

The crystal structure of a dimethylsulfide monooxygenase from Hyphomicrobium sulfonivorans has been solved, and comparison with its structural homologues revealed that it has a different substrate-binding pocket.
Keywords: dimethylsulfide monooxygenase, sulfur cycle, TIM-barrel fold, substrate-binding pocket, Hyphomicrobium sulfonivorans
Abstract
DmoA is a monooxygenase which uses dioxygen (O2) and reduced flavin mononucleotide (FMNH2) to catalyze the oxidation of dimethylsulfide (DMS). Although it has been characterized, the structure of DmoA remains unknown. Here, the crystal structure of DmoA was determined to a resolution of 2.28 Å and was compared with those of its homologues LadA and BdsA. The results showed that their overall structures are similar: they all share a conserved TIM-barrel fold which is composed of eight α-helices and eight β-strands. In addition, they all have five additional insertions. Detailed comparison showed that the structures have notable differences despite their high sequence similarity. The substrate-binding pocket of DmoA is smaller compared with those of LadA and BdsA.
1. Introduction
Dimethylsulfide (DMS), a volatile organosulfur compound, is considered to play an important role in the global sulfur cycle. It is the primary volatile sulfur compound in surface seawater and provides the main form of natural sea-to-air emission of sulfur (Andreae, 1990 ▸; Quinn & Bates, 2011 ▸; Bates et al., 1992 ▸; Amrani et al., 2013 ▸). In the air, DMS can be oxidized to sulfate aerosols, which are involved in the formation of cloud condensation nuclei and thus affect the formation of clouds (Curson et al., 2011 ▸; Quinn & Bates, 2011 ▸; de Zwart & Kuenen, 1992 ▸). DMS is mainly produced by the cleavage of dimethylsulfoniopropionate (DMSP) by marine bacteria and algae (Curson et al., 2011 ▸; Kiene et al., 2000 ▸; Alcolombri et al., 2015 ▸). The consumption of DMS in seawater is mainly achieved by ventilation to the air (under strong wind conditions), photolysis and microbial consumption (Kiene & Bates, 1990 ▸; Simó & Pedrós-Alió, 1999 ▸; Kieber et al., 1996 ▸). Although a few bacteria have been reported to utilize DMS (Hayes et al., 2010 ▸; Pol et al., 1994 ▸; Zwart et al., 1996 ▸; Vila-Costa et al., 2006 ▸), the structures of the enzymes involved in this process remain unknown.
Hyphomicrobium sulfonivorans, a methylotrophic bacterium that can metabolize DMS, has been reported and its DMS monooxygenase has been identified (Borodina et al., 2000 ▸, 2002 ▸). This DMS monooxygenase is composed of two subunits: DmoA and DmoB. DmoB is an oxidoreductase which can produce reduced flavin mononucleotide (FMNH2) using reduced nicotinamide adenine dinucleotide (NADH). DmoA, the main subunit of the DMS monooxygenase, is an FMNH2-dependent monooxygenase and can catalyze the oxidation of DMS with the participation of dioxygen (O2) to produce methanethiol and formaldehyde (Boden et al., 2011 ▸). Sequence analysis showed that DmoA consists of 480 amino acids. It shares 50% sequence identity with LadA, a long-chain alkane monooxygenase, and shares 33% sequence identity with BdsA, a dibenzothiophene sulfone monooxygenase. Here, we determined the crystal structure of DmoA from H. sulfonivorans to 2.28 Å resolution. Structure comparison with homologous structures revealed that they all share a conserved TIM-barrel fold, which is one of the most common enzyme folds (Wierenga, 2001 ▸). However, detailed comparison showed that the substrate-binding pocket of DmoA is smaller, which is consistent with its smaller substrate (DMS).
2. Materials and methods
2.1. Protein expression and purification
The dmoA gene was amplified from the genome of H. sulfonivorans by PCR using the FastPfu DNA polymerase. The PCR primers were designed with NdeI and XhoI restriction sites. The nucleotide sequence encoding DmoA was recombined into a pET-22b expression vector (Novagen) and was verified by sequencing. This construct contains an –LEHHHHHH tag at the C-terminus for purification. The recombinant plasmid was transformed into Escherichia coli BL21 (DE3) cells. The cells were grown at 310 K in LB medium in the presence of 0.1 mg ml−1 ampicillin to an OD600 of 0.8–1.0. The culture was then induced at 293 K overnight with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). The cells were harvested by centrifugation at 8000g for 10 min. The pellet was resuspended in lysis buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl) and lysed by sonication. The lysate was centrifuged at 16 000g for 40 min to remove the insoluble debris. The supernatant was loaded onto an Ni2+–NTA resin column (2 ml column volume; GE Healthcare) pre-equilibrated with lysis buffer. The column was washed with 20 ml wash buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 20 mM imidazole) and eluted with 10 ml elution buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 250 mM imidazole). The protein was then purified by anion-exchange chromatography using a Source 15Q column (GE Healthcare) with a linear gradient of 0–500 mM NaCl in 50 mM Tris–HCl pH 8.0. DmoA was further purified by gel filtration using a Superdex 200 column (GE Healthcare) pre-equilibrated with a buffer consisting of 10 mM Tris–HCl pH 8.0, 100 mM NaCl. The protein was finally concentrated to about 15 mg ml−1. Macromolecule-production information is summarized in Table 1 ▸.
Table 1. Macromolecule-production information.
| Source organism | H. sulfonivorans |
| DNA source | Genomic DNA |
| Cloning vector | pET-22b |
| Expression vector | pET-22b |
| Expression host | E. coli BL21 (DE3) |
| Complete amino-acid sequence of the construct produced | MKKRIVLNAFDMTCVSHQSAGTWRHPSSQAARYNDLEYWTNMAMELERGCFDCLFIADVVGVYDVYRGSAEMALRDADQVPVNDPFGAISAMAAVTEHVGFGVTAAITFEQPYLLARRLSTLDHLTKGRVAWNVVSSYLNSAALNIGMDQQLAHDERYEMADEYMEVMYKLWEGSWEDDAVKRDKKSGVFTDGSKVHPINHQGKYYKVPGFHICEPSPQRTPVIFQAGASGRGSKFAASNAEGMFILTTSVEQARQITTDIRNQAEAAGRSRDSIKIFMLLTVITGDSDEAAEAKYQEYLSYANPEGMLALYGGWTGIDFAKLDPDEPLQAMENDSLRTTLESLTHGENAKKWTVRDVIRERCIGGLGPVLVGGPQKVADELERWVDEGGVDGFNLAYAVTPGSVTDFIDYIVPELRKRGRAQDSYKPGSLRRKLIGTNDGRVESTHPAAQYRDAYVGKESVADRTQPSPFANAKAPVAELEHHHHHH |
2.2. Crystallization and data collection
The initial crystallization conditions were screened by the sitting-drop vapor-diffusion method using crystallization screening kits from Hampton Research at 293 K. Crystals of DmoA appeared in several conditions after 24 h and the best condition consisted of 0.1 M bis-Tris propane pH 8.5, 20%(w/v) PEG 3350, 0.2 M sodium iodide. Further optimization was performed by the hanging-drop vapor-diffusion method in 24-well plates. Equal volumes of protein and crystallization solution were mixed to give a total volume of 4 µl. High-quality crystals were obtained in a condition consisting of 0.1 M bis-Tris propane pH 8.3, 18%(w/v) PEG 3350, 0.2 M sodium iodide. The crystals were harvested using nylon loops and soaked in cryoprotectant consisting of 20% glycerol and 80% mother liquor. The crystals were then immediately cooled in liquid nitrogen. X-ray diffraction data were collected on beamline BL17U1 at the Shanghai Synchrotron Radiation Facility (SSRF), Shanghai, People’s Republic of China (Wang et al., 2016 ▸). The data were collected using an ADSC Q315r CCD detector at 100 K. The crystal-to-detector distance was 250 mm and the wavelength was 0.9791 Å. The diffraction data were indexed and integrated to 2.28 Å resolution with HKL-2000 (Otwinowski & Minor, 1997 ▸). Data-collection and processing statistics are summarized in Table 2 ▸.
Table 2. Data collection and processing.
Values in parentheses are for the outer shell.
| Diffraction source | BL17U1, SSRF |
| Wavelength (Å) | 0.9791 |
| Temperature (K) | 100 |
| Detector | ADSC Q315r |
| Crystal-to-detector distance (mm) | 250 |
| Rotation range per image (°) | 1 |
| Total rotation range (°) | 90 |
| Exposure time per image (s) | 0.5 |
| Space group | C2221 |
| a, b, c (Å) | 113.19, 163.96, 116.67 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution range (Å) | 47.53–2.28 (2.38–2.28) |
| Total No. of reflections | 86438 |
| No. of unique reflections | 47759 |
| Completeness (%) | 97.5 (98.8) |
| 〈I/σ(I)〉 | 18.2 (4.7) |
| R merge † | 0.092 (0.279) |
| Overall B factor from Wilson plot (Å2) | 27.2 |
R
merge = 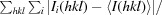
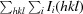 .
.
2.3. Structure determination and refinement
The structure of DmoA was determined by molecular replacement with Phaser (McCoy et al., 2007 ▸) in the PHENIX software package (Adams et al., 2002 ▸). The search model was taken from the structure of the long-chain alkane monooxygenase LadA (PDB entry 3b9o; Li et al., 2008 ▸; 50% sequence identity). Automated model building was performed with AutoBuild in PHENIX. Several rounds of refinement and manual building were then performed with phenix.refine (Afonine et al., 2012 ▸) and Coot (Emsley et al., 2010 ▸), respectively. The Ramachandran plot was analyzed by MolProbity (Chen et al., 2010 ▸). The refinement statistics are summarized in Table 3 ▸. The coordinates have been deposited in the Protein Data Bank with accession code 6ak1. Figures illustrating the structures and the electrostatic surfaces were prepared using PyMOL (Schrödinger; https://pymol.org/).
Table 3. Structure refinement.
Values in parentheses are for the outer shell.
| Resolution range (Å) | 47.53–2.28 (2.38–2.28) |
| Completeness (%) | 97.5 (98.8) |
| Final R work † | 0.158 (0.177) |
| Final R free | 0.202 (0.229) |
| No. of non-H atoms | 7797 |
| R.m.s. deviations | |
| Bonds (Å) | 0.008 |
| Angles (°) | 0.84 |
| Average B factor (Å2) | 30.37 |
| Ramachandran plot | |
| Favored regions (%) | 98.81 |
| Additionally allowed regions (%) | 1.19 |
| Outliers (%) | 0 |
R
work = 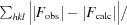
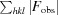 .
.
3. Results and discussion
3.1. Overall structure
The structure of DmoA was determined to 2.28 Å resolution using LadA as a search model. Molecular replacement generated a solution in space group C2221 with a solvent content of 52%. After refinement, the structure of DmoA was refined to an R work of 15.8% and an R free of 20.2%. The asymmetric unit consisted of two DmoA molecules (chain A and chain B) as a result of the noncrystallographic symmetry (Fig. 1 ▸ a). During refinement, it was observed that electron density for residues 1–2, 347–350 and 473–480 was absent in both chain A and chain B, and these residues were not modeled. The root-mean-square deviation (r.m.s.d.) between chain A and chain B is 0.156 Å, indicating that the two monomers were nearly identical. The final model is composed of 932 residues and 537 water molecules.
Figure 1.

(a) Overall structure of DmoA. There are two DmoA molecules in the asymmetric unit. Chain A is colored green and chain B is colored yellow. (b) Ribbon diagram of the DmoA monomer. The eight α-helices and eight β-strands of the TIM-barrel fold are labeled; the α-helices are colored blue and the β-strands are colored yellow. Other parts of the structure are colored green. (c) Ribbon diagram of the five additional insertions. AI1, AI2, AI3, AI4 and AI5 are colored red, yellow, blue, pink and cyan, respectively. Other parts are colored gray. (d) Cartoon diagram of the structural elements. AI1, AI2, AI3, AI4 and AI5 are marked with straight lines.
There are eight parallel β-strands in the structure of DmoA. Outside the β-strands, there are eight α-helices (Fig. 1 ▸ b). The eight parallel β-strands and the eight surrounding α-helices compose a typical TIM-barrel fold, which is a very common enzyme fold and was first reported in a triosephosphate isomerase (Wierenga, 2001 ▸). In addition to the basic (βα)8 structure, there are five additional insertions (AIs; Fig. 1 ▸ c), which are all located between the inside β-strands and the outside α-helices. These additional insertions are also observed in the long-chain alkane monooxygenase LadA (Li et al., 2008 ▸) and the dibenzothiophene sulfone monooxygenase BdsA (Okai et al., 2017 ▸; PDB entry 5tlc). The five additional insertions in DmoA are named AI1 to AI5. AI1, which is composed of 24 residues (Met12–Asp35), is located between the β1 strand and the α1 helix (Fig. 1 ▸ d). AI2 and AI3 both contain an α-helix. AI2 consists of 29 residues (Ala57–Pro85) and is positioned between β2 and α2, while AI3 consists of 21 residues (Ser136–Glu156) and is positioned between β4 and α4 (Fig. 1 ▸ d). AI4 (47 residues), which is located between α4 and β5 (Fig. 1 ▸ d), is mainly a long loop. Although it is positioned in the middle of the protein sequence (Trp176–Pro222), structurally AI4 is close to the C-terminal end of DmoA. AI5, which consists of 89 residues (Thr285–Gly374), is the longest of these five additional insertions and is mainly composed of four α-helices and one β-strand.
3.2. Comparison with homologues
Superposition of the DmoA (chain A) monomer with the LadA (chain A) and BdsA (chain A) monomers showed that their overall structures are similar (Fig. 2 ▸ a). The backbone r.m.s.d. between the DmoA monomer and the LadA monomer is 0.986 Å (over 386 aligned Cα positions; 50% sequence identity). The backbone r.m.s.d. between the DmoA monomer and the BdsA monomer is 1.582 Å (over 349 aligned Cα positions; 33% sequence identity). The backbone r.m.s.d. between DmoA and the other structural homologues RcaE (PDB entry 5w4y; D. M. Bhandari, Y. Chakrabarty, B. Zhao, J. Wood, P. Li & T. P. Begley, unpublished work), YtnJ (PDB entry 1yw1; New York SGX Research Center for Structural Genomics, unpublished work) and CmoJ (PDB entry 6asl; D. M. Bhandari, K. Krishnamoorthy, B. Zhao, P. Li & T. P. Begley, unpublished work) are 1.588, 1.659 and 2.011 Å, respectively, indicating that LadA is the most similar structure to DmoA among known structural homologues. DmoA, LadA and BdsA are all composed of a TIM-barrel structure with five additional insertions. The TIM-barrel structures are basically conserved except for the fourth and fifth α-helices. Both α4 and α5 of DmoA are shorter compared with those in LadA and BdsA (Fig. 2 ▸ a). Obvious differences between the additional insertions, especially AI5, were observed. AI5 in DmoA is composed of four α-helices (AI5α1, AI5α2, AI5α3 and AI5α4) and one β-strand, while the corresponding insertion in LadA is composed of only three α-helices (α7a, α7b and α7c) and one β-strand. The helix corresponding to AI5α3 is absent in the structure of LadA. Although the corresponding helix can be found in the structure of BdsA, AI5α3 in DmoA is closer to the putative position of FMN compared with that in BdsA (Fig. 2 ▸ a).
Figure 2.

(a) Superposition of DmoA, LadA and BdsA monomers (colored green, blue and gray, respectively). AI3, AI5α3, α4 and α5 of DmoA are marked by circles. (b) Conserved residues that interact with the isoalloxazine ring of FMN. The residues of DmoA, LadA and BdsA are colored green, blue and gray, respectively. The FMN molecule in LadA is colored yellow. The FMN molecule in BdsA is colored pink. (c) Electrostatic surface representation of the substrate-binding pocket of LadA. Negatively charged surfaces are red and positively charged surfaces are blue. The FMN molecule in LadA is colored yellow. The helices corresponding to α4 and α5 of DmoA are labeled. (d) Electrostatic surface representation of the putative substrate-binding pocket of DmoA. AI3, AI5α3, α4 and α5 under the transparent surface are labeled. The FMN molecule in LadA is shown to represent the putative position of FMN in DmoA. (e) Electrostatic surface representation of the substrate-binding pocket of BdsA. The FMN molecule in BdsA is colored pink. The helices corresponding to α4 and α5 of DmoA are labeled.
To obtain the structure of DmoA in complex with FMN, we performed co-crystallization and soaking experiments, but these both failed. It may be that the FMN molecule is not tightly bound in the structure of DmoA owing to the shorter α4 and α5. To identify the residues that interact with FMN, the structures of DmoA, LadA (in complex with FMN) and BdsA (in complex with FMN; PDB entry 5xkd; L. Gu, T. Su, S. Liu & J. Su, unpublished work) were superimposed. A detailed structural comparison showed that the residues of LadA and BdsA that interact with the isoalloxazine ring of FMN can also be found in the structure of DmoA and that their positions are nearly identical (Fig. 2 ▸ b). This indicates that the position of FMN in the structure of DmoA may be very similar to that in LadA and BdsA.
Despite their similar overall structures, the substrate-binding pockets of DmoA, LadA and BdsA have obvious differences. The substrate-binding pocket of DmoA is smaller compared with those of LadA and BdsA (Figs. 2 ▸ c, 2 ▸ d and 2 ▸ e). AI3 and AI5α3 of DmoA are located in the upper part of the substrate-binding pocket, making it hard for large substrates to enter (Fig. 2 ▸ d). AI3 of DmoA is composed of one helix and two loops, which can also be found in LadA and BdsA. However, in the structure of BdsA the loop that is near β4 is further from FMN, thus making the entrance to the substrate-binding pocket of BdsA wider (Figs. 2 ▸ a, 2 ▸ d and 2 ▸ e). Compared with LadA and BdsA, α4 and α5 of DmoA are both shorter, making the entrance to the substrate-binding pocket more open. These features are adapted to their different substrates. The substrate of LadA is a long-chain alkane and the substrate of BdsA is dibenzothiophene sulfone. The substrate of DmoA is DMS, which is much smaller. A smaller substrate-binding pocket could block large substrates and may assist the entry of DMS.
Supplementary Material
PDB reference: dimethylsulfide monooxygenase, 6ak1
Acknowledgments
We thank the staff of the BL19U1 and BL17U1 beamlines at the National Center for Protein Sciences Shanghai and Shanghai Synchrotron Radiation Facility, Shanghai, People’s Republic of China for assistance during data collection.
Funding Statement
This work was funded by National Key Research and Development Program of China grant 2016YFA0601303. National Natural Science Foundation of China grants 31630012, 41706152, and 31800107. Taishan Scholar Foundation of Shandong Province grant TS20090803. Qingdao National Laboratory for Marine Science and Technology grant 2017ASTCP-OS14. National Postdoctoral Program for Innovative Talents grants BX201600095 and 2018M632667. Natural Science Foundation of Jiangsu Province grant BK20170397. Natural Science Foundation of Shandong Province grant ZR2017BC079. Shandong Province Postdoctoral Innovation Projects grant 201701002.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Alcolombri, U., Ben-Dor, S., Feldmesser, E., Levin, Y., Tawfik, D. S. & Vardi, A. (2015). Science, 348, 1466–1469. [DOI] [PubMed]
- Amrani, A., Said-Ahmad, W., Shaked, Y. & Kiene, R. P. (2013). Proc. Natl Acad. Sci. USA, 110, 18413–18418. [DOI] [PMC free article] [PubMed]
- Andreae, M. O. (1990). Mar. Chem. 30, 1–29.
- Bates, T. S., Lamb, B. K., Guenther, A., Dignon, J. & Stoiber, R. E. (1992). J. Atmos. Chem. 14, 315–337.
- Boden, R., Borodina, E., Wood, A. P., Kelly, D. P., Murrell, J. C. & Schäfer, H. (2011). J. Bacteriol. 193, 1250–1258. [DOI] [PMC free article] [PubMed]
- Borodina, E., Kelly, D. P., Rainey, F. A., Ward-Rainey, N. L. & Wood, A. P. (2000). Arch. Microbiol. 173, 425–437. [DOI] [PubMed]
- Borodina, E., Kelly, D. P., Schumann, P., Rainey, F. A., Ward-Rainey, N. L. & Wood, A. P. (2002). Arch. Microbiol. 177, 173–183. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Curson, A. R., Todd, J. D., Sullivan, M. J. & Johnston, A. W. (2011). Nature Rev. Microbiol. 9, 849–859. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Hayes, A. C., Liss, S. N. & Allen, D. G. (2010). Appl. Environ. Microbiol. 76, 5423–5431. [DOI] [PMC free article] [PubMed]
- Kieber, D. J., Jiao, J., Kiene, R. P. & Bates, T. S. (1996). J. Geophys. Res. 101, 3715–3722.
- Kiene, R. P. & Bates, T. S. (1990). Nature (London), 345, 702–705.
- Kiene, R. P., Linn, L. J. & Bruton, J. A. (2000). J. Sea Res. 43, 209–224.
- Li, L., Liu, X., Yang, W., Xu, F., Wang, W., Feng, L., Bartlam, M., Wang, L. & Rao, Z. (2008). J. Mol. Biol. 376, 453–465. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Okai, M., Lee, W. C., Guan, L.-J., Ohshiro, T., Izumi, Y. & Tanokura, M. (2017). Proteins, 85, 1171–1177. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pol, A., Op den Camp, H. J., Mees, S. G., Kersten, M. A. & van der Drift, C. (1994). Biodegradation, 5, 105–112. [DOI] [PubMed]
- Quinn, P. K. & Bates, T. S. (2011). Nature (London), 480, 51–56. [DOI] [PubMed]
- Simó, R. & Pedrós-Alió, C. (1999). Global Biogeochem. Cycles, 13, 1173–1181.
- Vila-Costa, M., Del Valle, D. A., González, J. M., Slezak, D., Kiene, R. P., Sánchez, O. & Simó, R. (2006). Environ. Microbiol. 8, 2189–2200. [DOI] [PubMed]
- Wang, Z., Pan, Q., Yang, L., Zhou, H., Xu, C., Yu, F., Wang, Q., Huang, S. & He, J. (2016). J. Synchrotron Rad. 23, 1323–1332. [DOI] [PubMed]
- Wierenga, R. K. (2001). FEBS Lett. 492, 193–198. [DOI] [PubMed]
- Zwart, J. M. M. de & Kuenen, J. G. (1992). Biodegradation, 3, 37–59.
- Zwart, J. M. M. de, Nelisse, P. N. & Kuenen, J. G. (1996). FEMS Microbiol. Ecol. 20, 261–270.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: dimethylsulfide monooxygenase, 6ak1