chiara et al., 2013; Jayakar et al., 2014), raising a possibility that the number of functionally apparent binding sites is dependent on the concentration of the drug. One implication of this, and a caveat to the data and conclusions in the present study, is that the apparent effect of coapplication of GABAergic anesthetics can be dependent on the concentrations of individual drugs.
The findings have potential clinical relevance since they can predict the efficacy of drug combinations in combination therapy. For example, propofol is predicted to be highly synergistic when combined with neuroactive steroids because the two classes of drugs interact with distinct sites. On the other hand, the combination of propofol with pentobarbital is expected to show less synergy, due to partial overlap between the binding sites. It has been shown previously that coapplication of the neuroactive steroid alfaxalone enhances the GABAergic effects of etomidate (Li et al., 2014). In the present work, we find that the endogenous neurosteroid 3α5αP is a low efficacy agonist that interacts with the same sites as alfaxalone. Based on our analysis, we would predict that an increase in the concentration of endogenous 3α5αP would enhance the effects of etomidate in the absence of exogenous alfaxalone, but would actually reduce the ability of coadministered alfaxalone to enhance the sedative effect of etomidate. The extent of this effect would, of course, depend on the precise relationship between the open probability of the GABAA receptor and behavior, which is not well understood at present.
In summary, we have shown that the coagonist activation model predicts widely different magnitudes of potentiation depending on whether the individual agonists in a combination interact with the same or distinct sites. The experimental data confirm previous views on the overlap of binding sites for specific GABAergic agents including several orthosteric agonists, intravenous anesthetics, and steroids. We provide functional evidence that 5α- and 5β-reduced steroids, and natural and enantiomeric steroids, interact with the same sites on the GABAA receptor, and support for the notion that propofol and barbiturates share some of their binding sites. We also propose that the approach can be exploited to determine whether a novel drug shares a binding site with a known drug in a combination.
Abbreviations
- DMSO
dimethyl sulfoxide
- ent-3α5βP
enantiomer of 5β-pregnan-3α-ol-20-one
- Popen
open probability of the receptor
- Popen,const
open probability of a constitutively active receptor
- P4S
piperidine-4-sulfonic acid
- 3α5αP
5α-pregnan-3α-ol-20-one (allopregnanolone)
- 3α5βP
5β-pregnan-3α-ol-20-one (pregnanolone)
- RSS
residual sum of squares
Authorship Contributions
Participated in research design: Steinbach, Akk.
Conducted experiments: Shin, Germann.
Contributed new reagents or analytic tools: Covey.
Performed data analysis: Shin, Germann, Steinbach, Akk.
Wrote or contributed to the writing of the manuscript: Shin, Germann, Covey, Steinbach, Akk.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM108580]; and funds from the Taylor Family Institute for Innovative Psychiatric Research.
References
- Akk G, Shin DJ, Germann AL, Steinbach JH. (2018) GABA type A receptor activation in the allosteric coagonist model framework: relationship between EC50 and basal activity. Mol Pharmacol 93:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin J, Weiss DS. (1993) GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by pentobarbital. Nature 366:565–569. [DOI] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E. (2003) Individual properties of the two functional agonist sites in GABAA receptors. J Neurosci 23:11158–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat C. (2012) New insights into the structural bases of activation of Cys-loop receptors. J Physiol Paris 106:23–33. [DOI] [PubMed] [Google Scholar]
- Bracamontes J, McCollum M, Esch C, Li P, Ann J, Steinbach JH, Akk G. (2011) Occupation of either site for the neurosteroid allopregnanolone potentiates the opening of the GABAA receptor induced from either transmitter binding site. Mol Pharmacol 80:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracamontes JR, Steinbach JH. (2009) Steroid interaction with a single potentiating site is sufficient to modulate GABA-A receptor function. Mol Pharmacol 75:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR. (2004) Multimodel inference. Understanding AIC and BIC in model selection. Sociol Methods Res 33:261–304. [Google Scholar]
- Burnham KP, Anderson DR, Huyvaert KP. (2011) AIC model selection and multimodel inference in behavioral ecology: some background, observations, and comparisons. Behav Ecol Sociobiol 65:23–35. [Google Scholar]
- Cao LQ, Montana MC, Germann AL, Shin DJ, Chakrabarti S, Mennerick S, Yuede CM, Wozniak DF, Evers AS, Akk G. (2018) Enhanced GABAergic actions resulting from the coapplication of the steroid 3α-hydroxy-5α-pregnane-11,20-dione (alfaxalone) with propofol or diazepam. Sci Rep 8:10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Weiss DS. (1999) Allosteric activation mechanism of the α1β2γ2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J 77:2542–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, Cohen JB. (2013) Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor. J Biol Chem 288:19343–19357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua HC, Chebib M. (2017) GABAA receptors and the diversity in their structure and pharmacology. Adv Pharmacol 79:1–34. [DOI] [PubMed] [Google Scholar]
- Eaton MM, Cao LQ, Chen Z, Franks NP, Evers AS, Akk G. (2015) Mutational analysis of the putative high-affinity propofol binding site in human β3 homomeric GABAA receptors. Mol Pharmacol 88:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton MM, Germann AL, Arora R, Cao LQ, Gao X, Shin DJ, Wu A, Chiara DC, Cohen JB, Steinbach JH, et al. (2016) Multiple non-equivalent interfaces mediate direct activation of GABAA receptors by propofol. Curr Neuropharmacol 14:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman SA. (2012) Monod-Wyman-Changeux allosteric mechanisms of action and the pharmacology of etomidate. Curr Opin Anaesthesiol 25:411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman SA, Stewart D. (2012) Mutations in the GABAA receptor that mimic the allosteric ligand etomidate. Methods Mol Biol 796:317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. (2015) Structural comparisons of ligand-gated ion channels in open, closed, and desensitized states identify a novel propofol-binding site on mammalian γ-aminobutyric acid type A receptors. Anesthesiology 122:787–794. [DOI] [PubMed] [Google Scholar]
- Germann AL, Shin DJ, Manion BD, Edge CJ, Smith EH, Franks NP, Evers AS, Akk G. (2016) Activation and modulation of recombinant glycine and GABAA receptors by 4-halogenated analogues of propofol. Br J Pharmacol 173:3110–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. (2006) Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444:486–489. [DOI] [PubMed] [Google Scholar]
- Jayakar SS, Zhou X, Chiara DC, Dostalova Z, Savechenkov PY, Bruzik KS, Dailey WP, Miller KW, Eckenhoff RG, Cohen JB. (2014) Multiple propofol-binding sites in a γ-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J Biol Chem 289:27456–27468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Sahara Y, Dzubay JA, Westbrook GL. (1998) Defining affinity with the GABAA receptor. J Neurosci 18:8590–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogsgaard-Larsen P, Falch E, Schousboe A, Curtis DR, Lodge D. (1980) Piperidine-4-sulphonic acid, a new specific GABA agonist. J Neurochem 34:756–759. [DOI] [PubMed] [Google Scholar]
- Li P, Bracamontes J, Katona BW, Covey DF, Steinbach JH, Akk G. (2007) Natural and enantiomeric etiocholanolone interact with distinct sites on the rat α1β2γ2L GABAA receptor. Mol Pharmacol 71:1582–1590. [DOI] [PubMed] [Google Scholar]
- Li P, Bracamontes JR, Manion BD, Mennerick S, Steinbach JH, Evers AS, Akk G. (2014) The neurosteroid 5β-pregnan-3α-ol-20-one enhances actions of etomidate as a positive allosteric modulator of α1β2γ2L GABAA receptors. Br J Pharmacol 171:5446–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldifassi MC, Baur R, Sigel E. (2016) Functional sites involved in modulation of the GABAA receptor channel by the intravenous anesthetics propofol, etomidate and pentobarbital. Neuropharmacology 105:207–214. [DOI] [PubMed] [Google Scholar]
- Miller PS, Scott S, Masiulis S, De Colibus L, Pardon E, Steyaert J, Aricescu AR. (2017) Structural basis for GABAA receptor potentiation by neurosteroids. Nat Struct Mol Biol 24:986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12:88–118. [DOI] [PubMed] [Google Scholar]
- Nilsson KR, Zorumski CF, Covey DF. (1998) Neurosteroid analogues. 6. The synthesis and GABAA receptor pharmacology of enantiomers of dehydroepiandrosterone sulfate, pregnenolone sulfate, and (3α,5β)-3-hydroxypregnan-20-one sulfate. J Med Chem 41:2604–2613. [DOI] [PubMed] [Google Scholar]
- Nourmahnad A, Stern AT, Hotta M, Stewart DS, Ziemba AM, Szabo A, Forman SA. (2016) Tryptophan and cysteine mutations in M1 helices of α1β3γ2L γ-aminobutyric acid type A receptors indicate distinct intersubunit sites for four intravenous anesthetics and one orphan site. Anesthesiology 125:1144–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW. (2018) GABAA receptor: positive and negative allosteric modulators. Neuropharmacology 136:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea SM, Wong LC, Harrison NL. (2000) Propofol increases agonist efficacy at the GABAA receptor. Brain Res 852:344–348. [DOI] [PubMed] [Google Scholar]
- Ruesch D, Neumann E, Wulf H, Forman SA. (2012) An allosteric coagonist model for propofol effects on α1β2γ2L γ-aminobutyric acid type A receptors. Anesthesiology 116:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DJ, Germann AL, Johnson AD, Forman SA, Steinbach JH, Akk G. (2018) Propofol is an allosteric agonist with multiple binding sites on concatemeric ternary GABAA receptors. Mol Pharmacol 93:178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DJ, Germann AL, Steinbach JH, Akk G. (2017) The actions of drug combinations on the GABAA receptor manifest as curvilinear isoboles of additivity. Mol Pharmacol 92:556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieghart W. (2015) Allosteric modulation of GABAA receptors via multiple drug-binding sites. Adv Pharmacol 72:53–96. [DOI] [PubMed] [Google Scholar]
- Ziemba AM, Forman SA. (2016) Correction for inhibition leads to an allosteric co-agonist model for pentobarbital modulation and activation of α1β3γ2L GABAA receptors. PLoS One 11:e0154031. [DOI] [PMC free article] [PubMed] [Google Scholar]