

Abstract
Cannabinoid receptor 1 (CB1) is a G-protein–coupled receptor that is abundant in the central nervous system. It binds several compounds in its orthosteric site, including the endocannabinoids, arachidonoyl ethanolamide (anandamide) and 2-arachidonoyl glycerol, and the plant-derived Δ9-tetrahydrocannabinol, one of the main psychoactive components of marijuana. It primarily couples to Gi/o proteins to inhibit adenylate cyclase activity and typically induces downstream signaling that is Gi-dependent. Since this receptor is implicated in several maladies, such as obesity, pain, and neurodegenerative disorders, there is interest in developing therapeutics that selectively target this receptor. Allosteric modulators of CB1 offer one new approach that has tremendous therapeutic potential. Here, we reveal receptor- and cellular-level properties consistent with receptor activation by a series of pyrimidinyl biphenylureas (LDK1285, LDK1288, LDK1305, and PSNCBAM1), including promoting binding of the agonist CP55940 with positive cooperativity and inhibiting binding of the inverse agonist SR141716A with negative cooperativity, demonstrated via radioligand binding studies. Consistent with these findings, the allosteric modulators induced cellular internalization of the receptor and recruitment of β-arrestin 2 in human embryonic kidney cell line 293 cells monitored with confocal and total internal reflective fluorescence microscopy, respectively. These allosteric modulators, however, caused G-protein–independent but β-arrestin 1–dependent phosphorylation of the downstream kinases extracellular signal-regulated kinase 1/2, mitogen-activated protein kinase, and Src, shown by immunoblotting studies. These results are consistent with the involvement of β-arrestin and suggest that these allosteric modulators induce biased signaling.
Introduction
The cannabinoid receptor 1 (CB1) is a G-protein–coupled receptor (GPCR) expressed ubiquitously throughout the central nervous system (Mackie, 2006). CB1 and the subtype, cannabinoid receptor 2 (CB2), comprise the endocannabinoid system (ECS), along with their endogenous ligands (2-arachidonoylglycerol, N-arachidonoylethanolamine) and related biosynthetic and degradative enzymes (Pertwee, 2015). The ECS is fundamental to many physiologic processes; consequently, CB1 is a target for treatment of a wide range of pathologies including obesity, addiction, pain, anxiety, depression, and neurodegenerative disorders such as Alzheimer and Parkinson disease (Pacher et al., 2006; Skaper and Di Marzo, 2012; Khurana et al., 2017b).
To date, a number of exogenous ligands have been discovered, including the phytocannabinoid Δ9-tetrahydrocannabinol, a main psychoactive constituent of marijuana (Cannabis sativa). Although its role in many pathologic states makes CB1 a promising therapeutic target, these physiologic intricacies also pose significant challenges to the drug discovery and development process. A major obstacle in therapeutic development has been the incidence of side effects, some of which are quite extreme (e.g., depression associated with rimonabant) (Barth and Rinaldi-Carmona, 1999)). Furthermore, the vast majority of compounds pursued as potential therapies have been orthosteric ligands, which have affinity for the receptor’s endogenous ligand-binding pocket. Whereas therapeutics targeting orthosteric sites of GPCRs have certainly been successful overall, this pharmacologic approach is possibly untenable for certain targets owing to the risks of interfering with complex endogenous regulatory systems. Thus, a new avenue of approach is necessary.
A more nuanced view of the molecular mechanisms behind GPCR activation and signaling has emerged in recent years. In addition to the orthosteric site, many GPCRs contain one or more additional binding sites, known as allosteric sites (Changeux and Christopoulos, 2016). Developing therapies to target these sites has the potential to improve drug safety and efficacy by allowing spatiotemporal fine-tuning of the endogenous signaling systems already functioning within the body (Burford et al., 2013). Additionally, allosteric sites are generally less conserved than orthosteric sites, leading to increased subtype specificity (Conn et al., 2009). This approach to drugging GPCRs has already borne fruit, as evidenced by the clinical successes of miraviroc, a negative allosteric modulator of the chemokine receptor CCR5 approved for the treatment of HIV infection (Swinney et al., 2014), as well as cinacalcet, a positive allosteric modulator of the calcium-sensing receptor used for the treatment of secondary hyperparathyroidism (Davey et al., 2012).
Functional selectivity, also known as biased signaling, adds a further layer of complexity to GPCR signaling and, subsequently, pharmacology. Some orthosteric ligands are capable of promoting certain active receptor conformations with higher affinities for specific coupling proteins (e.g., Gi, Gs, β-arrestin 1), leading to “biased” induction of downstream cascades with “functional selectivity” (Mallipeddi et al., 2017). Induction of noncanonical signaling pathways has been validated as a therapeutic strategy, as demonstrated by using nebivolol, a biased antagonist of the β1-adrenergic receptor, for lowering blood pressure (Erickson et al., 2013). This paradigm recognizes the complex nature of intracellular signaling networks, leveraging the ability of receptors to induce multiple signal cascades and modulate a variety of cellular processes.
Logically, applying functionally selective allosteric modulators to GPCR pharmacology could lead to a greater capacity to fine-tune receptor responses to drugs, generating substantially more specific therapeutic impacts while decreasing risk; however, compounds that target CB1 in this manner have not been substantially characterized at the biochemical or cellular level, although we (Ahn et al., 2012, 2013b; Delgado-Peraza et al., 2016) and others (Baillie et al., 2013) have previously investigated the mechanisms of ORG27569. To that end, we have undertaken receptor-level studies on PSNCBAM1, a CB1 allosteric modulator first described by Horswill et al. (2007), and several structural analogs— LDK1285, LDK1288, and LDK1305—which have been previously reported as compounds 7d and 8d (Khurana et al., 2017a) and compound 29 (German et al., 2014). Notably, the compounds reported by our group, LDK1285 and LDK1288, differ from the PSNCBAM1 scaffold by both a cyano substitution on the 4-cholorophenyl position, as well as by the substitution of a pyrimidinyl ring for the pyridine ring, whereas LDK1305 is an intermediate, having just the cyano substitution. We report here an in-depth analysis of LDK1285, LDK1288, and LDK1305 that yields several outcomes consistent with activation of the receptor, including their impact on agonist and inverse agonist binding to CB1, cellular localization of the receptor, and downstream signaling.
Materials and Methods
Ligands.
The radiolabeled compounds, [3H]CP55940 and [3H]SR141716A, were both from PerkinElmer Life Sciences (Boston, MA). Nonlabeled compounds CP55940, SR141716A, and PSNCBAM1 were from Tocris Biosciences (Minneapolis, MN). The compounds LDK1285 and LDK1288 are equivalent to 7d and 8d, respectively, and synthesized as described previously (Khurana et al., 2017a). Compound LDK1305 was previously reported as compound 29 (German et al., 2014).
Cell Culture.
Human embryonic kidney 293 (HEK293) cells were maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% v/v fetal bovine serum and 3.5 mg/ml glucose.
Membrane Preparation and Radioligand Binding.
HEK293T cells were seeded at a density of 8 × 105 cells per 100-mm plate and incubated for approximately 24 hours before transient transfection with human CB1 in a pcDNA3.1 vector via the calcium phosphate precipitation method (Chen and Okayama, 1987). After 24 hours, cells were washed twice with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7) and harvested. After resuspension in PBS containing protease inhibitor cocktail (4-(2-aminoethyl) benzenesulfonyl fluoride, pepstatin A, E-64, bestatin, leupeptin, and aprotinin; Sigma-Aldrich, St. Louis, MO), cells were lysed via nitrogen cavitation using a Parr cell disruption bomb at 750 psi for 5 minutes, then centrifuged for 10 minutes at 500g and 4°C. The supernatant was collected and further spun for 45 minutes at 100,000g and 4°C. The resulting pellet was resuspended in TME buffer (25 mM Tris-HCl, 5 mM MgCl2, and 1 mM EDTA, pH 7.4) supplemented with 7% w/v sucrose, and protein concentration was determined using the Bradford assay (Bradford, 1976). Radioligand binding assays were performed using nine different concentrations (100 pM–100 μM) of nonradiolabeled compound and a fixed concentration of radiolabeled tracer ([3H]CP55940, 0.5 nM; [3H]SR141716A, 2 nM). Nonspecific binding was determined using 1 μM of the nonradiolabeled analog corresponding to each tracer compound. Approximately 3–5 μg of membrane fraction expressing CB1 were incubated with test and tracer compounds at 30°C for 60 minutes. TME buffer supplemented with 0.2% bovine serum albumin (BSA) was used to bring the final assay volume to 200 μl. The reaction was terminated by adding 300 μl of TME buffer containing 5% BSA and subsequent rapid vacuum filtration through Whatman GF/C filter paper with a 24-well sampling manifold (Brandel, Gaithersburg, MD). After two washes with ice-cold TME buffer, radioactivity was measured using liquid scintillation counting. Assays were carried out in duplicate, with at least three independent experiments performed. Data were analyzed by nonlinear regression using GraphPad Prism version 7.02 (GraphPad Software, La Jolla, CA). Data are represented as mean ± S.E.M. or with 95% confidence intervals.
Cooperativity between allosteric modulators and each radiolabeled tracer was evaluated using the allosteric ternary complex model (Christopoulos and Kenakin, 2002) as previously described (Price et al., 2005) using eq. 1:
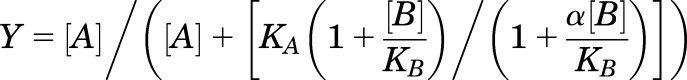 |
(1) |
Y represents the fractional specific binding of the radiolabeled orthosteric ligand, and [A] and [B] denote the concentration of the orthosteric and allosteric compound, respectively. KA and KB indicate the equilibrium dissociation constant of the orthosteric ligand and allosteric modulator, respectively. The relationship between the binding affinities of the allosteric modulator and orthosteric ligand is described by α, the antilogarithm of the cooperativity factor. An α value between 0 and 1 indicates negative cooperativity, an α = 1 indicates no cooperativity, and an α value > 1 is indicative of positive cooperativity.
Confocal Microscopy.
HEK293 cells were seeded at a density of 4 × 104 cells per well in eight-chamber slides (ThermoFisher Scientific, Rockford, IL) and incubated for 16–18 hours before transient transfection using Lipofectamine 2000 (ThermoFisher Scientific). Human CB1T210A- green fluorescent protein (GFP), a C terminally tagged receptor variant that constitutively localizes to the cell surface in the absence of ligand, was used to clearly distinguish ligand-induced internalization from the constitutive internalization found in wild-type CB1 (D’Antona et al., 2006; Ahn et al., 2012, 2013b; Simon et al., 2013).
Approximately 16 hours post-transfection, cells were treated with vehicle [0.03% dimethylsulfoxide (DMSO)] or 10 μM compound for 1, 2, or 3 hours. Cells designated 0-hour were fixed without treatment. Drug or vehicle treatment was followed by three washes in 37°C PBS, then fixation with 37°C 4% paraformaldehyde in PBS for 15 minutes at room temperature. After fixation, slides were washed twice in 37°C PBS, then air-dried for 15 minutes at room temperature in the dark before mounting in ProLong Diamond AntiFade mounting medium (ThermoFisher Scientific). The mounted slides were cured overnight at room temperature in the dark before imaging with a Leica SP8 spectral confocal microscope (Leica Microsystems, Buffalo Grove, IL) using a 40×/1.30 HC PL APO OIL CS2 objective. At least two fields of view containing approximately 10 cells each were imaged per condition for each of three independent experiments.
The FIJI (Schindelin et al., 2012) distribution package of NIH ImageJ (Schneider et al., 2012) was used to process and analyze images (National Institutes of Health, Bethesda, MD). Raw images were quantified using the Quantitative Imaging of Membrane Proteins version 1.1b (QUIMP11b) plugin (Dormann et al., 2002; Bosgraaf et al., 2009) for ImageJ/FIJI to segment cells and measure cortical fluorescence of each cell by detecting pixel intensity values, normalized to the cell interior, within 1.5 μm of the edge of the cell segment contour. Per-cell values of internal or membrane fluorescence as a percentage of total fluorescence (n = 3 independent transfections) were used to calculate mean and S.E. for each condition, expressed as fold change over vehicle at individual time points. The statistical significance of the differences between conditions was assessed by one-way analysis of variance, followed by Dunnett’s multiple comparison tests (Dunnett, 1955) using GraphPad Prism version 7.02 (GraphPad Software). For visualization purposes only, uniform linear adjustments of brightness and contrast were applied to representative images for each condition.
Total Internal Reflection Fluorescence Microscopy.
Total internal reflection fluorescence (TIRF) microscopy was performed as previously described (Roman-Vendrell and Yudowski, 2015). Briefly, HEK293 cells transiently expressing mRFP-β arrestin 1 or 2 were imaged using a Motorized Nikon Ti-E inverted microscope with a CFI-Apo 100 1.49 oil TIRF objective lens and a motorized stage with perfect focus (Melville, NY). The microscope was coupled to an iXonEM 1 DU897 back-illuminated electron multiplying charged coupled device (EMCCD camera; Andor, Belfast, UK). Imaging settings were kept constant throughout our imaging with exposure time of 300 milliseconds every 3 seconds. Cells were kept at 37°C with a Stable Z stage and objective warmer (Bioptechs, Butler, PA). Cells were gently rinsed three times with OptiMem supplemented with 20 mM HEPES (Life Technologies, Carlsbad, CA) and kept in the incubator for 10–30 minutes to acclimate before imaging. TIRF microscopy recording was conducted in the same imaging media for 1–3 minutes under basal condition (without any treatment) and followed by bath application of selected ligand using a custom-built perfusion chamber as previously described (Flores-Otero et al., 2014; Roman-Vendrell and Yudowski, 2015). Total time of live-imaging visualization and recording was less than 30 minutes. Image processing and analysis were performed using the public domain National Institutes of Health Image program ImageJ/FIJI software, which is freely available at http://fiji.sc/Fiji, as previously described (Flores-Otero et al., 2014; Roman-Vendrell and Yudowski, 2015). N ≥ 5 cells in independent experiments.
Immunoblotting Studies.
CB1-expressing cells were transfected with siRNA targeting β-arrestin 1, β-arrestin 2, or nonsilencing RNA duplex washed twice with PBS and treated with the compounds indicated. To abrogate Gi signaling, pertussis toxin (PTX) was added at 5 ng/ml (Calbiochem, San Diego, CA) before compound treatment. After an 18-hour incubation in the presence of toxin, cells were treated with compounds as previously described (Khurana et al., 2017a). Cell lysates were acquired by then treating the cells with ice-cold lysis buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 7.5). Samples were resolved via SDS-PAGE in 10% gels, and proteins were transferred onto polyvinylidene fluoride membranes. After blocking with blocking buffer (Thermo Fisher Scientific), the membrane was incubated for 1 hour with the corresponding antibody (1:3000 phospho-p44/42 and p44/42 MAPK antibodies; 1:3000 phospho-mitogen-activated protein kinase (MEK) and MEK antibodies; 1:3000 phospho-Src and Src antibodies; Cell Signaling Technology, Danvers, MA). The membranes were washed with Tris-buffered saline and 0.1% Tween 20 and then incubated with anti-rabbit peroxidase-conjugated secondary antibody (1:6000; Cell Signaling Technology) for 1 hour at room temperature. The immunoreactive proteins were detected using the SuperSignal West Femto Chemiluminescent Substrate System (ThermoFisher Scientific) using the manufacturer’s protocol. Immunoblots were quantified with the ImageJ/FIJI program.
Results
We have used a selection of compounds (Fig. 1) based on a pyrrodinyl biphenylurea scaffold, PSNCBAM1, which has previously been reported as an allosteric modulator of CB1 (Horswill et al., 2007). Follow-up SAR studies by Zhang and colleagues reported that substitution of the chloro group on ring A with a cyano group generated a variant with similar allosteric properties, identified in their work as compound 29, shown here as LDK1305 (German et al., 2014). We further extended this structural class by replacing the pyridine ring found on PSNCBAM1 and LDK1305 with a pyrimidinyl ring, producing LDK1285 and LDK1288, which differ in the position of the nitrogen within said ring (Khurana et al., 2017a). Our initial characterization of these compounds within that larger SAR study indicated that they held substantial promise, and so they were selected for analysis here.
Fig. 1.

Structures of allosteric modulators of CB1: PSNCBAM1, 1-(4-chlorophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea; LDK1285, 1-(4-cyanophenyl)-3-(3-(2-(pyrrolidin-1-yl)pyrimidine-4-yl)-phenyl)urea; LDK1288, 1-(4-cyanophenyl)-3-(3-(4-(pyrrolidin-1-yl)pyrimidine-2-yl)-phenyl)urea; and LDK1305, 1-(4-cyanophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea.
Effects of Allosteric Modulators on Orthosteric Ligand Binding.
Homologous competitive inhibition of tracer CP55940 binding with unlabeled CP55940, both orthosteric agonists, is shown for comparison and indicates the high affinity (Ki = 1.24 nM) this orthosteric compound has for its binding site (Fig. 2A; Table 1). A marked increase in binding of the orthosteric agonist tracer CP55940 to the receptor was induced by LDK1285, LDK1288, LDK1305, and PSNCBAM1 (Fig. 2, B–E), consistent with activation of the receptor by an allosteric modulator. Although no substantial difference in allosteric modulator binding affinities (KB) was found, the three LDK compounds have somewhat higher cooperativity factors (α) than PSNCBAM-1, indicative of stronger positive allosteric modulation of CP55940 binding to CB1 (Table 1).
Fig. 2.

Orthosteric ligand binding to CB1 in the presence of allosteric modulators. Left panels show binding assays using [3H]CP55940 against (A) CP55940 (i.e., orthosteric agonist shown for comparison), (B) LDK1285, (C) LDK1288, (D) LDK1305, and (E) PSNCBAM1. Right panels show binding assays using [3H]SR141716A against (F) SR141716A (i.e., orthosteric inverse agonist shown for comparison), (G) LDK1285, (H) LDK1288, (I) LDK1305, and (J) PSNCBAM1. Each data point represents the mean ± S.E. of three independent experiments performed in duplicate.
TABLE 1.
Binding parameters of orthosteric ligands and allosteric modulators
Ki, KB, and α values are shown with corresponding 95% confidence intervals in parentheses. For the first row, [3H]CP55940 and for the second row [3H]SR141716A are used as the orthosteric tracers. Three independent experiments, each done in duplicate, were used in each case.
| CP55940 | SR141716A | LDK1285 |
LDK1288 |
LDK1305 |
PSNCBAM1 |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ki | Ki | KB | α | KB | α | KB | α | KB | α | |
| [3H]CP55940 | nM | nM | nM | 11.5 | nM | 5.3 | nM | 4.3 | nM | 2.7 |
| 1.24 | ND | 198 | 62.5 | 20.0 | 41.4 | |||||
| (0.62–2.48) | (141–277) | (7.7–17.2) | (41.6–94.1) | (3.9–7.2) | (13.5–29.5) | (3.4–5.6) | (18.8–91.1) | (2.1–3.5) | ||
| [3H]SR141716A | ND | 2.13 | 473 | 0.1 | 224 | 0.1 | 115 | 0.3 | 156 | 0.1 |
| (1.37–3.32) | (160–1350) | (0.0–0.3) | (64.4–722) | (0.0–0.3) | (29.9–454) | (0.1–0.4) | (14.2–983) | (0.0–0.4) | ||
Ki, equilibrium dissociation constant of orthosteric ligand; KB, equilibrium dissociation constant of allosteric modulator; α, cooperativity factor between orthosteric and allosteric ligand binding; ND, not done.
As indicated in Fig. 2F and shown for comparison, homologous competition binding of orthosteric inverse agonist using tracer SR141716A with unlabeled SR141716A,showed this orthosteric compound bound with a Ki = 2.13 nM (Fig. 2F; Table 1). In contrast to their effects on agonist binding, all four allosteric modulators inhibited the binding of orthosteric inverse agonist SR141716A (Fig. 2, G–J). Complete inhibition of SR141716A binding was not observed up to 10 μM of any allosteric modulator, the highest concentration tested. The binding affinities and cooperativity factors of all four compounds were largely equivalent with respect to SR141716A (Table 1). It is notable that all four allosteric modulators exhibited negative cooperativity (α < 1) with the inverse agonist. Taken together, the ability of these compounds to cause an increase in agonist binding, yet a decrease in inverse agonist binding indicates their capacity to act as positive allosteric modulators (PAMs) of CB1.
Receptor Internalization in the Presence of Allosteric Modulators.
Despite their apparent positive allosteric modulation of orthosteric ligand binding affinity (Fig. 2; Table 1), we have previously reported that PSNCBAM1, LDK1285, and LDK1288 exert antagonistic effects on agonist-induced G-protein coupling (Khurana et al., 2017a). Similar inhibition of G-protein–mediated coupling by PSNCBAM1 has also been reported by others (Horswill et al., 2007; Baillie et al., 2013). To investigate these seemingly contradictory results further, we used confocal microscopy to examine the effect of these compounds in the absence of orthosteric ligand on subcellular localization of the receptor (Fig. 3, A–C). Activation of a GPCR typically results in internalization for subsequent quality-control checks (Serge et al., 2011); however, since wild-type CB1 shows some constitutive activity in the absence of ligand and is consequently internalized, we used a more inactive mutant of CB1 (that nonetheless still bound all ligands tested) to clearly analyze results. Consistent with previous findings (D’Antona et al., 2006; Ahn et al., 2012, 2013b; Simon et al., 2013), CB1T210A-GFP localized mainly to the cell surface in untreated and vehicle-treated cells (Fig. 3A, left-most column and top row, respectively). Vehicle treatment of 1, 2, or 3 hours induced no significant change in receptor internalization over untreated cells (P = 0.541, P = 0.759, P = 0.955, respectively). In comparison with cells treated with vehicle alone, prolonged exposure to all four test allosteric modulators elicited significant CB1 internalization (Fig. 3A, lower four rows), a pattern indicative of receptor activation (P < 0.001 for all test compounds at all time points, except PSNCBAM1 at 1 hour, P < 0.05). Quantification is given in Fig. 3, B and C.
Fig. 3.

Internalization of CB1 upon treatment with LDK1285, LDK1288, LDK1305, or PSNCBAM1. (A) Cells expressing CB1T210A-GFP were incubated with either vehicle alone (0.03% DMSO) or 10 μM of the indicated compound for 1, 2, or 3 hours before fixation. The 0-hour time point indicates untreated cells expressing CB1T210A-GFP. Images are representative of three independent transfections. Scale bar, 15 μm (white bar; see bottom-right image). (B) Internal CB1T210A-GFP was quantified as described in Materials and Methods. Percent internal fluorescence is expressed as the mean ± S.E. fold change over vehicle (n = 3 independent experiments). Statistical significance was assessed using one-way analysis of variance and Dunnett’s multiple comparisons test; *P < 0.05; ***P < 0.001. (C) Membrane fluorescence, derived from the experiment in frame A, is expressed as the mean ± S.E. fold change over vehicle (n = 3 independent experiments). Orange line indicates LDK1285; gray, LDK1288; yellow, LDK1305; blue, PSNCBAM1. Statistical significance was assessed using one-way analysis of variance and Dunnett’s multiple-comparisons test; ††P < 0.01 in comparison with PSNCBAM1. (D) Cells expressing CB1-GFP and β-arrestin 2-mcherry were treated with PSNCBAM1 (5 μM; blue curve) or CP55940 (0.5 μM; gray curve) or PSCNBAM1 and CP55940 (5, 0.5 μM, respectively; black curve). β-arrestin-2 recruitment to CB1 was measured by TIRF microscopy and is displayed as percent change in fluorescence intensity over time. CP55940 t1/2 = 36 seconds; CP55940 plus PSNCBAM1 t1/2 = 42 seconds. (E) Cells expressing CB1-GFP and β-arrestin 2-mcherry were treated with LDK1288 (5 μM; orange curve) or CP55940 (0.5 μM; red curve) or LDK1288 and CP55940 (5, 0.5 μM, respectively; black curve). β-arrestin 2 recruitment to CB1 was measured by TIRF microscopy and is displayed as percent change in fluorescence intensity over time. CP55940 t1/2 = 33 second; CP55940 plus LDK1288 = 36 second.
Two of the three LDK compounds (LDK1288 and LDK1305) induced internalization initially more rapidly than PSNCBAM1 (Fig. 3C; ††P < 0.01 in comparison with PSNCBAM1). Although no statistically significant differences were found between the maximal internalization induced by 3 hours of treatment of any of the four compounds tested, LDK1288 appeared to have the greatest effect; therefore, the impact of both LDK1288 and PSNCBAM1 on CB1 trafficking was further examined using TIRF microscopy, studying 7–10 cells per trace (Fig. 3, D and E). This provided an avenue to examine wild-type CB1 (as opposed to T210A) in the presence of orthosteric agonists, which occurs on a more rapid timescale. In cells expressing wild-type CB1, test compound treatment recruited β-arrestin 2 to the receptor at the cell membrane within a rapid timescale. Since β-arrestin 2 is involved in internalization of activated CB1 (Flores-Otero et al., 2014), these results further suggest that these compounds are positive allosteric modulators (PAMs), and after activation, internalization ensues. Furthermore, this indicates that these allosteric modulators recruited β-arrestin 2, much like the orthosteric agonist alone did, and indicates that internalization occurs in the wild-type as well as the mutant CB1. Failure to observe β-arrestin 2 recruitment in the absence of CP55940 is consistent with slower arrestin recruitment kinetics under these conditions, as TIRF experiments stop at 90 seconds, yet internalization is also observed at later time points. In future studies, it would be good to pursue these experiments further and use siRNA separately to β-arrestin 1 and β-arrestin 2.
Distinct Temporal Patterns of ERK1/2 Phosphorylation Induced by Allosteric Modulators.
Whereas treatment with the orthosteric agonist CP55940 (Fig. 4A) induced a more robust ERK1/2 phosphorylation response (pERK1/2), treatment with the allosteric modulators LDK1285, LDK1288, or LDK1305 (Fig. 4, B–D) produced a similar time-dependent response with a peak of ERK1/2 phosphorylation at 5–10 minutes and a decline observed to 20 minutes. At 20 minutes, the allosteric modulator-induced ERK1/2 phosphorylation level was near or at basal levels for the receptor (no compound treatment). Since ERK1/2 phosphorylation peaked at 5 minutes, this time point was used subsequently.
Fig. 4.

Effect of CP55940, LDK1285, LDK1288, or LDK1305 on ERK1/2 phosphorylation over time. HEK293 cells expressing CB1 were untreated (DMSO only) or treated with (A) CP55,940 (0.5 μM), (B) LDK1285 (10 μM), (C) LDK1288 (10 μM), or (D) LDK1305 (10 μM), for 2, 5, 10, 15, or 20 minutes. (E) Cells expressing CB1 and either β-arrestin 1 siRNA or control siRNA were treated with DMSO vehicle only, LDK1285, LDK1288, or LDK1305 (all at 10 μM) for 5 minutes. Cell lysates were separated by SDS-PAGE and analyzed by Western blots probed for phospho-ERK1/2 (pERK1/2). The total level of ERK1/2 was detected for comparison. Note that the two bands correspond to the predominant isoforms of ERK, ERK1 (p44) and ERK2 (p42). Cells not expressing CB1 and treated with LDK1288 showed no kinase phosphorylation relative to vehicle alone treatment (data not shown). Quantification of at least three independent experiments is displayed in the right-hand column and expressed as mean  S.E.-fold increase above the basal level of phosphorylation.
S.E.-fold increase above the basal level of phosphorylation.
In conjunction with our previous findings (Khurana et al., 2017a), these data show that the tested allosteric modulators may bias the receptor toward a non-canonical, non-G protein–dependent mechanism of signal transduction (i.e., functional selectivity). We explored this hypothesis by interfering with various signaling effectors before compound treatment and then assessing the phosphorylation levels of downstream kinases at 5 minutes. LDK1285, LDK1288, and LDK1305 demonstrated reliance on β-arrestin 1 for induction of ERK1/2 phosphorylation (Fig. 4E). In CB1-expressing cells cotransfected with siRNA against β-arrestin 1, all three allosteric modulators lacked the capacity to induce pERK1/2 levels substantially higher than basal; however, β-arrestin 2 siRNA, although knocked down, did not block phosphorylation of ERK1/2 by LDK1288 tested as a potent analog of the series (Supplemental Fig. 1).
Subsequently, we examined the ability of these LDK compounds to induce MEK phosphorylation at 5 minutes in CB1-expressing cells that were either pretreated with PTX to interfere with Gi/o protein function or cotransfected with β-arrestin 1, β-arrestin 2, or control siRNA (Fig. 5). LDK1285, LDK1288, or LDK1305 induced substantially more MEK phosphorylation levels relative to basal (vehicle alone-treated) levels. Knockdown of β-arrestin 1 prevented the LDK compounds from inducing MEK phosphorylation, whereas loss of either β-arrestin 2 or Gi/o protein function had little to no effect. No substantial differences were noted between the LDK compounds in their ability to induce MEK phosphorylation. As anticipated, since MEK is immediately upstream of ERK1/2, phosphorylation of these kinases was comparably dependent on β-arrestin 1 for signaling by these criteria.
Fig. 5.

Coupling partner dependency of MEK phosphorylation induced by LDK1285, LDK1288, or LDK1305. HEK293 cells expressing CB1 and β-arrestin 1 siRNA, CB1 and β-arrestin 2 siRNA, or CB1 and control siRNA, or expressing CB1 alone and pretreated with PTX were either untreated (DMSO only) or treated with (A) LDK1285 (10 μM), (B) LDK1288 (10 μM), or (C) LDK1305 (10 μM) for 5 minutes. Cell lysates were separated by SDS-PAGE and analyzed by Western blots probed for phospho-MEK (pMEK). The total level of MEK was detected for comparison. Quantification of at least three independent experiments is displayed in the right-hand column and expressed as mean  S.E.-fold increase above the basal level of phosphorylation.
S.E.-fold increase above the basal level of phosphorylation.
Due to the functional proximity of MEK and ERK1/2, as well as the similar nature of their LDK-induced phosphorylation by a β-arrestin 1–dependent mechanism, we also examined the effects of the LDK compounds on phosphorylation of Src, a comparatively distant signaling protein. Src phosphorylation in uninhibited cells was assessed with 5-minute treatment with each LDK compound. Cells expressing either CB1 alone or CB1 cotransfected with β-arrestin 1, β-arrestin 2, or control siRNA were also investigated (Fig. 6). After compound treatment, pSrc levels increased to comparable levels for each compound in control siRNA-treated cells and β-arrestin 2-knockdown cells. For both LDK1285 and LDK1305, this was a roughly 2-fold increase over basal (vehicle-treated), whereas LDK1288 treatment induced an approximately 4-fold increase over basal levels. Notably, the ability of all three LDK compounds to induce Src phosphorylation was diminished by knockdown of β-arrestin 1 (Fig. 6) but not PTX (data not shown).
Fig. 6.

Pathway dependency of Src phosphorylation induced by LDK1285, LDK1288, or LDK1305. HEK293 cells expressing CB1 alone, CB1 and β-arrestin 1 siRNA, CB1 and β-arrestin 2 siRNA, CB1 and control siRNA were either untreated (DMSO only) or treated with (A) LDK1285 (10 μM), (B) LDK1288 (10 μM), or (C) LDK1305 (10 μM) for 5 minutes. Cell lysates were separated by SDS-PAGE and analyzed by Western blots probed for phospho-Src (pSrc). The total level of Src was detected for comparison. Quantification of at least three independent experiments is displayed in the right-hand column and expressed as mean  S.E.-fold increase above basal level of phosphorylation.
S.E.-fold increase above basal level of phosphorylation.
Discussion
Our results are in general agreement with previous studies showing that PSNCBAM1 and LDK1305 caused dose-dependent increases in CP55940 binding (Horswill et al., 2007; Baillie et al., 2013; German et al., 2014). Using CP55940 as the orthosteric compound, we find that LDK1305 had the strongest binding affinity (KB = 20 nM) and LDK1285 the highest cooperativity factor (α = 11.5). The equilibrium dissociation constants indicate that the allosteric modulators have a strong affinity for CB1 in the presence of the orthosteric agonist CP55940. LDK1305 has an affinity that is 10-fold stronger than that of the CB1 allosteric modulator ORG27569 (KB = 217.3 nM) (Ahn et al., 2012). Similarly, two indole-2-carboxamide allosteric modulators, intercellular adhesion molecule (ICAM)-a and ICAM-b, have binding affinities that are 10–100 times weaker than LDK1305 for CB1 in the presence of CP55940 (KB = 5778 nM and KB = 469.9 nM, respectively) (Ahn et al., 2013a). The cooperativity factors of the LDK compounds are also comparable to those of known CB1 allosteric modulators. For example, the value of LDK1285 in the presence of CP55940 is similar to that of ORG27569 (α = 6.95) (Ahn et al., 2012) and ICAM-b (α = 17.9) (Ahn et al., 2013a), and it is 3- to 10-fold stronger than the corresponding values for ORG27759 (α = 3), ORG29647 (α = 2.8) (Price et al., 2005), and ICAM-a (α = 1.9) (Ahn et al., 2013a). Thus, the LDK compounds examined here, especially LDK 1285 and LDK1305, would act as effective allosteric modulators given the strong binding affinity and the high cooperativity factors in the presence of an orthosteric agonist.
Saturation and kinetic binding studies have previously suggested that PSNCBAM-1 affects CP55940 binding to CB1 by altering the proportion of available binding sites (Bmax), rather than the specific affinity (Kd) of the ligand for any one binding site (Baillie et al., 2013). Although we have not performed saturation binding assays to assess the Bmax under these conditions, the markedly greater increase in maximal agonist binding prompted by the LDK compounds in comparison with PSNCBAM1 may indicate that the cyanophenyl ring imparts an increased ability to induce an activated receptor state, thereby increasing sites available for CP55940 binding (Fig. 2; Table 1). That the allosteric modulators showed positive cooperativity for binding with the agonist CP55940 but negative cooperativity for binding with the inverse agonist SR141716A is consistent with these allosteric modulators promoting an active form of the receptor.
The ability of these compounds to activate CB1 is further supported by our results using microscopy to evaluate receptor internalization (Fig. 3). Whereas LDK1285, LDK1288, and PSNCBAM1 displayed progressive trends of increased internalization as exposure time increased, PSNCBAM1-induced internalization was markedly slower than for the LDK compounds at the earliest time point. This difference is especially pronounced in comparison with LDK1288 and LDK1305, which produced roughly double the internalization induced by PSNCBAM1 after 1 hour of exposure to compound. This further supports our previous conclusion that pyrimidinyl biphenylureas bearing the cyano group on the first phenyl ring induce a greater proportion of the receptor population into an activated state than the PSNCBAM1 scaffold (Khurana et al., 2017a).
TIRF experiments demonstrated that the positive allosteric modulators in the presence of agonist CP55940 resulted in β-arrestin 2 recruitment (Fig. 3, D and E). In our experience, this indicates that the initial recruitment kinetics are not affected by the PAMs and cannot be further increased beyond the agonist alone. This is likely due to the kinetics of early recruitment recorded in TIRF, and later, the difference is clear (Ahn et al., 2013b). We also cannot rule out that the engineered nature of CB1 impacts internalization. Figure 3 includes both CB1 T210A and wild-type CB1, both of which have fused GFP C-terminal to the receptor.
Additionally, we have found that the LDK compound–induced phosphorylation of ERK1/2, MEK, and Src is dependent on β-arrestin 1. Thus, the β-arrestin 1 dependency of CB1 signaling induced by this series of compounds appears to be a broad-spectrum characteristic, holding across a wide time span and multiple signaling proteins. This is strongly suggestive of functional selectivity originating at the receptor level, where the allosteric modulator promotes an alternative active conformation that precludes G-protein coupling while promoting β-arrestin recruitment. To the extent analyzed, whereas phosphorylation of these kinases is dependent on β-arrestin 1, internalization induced by ORG27569 is β-arrestin 2–dependent (Ahn et al., 2013b), and that is also indicated here (Fig. 3) for compounds PSNCBAM1, LDK1283, LDK1288, and LDK1305. GPCR signaling and internalization are dependent on different β-arrestin isoforms, which has been viewed previously for other GPCRs (Vibhuti et al., 2011; Abrisqueta et al., 2013; Srivastava et al., 2015). Thus, these allosteric modulators are not orthosteric agonists (e.g., they enhance CP55940 binding) but rather can function as PAMs (e.g., binding analysis) or ago-allosteric modulators (i.e., they can function in signaling without the orthosteric compound being present). In future studies, it would be valuable to determine whether cell signaling is enhanced for the endogenous cannabinoid plus the allosteric modulator over what is found for the endogenous cannabinoids or the allosteric modulator alone.
This study indicates that LDK1285, LDK1288, and LDK1305 are also examples of biased allosteric modulators for GPCRs. Rather than couple to Gi, the PAMs promote β-arrestin coupling by the criteria examined here and previously (Khurana et al., 2017b). Thus, these compounds could stabilize an active conformation that binds to β-arrestin, and this CB1 active structure is possibly distinct from the one that binds to the Gi protein. Several other known compounds induce biased signaling in GPCRs. For example, the pepducin ATI-2341 induces Gi-protein binding rather than G13 or β-arrestin binding induced by the natural ligand factor-1α (Quoyer et al., 2013). The μ-opioid receptor agonist TRV130 induces G-protein rather than β-arrestin signaling (DeWire et al., 2013). Furthermore, several examples of allosteric modulators of GPCRs are functionally selective. PDC113.824 is an allosteric modulator for the prostaglandin F2α receptor (PGF2α). This compound positively modulates Gq binding and subsequent activation while inhibiting G12 protein binding to PGF2α (Goupil et al., 2010). Similarly, the activated calcium-sensing receptor binds to both the Gi and Gq proteins, but an acquired hypocalciuric hypercalcemia autoantibody serves as an allosteric modulator that activates the calcium-sensing receptor–mediated Gq pathway (Makita et al., 2007). For the same receptor, cinacalcet and NPS-R568 both increased the potency of Ca2+-mediated Ca2+ mobilization, which is indicative of Gq signaling, whereas NPS-2143 negatively impacted the potency of Ca2+ mobilization. On the other hand, plasma membrane ruffling, which is indicative of β-arrestin or G12/13 protein binding, corresponded to a higher affinity for NPS-R568 rather than NPS-2143 and cinacalet (Davey et al., 2012). Finally, osmotic membrane stretch allosterically modulates angiotensin II type 1 receptor binding to β-arrestin 2 (Tang et al., 2014).
Biased signaling of allosteric modulators is just beginning to be applied to therapeutic strategies. For example, PDC113.824 induces Gq coupling to PGF2α, which results in the inhibition of G12-mediated Rho/ROCK signaling pathways (Goupil et al., 2010). Animal experiments show that blocking the ROCK signaling pathway prevents preterm labor in mice (Tahara et al., 2005; Lartey et al., 2007; Lartey and López Bernal, 2009). Knowledge of the pharmacology and mechanisms of allosteric modulators have opened new avenues of research for therapeutic discoveries. In this study, we have shown the biased impact of LDK1285, LDK1288, and LDK1305 compounds on downstream signaling. In the future, we hope their physiologic effects and medicinal promise can be determined.
Acknowledgments
We thank Yu-Hsien Liao for experimental contributions.
Abbreviations
- CB1
cannabinoid receptor 1
- DMSO
dimethylsulfoxide
- ERK
extracellular signal-regulated kinase
- GFP
green fluorescent protein
- GPCR
G-protein–coupled receptor
- HEK
human embryonic kidney (cells)
- ICAM
intercellular adhesion molecule
- MEK
mitogen-activated protein kinase
- PAM
positive allosteric modulator
- PTX
pertussis toxin
- TIRF, total internal reflection fluorescence. Chemical names
CP55940, (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)-phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol
- LDK1285
1-(4-cyanophenyl)-3-(3-(2-(pyrrolidin-1-yl)pyrimidine-4-yl)-phenyl)urea
- LDK1288
1-(4-cyanophenyl)-3-(3-(4-(pyrrolidin-1-yl)pyrimidine-2-yl)-phenyl)urea
- LDK1305
1-(4-cyanophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea
- PSNCBAM1
1-(4-chlorophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea
- SR141716A
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
Authorship Contributions
Participated in research design: Jagla, Tang, Yudowski, Lu, Kendall.
Conducted experiments: Jagla, Scott, Tang, Mateo-Semidey.
Contributed new reagents or analytic tools: Qiao, Lu.
Performed data analysis: Jagla, Scott, Tang, Yudowski, Kendall.
Wrote or contributed to the writing of the manuscript: Jagla, Scott, Yudowski, Kendall.
Footnotes
This work was supported in part by the National Institutes of Health, National Institute on Drug Abuse [Grant DA039942].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Abrisqueta M, Herraiz C, Pérez Oliva AB, Sanchez-Laorden BL, Olivares C, Jiménez-Cervantes C, García-Borrón JC. (2013) Differential and competitive regulation of human melanocortin 1 receptor signaling by β-arrestin isoforms. J Cell Sci 126:3724–3737. [DOI] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Kendall DA. (2012) Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem 287:12070–12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Samala S, Lu D, Kendall DA. (2013a) Profiling two indole-2-carboxamides for allosteric modulation of the CB1 receptor. J Neurochem 124:584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Shim JY, Kendall DA. (2013b) Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J Biol Chem 288:9790–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, McAllister S, Strange PG, Stephens GJ, Pertwee RG, et al. (2013) CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol 83:322–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth F, Rinaldi-Carmona M. (1999) The development of cannabinoid antagonists. Curr Med Chem 6:745–755. [PubMed] [Google Scholar]
- Bosgraaf L, van Haastert PJ, Bretschneider T. (2009) Analysis of cell movement by simultaneous quantification of local membrane displacement and fluorescent intensities using Quimp2. Cell Motil Cytoskeleton 66:156–165. [DOI] [PubMed] [Google Scholar]
- Bradford MM. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. [DOI] [PubMed] [Google Scholar]
- Burford NT, Clark MJ, Wehrman TS, Gerritz SW, Banks M, O’Connell J, Traynor JR, Alt A. (2013) Discovery of positive allosteric modulators and silent allosteric modulators of the μ-opioid receptor. Proc Natl Acad Sci USA 110:10830–10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7:2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. (2002) G protein-coupled receptor allosterism and complexing. Pharmacol Rev 54:323–374. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. (2009) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antona AM, Ahn KH, Kendall DA. (2006) Mutations of CB1 T210 produce active and inactive receptor forms: correlations with ligand affinity, receptor stability, and cellular localization. Biochemistry 45:5606–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A. (2012) Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153:1232–1241. [DOI] [PubMed] [Google Scholar]
- Delgado-Peraza F, Ahn KH, Nogueras-Ortiz C, Mungrue IN, Mackie K, Kendall DA, Yudowski GA. (2016) Mechanisms of biased β-arrestin-mediated signaling downstream from the cannabinoid 1 receptor. Mol Pharmacol 89:618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, et al. (2013) A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 344:708–717. [DOI] [PubMed] [Google Scholar]
- Dormann D, Libotte T, Weijer CJ, Bretschneider T. (2002) Simultaneous quantification of cell motility and protein-membrane-association using active contours. Cell Motil Cytoskeleton 52:221–230. [DOI] [PubMed] [Google Scholar]
- Dunnett CW. (1955) A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc 50:1096–1121. [Google Scholar]
- Erickson CE, Gul R, Blessing CP, Nguyen J, Liu T, Pulakat L, Bastepe M, Jackson EK, Andresen BT. (2013) The β-blocker nebivolol is a GRK/β-arrestin biased agonist. PLoS One 8:e71980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Otero J, Ahn KH, Delgado-Peraza F, Mackie K, Kendall DA, Yudowski GA. (2014) Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat Commun 5:4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German N, Decker AM, Gilmour BP, Gay EA, Wiley JL, Thomas BF, Zhang Y. (2014) Diarylureas as allosteric modulators of the cannabinoid CB1 receptor: structure-activity relationship studies on 1-(4-chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl)pyridin-2-yl]phenyl}urea (PSNCBAM-1). J Med Chem 57:7758–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goupil E, Tassy D, Bourguet C, Quiniou C, Wisehart V, Pétrin D, Le Gouill C, Devost D, Zingg HH, Bouvier M, et al. (2010) A novel biased allosteric compound inhibitor of parturition selectively impedes the prostaglandin F2alpha-mediated Rho/ROCK signaling pathway. J Biol Chem 285:25624–25636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C, Wong Kai In P. (2007) PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol 152:805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Fu BQ, Duddupudi AL, Liao YH, Immadi SS, Kendall DA, Lu D. (2017a) Pyrimidinyl biphenylureas: identification of new lead compounds as allosteric modulators of the cannabinoid receptor CB1. J Med Chem 60:1089–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Mackie K, Piomelli D, Kendall DA. (2017b) Modulation of CB1 cannabinoid receptor by allosteric ligands: pharmacology and therapeutic opportunities. Neuropharmacology 124:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartey J, López Bernal A. (2009) RHO protein regulation of contraction in the human uterus. Reproduction 138:407–424. [DOI] [PubMed] [Google Scholar]
- Lartey J, Smith M, Pawade J, Strachan B, Mellor H, López Bernal A. (2007) Up-regulation of myometrial RHO effector proteins (PKN1 and DIAPH1) and CPI-17 (PPP1R14A) phosphorylation in human pregnancy is associated with increased GTP-RHOA in spontaneous preterm labor. Biol Reprod 76:971–982. [DOI] [PubMed] [Google Scholar]
- Mackie K. (2006) Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol 46:101–122. [DOI] [PubMed] [Google Scholar]
- Makita N, Sato J, Manaka K, Shoji Y, Oishi A, Hashimoto M, Fujita T, Iiri T. (2007) An acquired hypocalciuric hypercalcemia autoantibody induces allosteric transition among active human Ca-sensing receptor conformations. Proc Natl Acad Sci USA 104:5443–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallipeddi S, Janero DR, Zvonok N, Makriyannis A. (2017) Functional selectivity at G-protein coupled receptors: advancing cannabinoid receptors as drug targets. Biochem Pharmacol 128:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, Kunos G. (2006) The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 58:389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. (2015) Endocannabinoids and their pharmacological actions. Handb Exp Pharmacol 231:1–37. [DOI] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, et al. (2005) Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol 68:1484–1495. [DOI] [PubMed] [Google Scholar]
- Quoyer J, Janz JM, Luo J, Ren Y, Armando S, Lukashova V, Benovic JL, Carlson KE, Hunt SW, III, Bouvier M. (2013) Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proc Natl Acad Sci USA 110:E5088–E5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman-Vendrell C, Yudowski GA. (2015) Real-time imaging of mu opioid receptors by total internal reflection fluorescence microscopy. Methods Mol Biol 1230:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergé A, de Keijzer S, Van Hemert F, Hickman MR, Hereld D, Spaink HP, Schmidt T, Snaar-Jagalska BE. (2011) Quantification of GPCR internalization by single-molecule microscopy in living cells. Integr Biol 3:675–683. [DOI] [PubMed] [Google Scholar]
- Simon AC, Loverdo C, Gaffuri AL, Urbanski M, Ladarre D, Carrel D, Rivals I, Leterrier C, Benichou O, Dournaud P, et al. (2013) Activation-dependent plasticity of polarized GPCR distribution on the neuronal surface. J Mol Cell Biol 5:250–265. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Di Marzo V. (2012) Endocannabinoids in nervous system health and disease: the big picture in a nutshell. Philos Trans R Soc Lond B Biol Sci 367:3193–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Gupta B, Gupta C, Shukla AK. (2015) Emerging functional divergence of β-arrestin isoforms in GPCR function. Trends Endocrinol Metab 26:628–642. [DOI] [PubMed] [Google Scholar]
- Swinney DC, Beavis P, Chuang KT, Zheng Y, Lee I, Gee P, Deval J, Rotstein DM, Dioszegi M, Ravendran P, et al. (2014) A study of the molecular mechanism of binding kinetics and long residence times of human CCR5 receptor small molecule allosteric ligands. Br J Pharmacol 171:3364–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara M, Kawagishi R, Sawada K, Morishige K, Sakata M, Tasaka K, Murata Y. (2005) Tocolytic effect of a Rho-kinase inhibitor in a mouse model of lipopolysaccharide-induced preterm delivery. Am J Obstet Gynecol 192:903–908. [DOI] [PubMed] [Google Scholar]
- Tang W, Strachan RT, Lefkowitz RJ, Rockman HA. (2014) Allosteric modulation of β-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem 289:28271–28283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibhuti A, Gupta K, Subramanian H, Guo Q, Ali H. (2011) Distinct and shared roles of β-arrestin-1 and β-arrestin-2 on the regulation of C3a receptor signaling in human mast cells. PLoS One 6:e19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten D, Christopoulos A, Sexton PM. (2013) Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Disc 12:630–644. [DOI] [PubMed] [Google Scholar]