

Abstract
Background
While lipoprotein(a) [Lp(a)] is associated with incident cardiovascular disease (CVD), its contribution to prediction remains controversial.
Objective
We examined the association and clinical utility of Lp(a) with incident CVD in women.
Methods
A turbidimetric assay assessed Lp(a) in three cohorts of women [the Women’s Health Study (WHS, N=24,558), a case-cohort sample from the Women’s Health Initiative Observational Study (WHI, 1815 cases, subcohort =1989), and the Justification for Use of Statins in Prevention (JUPITER) trial (N=2,569)] and men from JUPITER (N=5,161). A WHS derivation sample (N=16,400) determined the form of association with incident CVD. This was tested in WHS validation data (N=8,158) and the other study samples. Models including traditional CV risk factors but with and without Lp(a) were compared using risk reclassification.
Results
In the WHS there was a curvilinear association, with increased CVD risk among those with Lp(a) above 50 mg/dL, but only among women with total cholesterol (TC) above 220 mg/dL. In the WHS test sample there was a small but significant change in the c-statistic (0.790 to 0.797, p=0.035) but no improvement in measures of reclassification. This pattern was replicated among women in the WHI and JUPITER. In contrast, there was a strong association of Lp(a) with CVD among men with low TC levels in JUPITER.
Conclusion
In three cohorts of women, Lp(a) was associated with CVD only among those with high TC, and improvement in prediction was minimal. These data have implications for Lp(a) in clinical practice among women and for trials of Lp(a)-lowering agents.
Keywords: lipoproteins, cholesterol, cardiovascular disease, risk prediction, reclassification, gender
CONDENSED ABSTRACT
We examined the clinical utility of Lp(a) with incident cardiovascular disease in three cohorts of women from primary prevention. There was increased cardiovascular risk among those with Lp(a) above 50 mg/dL, but only among women with total cholesterol above 220 mg/dL. This pattern was replicated among women in all three cohorts. Improvement in prediction among women was minimal. In contrast, there was a strong association of Lp(a) with cardiovascular disease among men with low total cholesterol levels in JUPITER. This suggests that the role of screening for elevated Lp(a) may be limited in women without known vascular disease and with low total cholesterol. Further investigation is needed to assess potential gender differences.
Lipoprotein(a) [Lp(a)] consists of an apolipoprotein B (ApoB) particle covalently bound by a single disulfide bond to apolipoprotein(a). When measured with an isoform-independent assay, Lp(a) – a rightward skewed variable - is generally associated with incident vascular events in a curvilinear manner (1) and has modest effects on risk reclassification (2). Mendelian randomization studies suggest that genetic variants associated with elevated Lp(a) associate with vascular events (3,4). Conversely, in rare cases of null apolipoprotein(a) mutations where circulating Lp(a) is absent or extremely low, reduced vascular risk has been observed (5). These data contribute to an emerging view that Lp(a) may have a causal role in atherothrombosis (6). To date, clinical recommendations for those with elevated Lp(a) levels include the use of aspirin and statin therapy, even though the latter may mildly increase Lp(a) levels (7).
However, there remains controversy regarding the shape of the Lp(a) risk curve and whether elevated Lp(a) confers risk for cardiovascular disease (CVD) independent of traditional risk factors used in current risk equations, including total cholesterol (TC), blood pressure, and perhaps C-reactive protein (CRP). In particular, while Lp(a) predicts risk among those with low TC or low density lipoprotein cholesterol (LDLC) levels secondary to statin treatment (8,9), the role of Lp(a) as a determinant of risk among those with naturally low lipid levels remains uncertain. Further, in most studies, the risk conferred by Lp(a) itself is largely limited to those with the very highest levels.
In addition to risk reclassification, the potential for interaction between Lp(a) and lipid levels is important to understand as it has implications for the design and conduct of Lp(a) lowering clinical trials. This issue has taken on greater importance as novel strategies are emerging that employ anti-sense oligonucelotides to inhibit hepatic production of apo(a) and lower circulating Lp(a) levels (10,11). In addition, there is some limited evidence of a differential relationship of Lp(a) to risk in women vs. men (12), which may be partly mediated through hormone therapy (13), although this finding is not consistent (1). We therefore sought to address the shape of the relationship of Lp(a) with cardiovascular risk as well as any interaction between Lp(a) and lipids in three prospective cohorts of women, including a derivation sample from the Women’s Health Study (WHS) and validation samples from the WHS, the Women’s Health Initiative, and women from the Justification for the Use of Statins in Prevention (JUPITER) trial, along with a comparison sample of men from JUPITER.
METHODS
Women’s Health Study (WHS)
The WHS was a randomized trial of aspirin and vitamin E among 39,876 U.S. female health professionals beginning in 1992 that has been described previously (14,15). Eligible women were free of cardiovascular disease and cancer at study entry. Baseline questionnaires collected information on age, current blood pressure, smoking, diabetes, and parental history of myocardial infarction before age 60, along with other exposures. The randomized interventions ended in March 2004 for a median follow-up of 10.2 years (25th, 75th percentiles = 9.7, 10.6 years). Women were followed through the end of the trial for the development of CVD, here defined as incident myocardial infarction, ischemic stroke, coronary revascularization, and cardiovascular deaths. Self-reports of these events were adjudicated by an end-points committee after medical record review.
The current analysis includes those who provided a baseline blood sample and had complete data on blood biomarkers used as candidates in the development of the Reynolds Risk Score (RRS) (N=24,558) (16). The plasma samples were measured for Lp(a), other lipids, hemoglobin A1c, and high-sensitivity C-reactive protein in a core laboratory facility certified by the National Heart, Lung, and Blood Institute/Centers for Disease Control and Prevention Lipid Standardization Program. The concentration of Lp(a) was determined using a turbidimetric assay on the Hitachi 917 analyzer (Roche Diagnostics, Indianapolis, Ind), using reagents and calibrators from Denka Seiken (Tokyo, Japan). The day-to-day variability of Lp(a) concentrations of 17.6 and 58.1 mg/dL were 3.6% and 1.5%, respectively (12).
A total of 766 CVD events occurred during follow-up. During the development of the Reynolds Risk Score, two-thirds of the study participants (n=16,400) were randomly assigned to a model derivation data set and one-third (n=8,158) were reserved as an independent validation data set. The same derivation and test sets were used here to determine and validate the shape of the association of Lp(a) with CVD.
Women’s Health Initiative (WHI)
A sample of women from the WHI were included as a separate external validation set. A case-cohort sample of 4,000 women had previously been selected from the WHI-OS, including 2,000 cases, with over-sampling of under-represented minority groups (17). A subcohort of approximately 2,000 women were frequency matched to the cases by race/ethnicity and 10-year age groups. Eligible women for this analysis did not have any prior CVD conditions, leading to a final sample size of 1,815 cases and a subcohort of 1,989, of whom 131 also became cases. The primary outcome for this analysis was a combined end point of major CVD, including myocardial infarction and coronary death, ischemic stroke, and death resulting from cardiovascular causes. The median follow-up time among non-cases was 9.9 years (25th, 75th percentiles= 8.6, 11.8 years).
Laboratory assays for baseline levels of Lp(a) as well as other lipid measures, hemoglobin A1c, and high-sensitivity C-reactive protein (hsCRP) were previously assessed in this sample using the same core laboratory and assays as used for the WHS samples (17).
Justification for the Use of Statins in Prevention (JUPITER) trial
JUPITER was a primary prevention, randomized, double-blind, placebo-controlled trial investigating whether rosuvastatin 20 mg/d would decrease incident CVD in 17,802 asymptomatic men and women with LDLC <130 mg/dL and hsCRP level ≥2.0 mg/L (18). Exclusion criteria included previous or current use of lipid-lowering therapy and current use of postmenopausal hormone therapy. Baseline samples from 7,730 white participants were included in these analyses, with 2,569 women and 5,161 men (9). Participants were followed for a median 1.9 years for incidence of the primary composite CVD endpoint comprised of incident myocardial infarction, stroke, hospitalization for unstable angina, arterial revascularization, or cardiovascular death, with 210 cases occurring.
Lipid, apolipoprotein, and hsCRP values were assayed in a core laboratory. Lp(a) concentrations were measured in a blinded manner at Quest Diagnostics Nichols Institute (San Juan Capistrano, CA) with a commercially available assay (Randox Laboratories; Crumlin, Co. Antrim, United Kingdom) that is not affected by kringle IV type 2 repeats. Values were measured and reported in nanomoles per liter to reflect the molar concentration of Lp(a) particles (9). Conversion to milligrams per deciliter was approximated by dividing values in nanomoles per liter by 2.15, with a minimum value set to 10 mg/dL in these data.
Statistical Analysis
WHS
The goal of the current analysis was to determine whether Lp(a) can add to current risk prediction equations. We attempted to define and validate the best functional form (linear or nonlinear) for Lp(a) in predicting CVD, including its interactions. In previous analyses in the WHS (16), the best-fitting model (Model A) included a linear term for Lp(a) levels greater than 10 mg/dL only if ApoB was 100 mg/dL or higher. In the same WHS derivation sample, we further evaluated any non-linear association of Lp(a) with CVD, including linear and natural log terms for Lp(a), smoothing splines, and categories of Lp(a). We also examined interactions of Lp(a) with total cholesterol in addition to ApoB. This was repeated using the covariables in the RRS, the American College of Cardiology/ American Heart Association (ACC/AHA) model (19), and the Reynolds Model A (16). Models with the lowest value of the Bayes Information Criterion were selected as the final models for each set of covariables. Due to the exploratory nature of these analysis, we evaluated the same functional forms within the WHS test data set to determine whether the same patterns held. Finally, we estimated the relationships with CVD in the combined WHS sample (N=24,558).
Because differential effects of aspirin have been found by variations in the apolipoprotein gene (20), we evaluated the association of Lp(a) in subgroups defined by the randomized aspirin assignment. We also explored subgroup results by randomized vitamin E assignment, by hormone therapy, and among whites only. The WHS included too few black women to evaluate the relationship in that subgroup (18 events among 461 women.)
We also examined whether Lp(a) led to improvement in model fit when compared to a model with all other risk factors included in either the Reynolds Risk Scores or the ACC/AHA pooled cohort equations. We examined changes in the c-statistic and the integrated discrimination improvement (IDI) (21) as well as reclassification statistics (22), including the reclassification calibration test (23) and the categorical net reclassification improvement (NRI) with cut-points at 5, 10, and 20% 10-year risk, and the continuous NRI (21). Survival methods were used throughout, and bootstrapping was used to determine standard errors (24,25). Kaplan-Meier estimates were used to estimate the observed disease incidence within each reclassified cell.
WHI
The WHI data provided a separate external validation sample for the functional form for the relationship of Lp(a) with CVD. Overall population characteristics were estimated in the subcohort sample using weights equal to the inverse of the inclusion probabilities using the Horvitz-Thompson weighting approach (26). We examined trends in risk factors across categories using chi-square tests for trend or linear regression analysis for continuous variables.
Models for CVD were analyzed as a case-cohort study using previously described methods for proportional hazards regression (27), with stratum-specific weighting of the observations by race/ethnicity and age (28). Models included the same covariables as above with the addition of race. Corrected standard errors were obtained using robust sandwich estimators (29,30). The association of Lp(a) with CVD in subgroups of white and black women were also examined in the WHI. Estimates of absolute risk were obtained (31), and were used to compare predicted values from models with and without Lp(a). We examined whether Lp(a) could add clinical utility to risk prediction models for CVD using the same measures of model improvement described above, with stratum-specific weighting.
JUPITER
We additionally examined the association of Lp(a) with CVD risk among white women and men separately in JUPITER using similar approaches. Covariables from the final fitted models were included in Cox regression models. Analyses were repeated among the 3,855 women and men with Lp(a) measures randomly assigned to placebo.
In all three samples, we examined the crude associations of Lp(a) with CVD risk factors grouping the Lp(a) variables into categories with cut points at 10, 25, 50 and 75 mg/dL as well as with continuous terms for Lp(a). Due to the effects of hormone therapy on lipids, we also examined associations among users and non-users separately among women in the WHS and WHI. Since hormone therapy was an exclusion in JUPITER, all women in that cohort were non-users. Additional sensitivity analyses were run including triglycerides in the model, as well as a total cholesterol corrected for Lp(a)-cholesterol (by subtracting 0.3 or 0.45 times Lp[a] since total cholesterol included Lp(a) cholesterol). All analyses were conducted using SAS 9.3, except for smoothing spline plots which were estimated in R using the pspline function.
RESULTS
WHS
Among the 24,558 women in the full WHS sample, the average age was 54 years, 11% were current smokers, 2% had diabetes at baseline, and 44% of women used hormone therapy at baseline. Mean values of total and HDL cholesterol were 211 and 54 mg/dL, respectively. The distribution of Lp(a) was highly skewed, with a median of 10.5 mg/dL (25th, 75th percentiles = 4.4, 32.3). Baseline characteristics by level of Lp(a) are presented in Table 1. Age, percent black, and systolic blood pressure increased with Lp(a) level, as did total and LDL cholesterol, ApoB, and hsCRP. Use of hormone therapy was highest in those at the lowest Lp(a) level. HDL cholesterol and apolipoprotein A1 were not associated with Lp(a).
Table 1.
Baseline characteristics by Lp(a) in the Women’s Health Study.
| Lp(a) Category (mg/dL) | ||||||
|---|---|---|---|---|---|---|
| <10 | 10-<25 | 25-<50 | 50-<75 | 75+ | P for trend |
|
| N | 11,891 | 5,485 | 3,007 | 2,499 | 1,676 | |
| Age (yrs) | 53.9 | 54.3 | 54.4 | 54.3 | 54.8 | <0.0001 |
| Black (%) | 0.5 | 1.9 | 4.3 | 3.1 | 5.2 | <0.0001 |
| Current smoking (%) | 11.5 | 11.2 | 11.7 | 11.1 | 12.6 | 0.49 |
| SBP (mmHg) | 123.4 | 123.5 | 124.0 | 123.5 | 124.4 | 0.010 |
| DBP (mmHg) | 76.7 | 76.7 | 77.1 | 76.6 | 77.2 | 0.11 |
| Anti-hypertensive use (%) | 13.2 | 12.7 | 13.4 | 12.4 | 14.9 | 0.44 |
| Diabetes (%) | 2.5 | 2.2 | 2.2 | 2.1 | 3.2 | 0.81 |
| HbA1c (%) among diabetics | 7.41 | 7.46 | 7.50 | 7.96 | 7.58 | 0.95 |
| Current hormone therapy (%) | 47.0 | 40.0 | 41.2 | 42.2 | 41.7 | <0.0001 |
| Family history of MI (%) | 12.3 | 12.5 | 13.2 | 13.3 | 16.2 | <0.0001 |
| Total cholesterol (mg/dL) | 206.9 | 211.9 | 213.4 | 218.8 | 227.9 | <0.0001 |
| LDL cholesterol (mg/dL) | 118.8 | 125.7 | 126.7 | 130.4 | 138.8 | <0.0001 |
| HDL cholesterol (mg/dL) | 54.0 | 53.4 | 53.6 | 54.3 | 54.6 | 0.26 |
| ApoB (mg/dL) | 100.1 | 103.9 | 105.4 | 108.1 | 115.8 | <0.0001 |
| ApoA1 (mg/dL) | 151.9 | 149.6 | 149.9 | 151.0 | 152.7 | 0.28 |
| hsCRP (mg/L)* | 1.85 | 1.76 | 1.81 | 1.95 | 2.01 | 0.0068 |
| Randomized to aspirin (%) | 49.6 | 50.7 | 50.2 | 49.8 | 50.1 | 0.64 |
| Randomized to vitamin E (%) |
49.4 | 50.6 | 49.8 | 50.8 | 50.6 | 0.18 |
Geometric mean
In the development set, using the RRS covariables there was a direct linear relationship of Lp(a) with CVD (p<0.0001) (Online Table 1). While a log transformation normalized the distribution of Lp(a), the association with CVD became weaker, as evidenced by the smaller chi-squared value and higher Bayes Information Criterion (Online Figure 1). The association displayed some nonlinearity at lower levels, and a stronger linear association with CVD occurred among those with levels of Lp(a) greater than 10. This varied by level of total cholesterol, however, and was found to be strongest (lowest Bayes Information Criterion) among those with levels of total cholesterol greater than 220 mg/dL. There was a significant interaction between Lp(a) and total cholesterol (p=0.016), with an apparent association only among those with total cholesterol > 220 mg/dL (Online Table 2). The hazard ratio increased across categories of Lp(a) only among those with TC > 220 mg/dL (Online Table 3). These relationships held using either the RRS or ACC/AHA covariables. When substituting apolipoproteins A1 and B for the cholesterol measures, the previously determined interaction with ApoB also showed a strong association.
The above associations were replicated in the WHS test set data. Again, the association of Lp(a) was strongest among those with levels above 10 mg/dL and with TC above 220 mg/dL (Online Table 1). The slope for Lp(a) was null among those with low levels of TC (Online Table 2), with little trend over categories (Online Table 3). Among those with high TC, the estimated hazard ratios were similar, though slightly attenuated, compared to estimates in the derivation sample.
Final estimates in the total WHS sample are presented in Table 2 and the Central Illustration (A). Among those with high TC, the hazard of CVD increased by 8% per 10 mg/dL of Lp(a) using the RRS covariables (95% confidence interval [CI] = 6–10%, p<0.0001). Compared to those with Lp(a)<10 mg/dL, those with levels of 75 mg/dL or greater had more than twice the risk of CVD. Figure 1A and 1B show spline curves of the hazard ratio among those with TC above and below 220 mg/dL. Risk appeared to increase at levels of 50 mg/dL and higher among women with high lipids. Similar curves in those with high and low levels of ApoB are shown in Online Figures 2A-B. While the levels of Lp(a) were slightly higher among those with high TC or ApoB, there was substantial overlap, as shown in Online Figures 3 A-D on both the original and natural log scales. Similar results were found in subgroups defined by randomized assignment to the aspirin and vitamin E interventions and among whites only (Online Table 4), although the estimates were somewhat attenuated among those randomized to active aspirin (p for interaction = 0.096). Similar trends were noted among women who were or were not using hormone therapy at baseline (Online Table 4), and when including triglycerides or corrected total cholesterol in the equations (Online Table 5).
Table 2.
Association of Lp(a) with CVD using continuous terms (per 10 mg/dL) or categories, adjusting for RRS or ACC/AHA covariables in the full WHS sample.*
| Variable | Total | Cases | Beta | P | HR | CI |
|---|---|---|---|---|---|---|
| RRS Vars + | ||||||
| Lp(a) (if > 10 and TC<220) | 15174 | 344 | 0.0048 | 0.81 | 1.005 | 0.965–1.046 |
| Lp(a) (if > 10 and TC≥220) | 9384 | 422 | 0.0807 | <0.0001 | 1.084 | 1.060–1.108 |
| Lp(a) Categories for TC≥220 | ||||||
| Ref | 19188 | 497 | 0 | - | 1.00 | - |
| 10-<25 | 2079 | 71 | −0.102 | 0.45 | 0.903 | 0.694–1.176 |
| 25-<50 | 1197 | 48 | 0.091 | 0.57 | 1.095 | 0.803–1.494 |
| 50-<75 | 1145 | 71 | 0.636 | <0.0001 | 1.889 | 1.448–2.465 |
| 75+ | 949 | 79 | 0.813 | <0.0001 | 2.254 | 1.746–2.910 |
| ACC/AHA Vars + | ||||||
| Lp(a) (if > 10 and TC<220) | 15174 | 344 | 0.0061 | 0.77 | 1.006 | 0.967–1.047 |
| Lp(a) (if > 10 and TC≥220) | 9384 | 422 | 0.0829 | <0.0001 | 1.086 | 1.063–1.111 |
| Lp(a) Categories for TC≥220 | ||||||
| Ref | 19188 | 497 | 0 | - | 1.00 | - |
| 10-<25 | 2079 | 71 | −0.134 | 0.32 | 0.874 | 0.671–1.140 |
| 25-<50 | 1197 | 48 | 0.092 | 0.56 | 1.097 | 0.804–1.496 |
| 50-<75 | 1145 | 71 | 0.638 | <0.0001 | 1.892 | 1.451–2.467 |
| 75+ | 949 | 79 | 0.818 | <0.0001 | 2.266 | 1.756–2.925 |
| RRS Model A Variables + | ||||||
| Lp(a) (if > 10 and ApoB<100) | 12320 | 221 | 0.0061 | 0.83 | 1.006 | 0.952–1.064 |
| Lp(a) (if >10 and ApoB≥100) | 12238 | 545 | 0.0624 | <0.0001 | 1.064 | 1.042–1.087 |
| Lp(a) Categories for ApoB≥100 | ||||||
| Ref† | 17611 | 428 | 0 | - | 1.00 | - |
| 10-<25 | 2744 | 99 | −0.109 | 0.35 | 0.897 | 0.715–1.126 |
| 25-<50 | 1608 | 63 | 0.030 | 0.83 | 0.971 | 0.739–1.275 |
| 50-<75 | 1425 | 92 | 0.551 | <0.0001 | 1.735 | 1.373–2.192 |
| 75+ | 1170 | 84 | 0.566 | <0.0001 | 1.762 | 1.379–2.251 |
HR=hazard ratio; CI=confidence interval; Ref = those with TC<220 or Lp(a)<10.
Ref = those with ApoB<100 or Lp(a)<10.
Central Illustration.
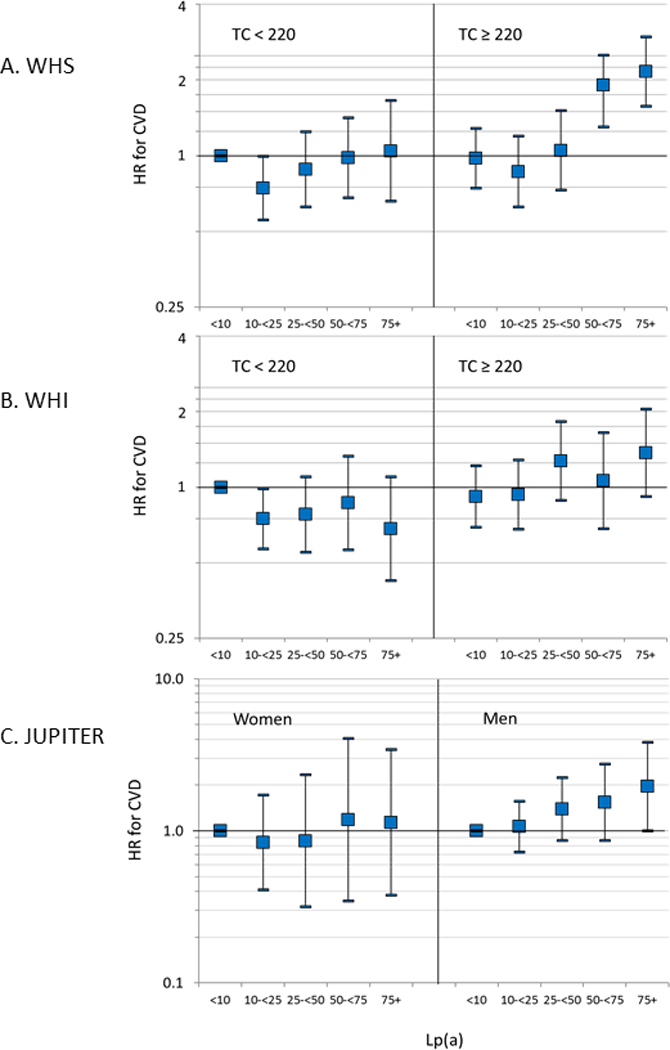
Association of lipoprotein(a) (Lp(a)) in categories with incident cardiovascular disease (CVD) A) by total cholesterol level (TC) in the Women’s Health Study (WHS), B) by TC in the Women’s Health Initiative (WHI), and C) among women and men with TC < 220 mg/dL in the Justification for Use of Statins in Prevention (JUPITER) trial. HR= hazard ratio. Risk appears to increase at levels of 50 mg/dL and higher only among women with high lipids in all three studies.
Figure 1.
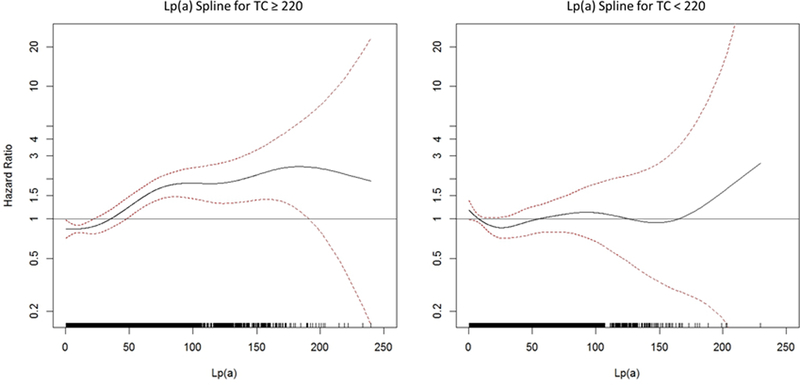
Spline plot for lipoprotein(a) (Lp(a)) and cardiovascular disease (CVD) in the Women’s Health Study (WHS) among those with A) total cholesterol (TC) ≥ 220 mg/dL and B) TC < 220 mg/dL. Risk appears to increase at levels of 50 mg/dL and higher only among women with high TC.
We also considered measures of model improvement when adding the Lp(a) terms to models with the RRS covariables both in the test sample only (Table 3) and in the full WHS data (Online Table 6). Results in the WHS test set only showed little improvement. While there was a small but significant improvement of 0.007 in the c-statistic when Lp(a) was added to the Reynolds or ACC/AHA variables, there was no improvement in reclassification. Values for the categorical NRI were negative, and were non-significant for the continuous NRI and IDI (Table 3). Similar results were obtained when only those with high lipid values were included (Online Table 7), though the continuous NRI was larger and significant, reflecting the stronger association in these women.
Table 3.
Model Improvement statistics in the WHS test sample (N=8,158).
| RRS | AHA/ACC Variables |
RRS Model A | |
|---|---|---|---|
| Lp(a) (if >10 and TC>=220) |
Lp(a) (if >10 and TC>= 220) |
Lp(a) (if >10 and ApoB >= 100) |
|
| RC | |||
| X12 | 8.76 | 10.89 | 8.11 |
| (p) | (0.56) | (0.37) | (0.42) |
| X22 | 5.49 | 8.35 | 10.30 |
| (p) | (0.86) | (0.60) | (0.24) |
| NRI cat* | −0.0023 | −0.0045 | −0.0309 |
| CI | −0.0589 to 0.0565 | −0.0564 to 0.0452 | −0.0789 to 0.2157 |
| (p) | (0.94) | (0.86) | (0.21) |
| NRI>0 | 0.0652 | 0.0989 | 0.0625 |
| CI | −0.0425 to 0.1954 | −0.0070 to 0.2378 | −0.0399 to 0.2017 |
| (p) | (0.31) | (0.13) | (0.32) |
| IDI | 0.0030 | 0.0025 | 0.0011 |
| CI | −0.0018 to 0.0094 | −0.0019 to 0.0095 | −0.0027 to 0.0065 |
| (p) | (0.30) | (0.33) | (0.61) |
| C1† | 0.7895 | 0.7863 | 0.7892 |
| C2 | 0.7967 | 0.7937 | 0.7913 |
| Diff C | 0.0071 | 0.0074 | 0.0021 |
| CI | 0.0005 to 0.0158 | 0.0012–0.0168 | −0.0023 to 0.0087 |
| (p) | (0.035) | (0.039) | (0.42) |
Categories based on 5, 10, and 20% 10-year risk (extrapolated from 8 years).
Based on survival to 8 years.
WHI
Baseline characteristics for the WHI case-cohort sample, reweighted to reflect the full cohort, are shown in Online Table 8. In the subcohort sample, the average age was 63 at baseline, 7% of the women were black, 6% current smokers, 4% diabetics, and 48% used hormone therapy. There was no association of Lp(a) with age in this sample, but significant trends with race, diastolic blood pressure, statin use, hormone therapy, total cholesterol and ApoB.
In the WHI sample, a similar interaction of Lp(a) with TC was found (p=0.005) (Table 4 and the Central Illustration [B]). There was no association of Lp(a) with CVD among those with low levels of TC or ApoB (Online Table 9), but a direct linear relationship across levels of Lp(a) above 10 mg/dL in those with high levels of TC or ApoB, with an increase of 4.3% per 10 mg/dL of Lp(a) using the RRS covariables. Compared to those with levels <10 mg/dL, those with levels of 75 mg/dL or higher had an increased risk of CVD. The same was true using the other covariable sets. There were no significant interactions by race or hormone therapy in these data (data not shown). When measures of model fit were examined, there was a significant continuous NRI, but little improvement in other measures in the WHI sample (Table 5). When restricted to those with high lipids (Online Table 10), the IDI, but not the categorical NRI or c-statistic, also suggested improvement.
Table 4.
Association of Lp(a) with CVD using continuous terms (per 10 mg/dL) or categories, adjusting for RRS or ACC/AHA covariables in the WHI sample.*
| Variable | Subcohort | Cases | Beta | P | HR | CI |
|---|---|---|---|---|---|---|
| RRS Vars + | ||||||
| Lp(a) (if > 10 and TC<220) | 881 | 820 | −0.0313 | 0.15 | 0.969 | 0.928–1.012 |
| Lp(a) (if > 10 and TC≥220) | 1108 | 995 | 0.0407 | 0.007 | 1.042 | 1.011–1.073 |
| Lp(a) Categories for TC≥220 | ||||||
| Ref | 1322 | 1191 | 0 | - | 1.00 | - |
| 10-<25 | 317 | 257 | 0.04166 | 0.73 | 1.043 | 0.825–1.318 |
| 25-<50 | 147 | 151 | 0.36133 | 0.016 | 1.435 | 1.070–1.925 |
| 50-<75 | 101 | 102 | 0.17286 | 0.376 | 1.189 | 0.811–1.743 |
| 75+ | 102 | 114 | 0.42690 | 0.014 | 1.532 | 1.091–2.152 |
| ACC/AHA Vars + | ||||||
| Lp(a) (if > 10 and TC<220) | 881 | 820 | −0.0300 | 0.17 | 0.970 | 0.930–1.013 |
| Lp(a) (if > 10 and TC≥220) | 1108 | 995 | 0.0419 | 0.0051 | 1.043 | 1.013–1.074 |
| Lp(a) Categories for TC≥220 | ||||||
| Ref | 1322 | 1191 | 0 | - | 1.00 | - |
| 10-<25 | 317 | 257 | 0.01801 | 0.88 | 1.018 | 0.809–1.282 |
| 25-<50 | 147 | 151 | 0.31708 | 0.034 | 1.373 | 1.025–1.840 |
| 50-<75 | 101 | 102 | 0.19965 | 0.26 | 1.221 | 0.860–1.734 |
| 75+ | 102 | 114 | 0.38929 | 0.022 | 1.476 | 1.057–2.061 |
| RRS Model A Variables + | ||||||
| Lp(a) (if >10 and ApoB<100) | 1073 | 929 | −0.0200 | 0.31 | 0.980 | 0.943–1.019 |
| Lp(a) (if >10 and ApoB≥100) | 916 | 885 | 0.0381 | 0.015 | 1.039 | 1.007–1.071 |
| Lp(a) Categories for ApoB≥100 | ||||||
| Ref† | 1429 | 1266 | 0 | - | 1.00 | - |
| 10-<25 | 259 | 210 | −0.06795 | 0.60 | 0.934 | 0.727–1.201 |
| 25-<50 | 123 | 144 | 0.24528 | 0.13 | 1.278 | 0.933–1.750 |
| 50-<75 | 87 | 90 | 0.10699 | 0.60 | 1.113 | 0.749–1.653 |
| 75+ | 91 | 104 | 0.36637 | 0.041 | 1.442 | 1.014–2.051 |
HR=hazard ratio; CI=confidence interval; Ref = those with TC<220 or Lp(a)<10.
Ref = those with ApoB<100 or Lp(a)<10.
Table 5.
Model Improvement statistics in the WHI sample (N=1,989).
| Lp(a) (if >10 and TC>=220) | Lp(a) (if >10 and ApoB >= 100) |
||
|---|---|---|---|
| RRS Vars | AHA/ACC Vars | Model A Vars | |
| NRI cat* | 0.0050 | 0.0141 | 0.0069 |
| CI | −0.0126 to 0.0234 | −0.0015 to 0.0268 | −0.0079 to 0.0218 |
| (p) | (0.55) | (0.052) | (0.35) |
| NRI>0 | 0.1006 | 0.1345 | 0.0523 |
| CI | 0.0160 to 0.1767 | 0.0668 to 0.2188 | −0.0143 to 0.1336 |
| (p) | (0.0089) | (0.0005) | (0.19) |
| IDI | 0.00074 | 0.00147 | 0.00059 |
| CI | −0.00057 to 0.00227 | 0.00049 to 0.00290 | −0.00051 to 0.00193 |
| (p) | (0.27) | (0.012) | (0.31) |
| C1‡ | 0.77420 | 0.77434 | 0.76544 |
| C2 | 0.77529 | 0.77548 | 0.76667 |
| Diff C | 0.00109 | 0.00114 | 0.00123 |
| CI | −0.00340 to 0.00231 | −0.00037 to 0.00240 | −0.00012 to 0.00269 |
| (p) | (0.12) | (0.12) | (0.090) |
Categories based on 5, 10, and 20% 10-year risk (extrapolated from 8 years).
At overall incidence rate of 0.0423.
Based on survival to 8 years.
JUPITER
Due to eligibility requirements on LDLC, most participants in JUPITER (94%) had a total cholesterol less than 220 mg/dL, though the proportion with higher levels differed between men (3%) and women (10%). None of the participants were using hormone therapy or were diabetic at baseline. Baseline characteristics of the Lp(a) cohort have previously been published.(9) Briefly, the average age was 66 years, 67% were male, and 14% were current smokers at baseline. Median baseline Lp(a) was 10.7 mg/dL (25th, 75th percentiles = (4.7, 23.3)), with slightly higher levels in women than men (median 12.1 vs. 10.2, p<0.0001).
Among men in JUPITER, there was a linear association of Lp(a) with CVD, as reported previously (Online Table 9).(9) Among women, the association was attenuated, particularly among those with total cholesterol < 220 mg/dL, though this gender difference was not statistically significant due to small numbers. While there was a sizeable trend in risk by Lp(a) category in men even with low TC levels, this was not seen in women (Central Illustration [C]). As seen in Online Table 9, the hazard ratios in the highest categories of Lp(a) were attenuated when those with high cholesterol were excluded, from 1.55 and 1.26 for Lp(a) 50-<75 and 74+, respectively, to 1.19 and 1.14. As in the WHS and WHI, CVD risk in the women appeared to be generally lower in the 10–25 mg/dL category of Lp(a) rather than in those with Lp(a) <10 mg/dL, though this was not statistically significant. Results were more variable among women assigned to placebo in JUPITER (Online Figure 4).
DISCUSSION
While Lp(a) is generally associated with incident CVD, with risk particularly increasing at levels greater than about 50 mg/dL(1), our data from three cohorts suggest that the relation may be somewhat more complex in women. During the development of the Reynolds risk score in women (16), we found a significant interaction of baseline Lp(a) and total cholesterol, which was replicated in women from the WHI and appeared similar in JUPITER. While an increase in risk with Lp(a) is apparent among women with higher lipid levels, here total cholesterol of 220 mg/dL or more, this is not seen among those with lower levels. This anomaly does not appear to be explained by hormone therapy use, and is not present in men in JUPITER. Previous analyses in JUPITER modeled the natural log of Lp(a), which was found to be less predictive in the WHS (Online Table 1 and Online Figure 1). These did not find a gender difference, but did not limit the sample to those with lower cholesterol (9). Among women with higher total cholesterol in the WHS and WHI, the nadir of risk is close to Lp(a) levels of 10–25 mg/dL, with increasing risk at levels of about 50 mg/dL and higher. The Emerging Risk Factors Collaboration found a similar increase starting near 50 mg/dL (1). While they found no difference between men and women, there was a suggestion of a lower effect in those with higher levels of HDL, which would be consistent with these results for women.
We believe our data have implications for the use of Lp(a) in risk prediction, suggesting a limited role for Lp(a), at least in lower risk primary prevention women. While there was some indication of improvement in the full data, the results in the derivation subset are subject to optimism. The changes in c-statistic observed here were very small (0.790 to 0.797 in the WHS) and we found no improvement in measures of reclassification in our validation samples from the WHS and WHI. While the continuous NRI suggested improvement in the WHI sample, this measure has been shown to reflect association with the outcome rather than model improvement (32). We recognize that others have found an improvement in risk stratification with Lp(a) among those in the intermediate risk group (33), but this estimate must be corrected for bias since there may be an apparent improvement even when the association is null (34,35). When applied to those published results, bias-correction reduced the intermediate NRI to −15.9% for the ACC/AHA model and to −4.91% for the SCORE model, indicating worse fit when Lp(a) was added, possibly due to model over-fitting. As such, at least based on currently available data, we do not believe a strong argument can be made to add Lp(a) to standard risk prediction algorithms for women.
We believe our data may also have implications for the design and conduct of trials evaluating Lp(a) reduction as a potential new method to reduce cardiovascular event rates. While it is clearly optimal to include both genders in any planned Lp(a) lowering trials, consideration of power based on gender specific enrollment may need to be taken into consideration if parallel trial designs are based upon “residual Lp(a) risk”. Relatively few individuals with elevated Lp(a) will not already be treated with statin therapy, so our data indicating little augmented risk among women without concomitant hyperlipidemia may be relevant in discussions of trial inclusion and exclusion criteria, at least in primary care populations. Our data indicating that HT did not modify the increase in CVD risk above Lp(a) levels of about 50 mg/dL and higher suggest that clinical trials do not need to exclude women taking HT.
Our analysis is limited by the fact that the WHS and WHI assays did not use the whole particle molar Lp(a) measures, which is the international standard. Assays from JUPITER, though, were expressed as nmol/L, and converted by a constant. These showed similar results, suggesting that assay type does not explain these effects. However, very few events occurred in women with low TC and high Lp(a) levels in JUPITER. Furthermore, while we examined whether Lp(a) improves risk prediction in generally lower risk primary prevention women, we cannot exclude a role for Lp(a) as a potentially useful clinical measure in women with established CVD or those with a strong family history of premature CVD.
CONCLUSIONS
In the WHS and WHI cohorts of women, we found that Lp(a) was associated with CVD only among those with high levels of total cholesterol. However, we also found no relationship for women in JUPITER, data that contrast to those in men. Further, improvement in risk prediction was minimal in in independent validation samples, suggesting that routine screening for Lp(a) may have limited utility in primary prevention women. These data also have potential implications for enrollment criteria of clinical trials of Lp(a)-lowering agents.
Supplementary Material
Acknowledgments
Funding: The WHS was supported by grants HL043851 from the National Heart, Lung, and Blood Institute (NHLBI) and CA047988 from the National Cancer Institute (Bethesda, Md). The WHS blood analysis was supported by the Donald W. Reynolds Foundation (Las Vegas, Nev) and the Leducq Foundation (Paris, France). The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C. The WHI blood analysis was supported by BAA award HHSN268200960011C and grant HL113080 from the NHLBI. The JUPITER trial was supported by AstraZeneca, which had no role in the design or conduct of the present study. Quest Diagnostics Nichols Institute in San Juan Capistrano, CA, performed the Lp(a) measurements in JUPITER at no additional cost to the study. The study was also funded by grants HL117861, HL136852, and K134811 from the NHLBI.
ABBREVIATIONS
- ACC/AHA
American College of Cardiology/ American Heart Association
- ApoB
apolipoprotein B
- CI
confidence interval
- CVD
cardiovascular disease
- IDI
integrated discrimination improvement
- JUPITER
Justification for the Use of Statins in Prevention
- LDLC
low-density lipoprotein cholesterol
- Lp(a)
lipoprotein (a)
- NRI
net reclassification improvement
- RRS
Reynolds Risk Score
- WHI
Women’s Health Initiative
- WHS
Women’s Health Study
Footnotes
COMPETENCY IN MEDICAL KNOWLEDGE
In women, there was an association of Lp(a) only among those with high levels of total cholesterol. This suggests that the role of screening for elevated Lp(a) may be limited in women.
TRANSLATIONAL OUTLOOK IMPLICATIONS
The utility of strategies to lower circulating Lp(a) levels, including novel anti-sense oligonucelotides, in reducing incidence of cardiovascular disease may be more limited in women. Further investigation is needed to assess potential gender differences.
Disclosures: Dr Ridker reports having received research funding support from multiple not-for-profit entities including the National Heart, Lung, and Blood Institute, the National Cancer Institute, the American Heart Association, the Doris Duke Charitable Foundation, the Leducq Foundation, and the Donald W Reynolds Foundation. Dr Ridker also reports having received investigator-initiated research support from Astra-Zeneca, Novartis, Pfizer, and Kowa, as well as non-financial research support from Amgen. Dr Ridker is listed as a co-inventor on patents held by the Brigham and Women’s Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease that have been licensed to Siemens and AstraZeneca, and has served during the past year as a research consultant to Sanofi, Quintiles, AstraZeneca, and TEVA. Dr Mora has received institutional research grant support from Atherotech Diagnostics and the National Institute of Diabetes and Digestive and Kidney Diseases (DK112940), and served as consultant to Amgen, Pfizer, and Quest Diagnostics.
Dr. Cook reports no disclosures.
REFERENCES
- 1.Emerging Risk Factors C, Erqou S, Kaptoge S et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. Jama 2009;302:412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willeit P, Kiechl S, Kronenberg F et al. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): prospective 15-year outcomes in the Bruneck Study. Journal of the American College of Cardiology 2014;64:851–60. [DOI] [PubMed] [Google Scholar]
- 3.Clarke R, Peden JF, Hopewell JC et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. The New England journal of medicine 2009;361:2518–28. [DOI] [PubMed] [Google Scholar]
- 4.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. Jama 2009;301:2331–9. [DOI] [PubMed] [Google Scholar]
- 5.Kyriakou T, Seedorf U, Goel A et al. A common LPA null allele associates with lower lipoprotein(a) levels and coronary artery disease risk. Arteriosclerosis, thrombosis, and vascular biology 2014;34:2095–9. [DOI] [PubMed] [Google Scholar]
- 6.Tsimikas S A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. Journal of the American College of Cardiology 2017;69:692–711. [DOI] [PubMed] [Google Scholar]
- 7.Yeang C, Hung MY, Byun YS et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). Journal of clinical lipidology 2016;10:594–603. [DOI] [PubMed] [Google Scholar]
- 8.Nestel PJ, Barnes EH, Tonkin AM et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arteriosclerosis, thrombosis, and vascular biology 2013;33:2902–8. [DOI] [PubMed] [Google Scholar]
- 9.Khera AV, Everett BM, Caulfield MP et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation 2014;129:635–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsimikas S, Viney NJ, Hughes SG et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015;386:1472–83. [DOI] [PubMed] [Google Scholar]
- 11.Viney NJ, van Capelleveen JC, Geary RS et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016;388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 12.Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. Jama 2006;296:1363–70. [DOI] [PubMed] [Google Scholar]
- 13.Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. Journal of the American College of Cardiology 2008;52:124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rexrode KM, Lee IM, Cook NR, Hennekens CH, Buring JE. Baseline characteristics of participants in the Women’s Health Study. J Womens Health Gend Based Med 2000;9:19–27. [DOI] [PubMed] [Google Scholar]
- 15.Ridker PM, Cook NR, Lee IM et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. The New England journal of medicine 2005;352:1293–304. [DOI] [PubMed] [Google Scholar]
- 16.Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women. Jama 2007;297:611–619. [DOI] [PubMed] [Google Scholar]
- 17.Cook NR, Paynter NP, Eaton CB et al. Comparison of the Framingham and Reynolds Risk scores for global cardiovascular risk prediction in the multiethnic Women’s Health Initiative. Circulation 2012;125:1748–56, S1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ridker PM, Danielson E, Fonseca FA et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. The New England journal of medicine 2008;359:2195–207. [DOI] [PubMed] [Google Scholar]
- 19.Goff DC Jr., Lloyd-Jones DM, Bennett G et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Journal of the American College of Cardiology 2014;63:2935–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chasman DI, Shiffman D, Zee RY et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis 2009;203:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pencina MJ, D’Agostino RBS, D’Agostino RBJ, Vasan RS. Evaluating the added predictive ability of a new biomarker: from area under the ROC curve to reclassification and beyond. Stat Med 2008;27:157–72. [DOI] [PubMed] [Google Scholar]
- 22.Cook NR. Use and misuse of the receiver operating characteristic curve in risk prediction. Circ 2007;115:928–35. [DOI] [PubMed] [Google Scholar]
- 23.Cook NR, Ridker PM. Advances in measuring the effect of individual predictors of cardiovascular risk: the role of reclassification measures. Ann Intern Med 2009;150:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chambless LE, Cummiskey CP, Cui G. Several methods to assess improvement in risk prediction models: Extension to survival analysis. Stat Med 2010;30:22–38. [DOI] [PubMed] [Google Scholar]
- 25.Pencina MJ, D’Agostino RB Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med 2011;30:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horvitz DG, Thompson DJ. A generalization of sampling without replacement from a finite universe. J Amer Statist Assoc 1952;47:663–85. [Google Scholar]
- 27.Barlow WE. Robust variance estimation for the case-cohort design. Biom 1994;50:1064–72. [PubMed] [Google Scholar]
- 28.Borgan Ø, Langholz B, Samuelson SO, Goldstein L, Pogoda J. Exposure stratified case-cohort designs. Lifetime Data Analy 2000;6:39–58. [DOI] [PubMed] [Google Scholar]
- 29.Therneau TM, Li H. Computing the Cox model for case cohort designs. Lifetime Data Analy 1999;5:99–112. [DOI] [PubMed] [Google Scholar]
- 30.Barlow WE, Ichikawa L, Rosner D, Izuma S. Analysis of case-cohort designs. J Clin Epidemiol 1999;52:1165–72. [DOI] [PubMed] [Google Scholar]
- 31.Langholz B, Jiao J. Computational methods for case-cohort studies. Comp Stats Data Analysis 2007;51:3737–48. [Google Scholar]
- 32.Pencina MJ, D’Agostino RB, Pencina KM, Janssens AC, Greenland P. Interpreting incremental value of markers added to risk prediction models. American journal of epidemiology 2012;176:473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verbeek R, Sandhu MS, Hovingh GK et al. Lipoprotein(a) Improves Cardiovascular Risk Prediction Based on Established Risk Algorithms. Journal of the American College of Cardiology 2017;69:1513–1515. [DOI] [PubMed] [Google Scholar]
- 34.Paynter NP, Cook NR. A bias-corrected net reclassification improvement for clinical subgroups. Medical decision making : an international journal of the Society for Medical Decision Making 2013;33:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paynter NP, Cook NR. Adding tests to risk based guidelines: evaluating improvements in prediction for an intermediate risk group. Bmj 2016;354:i4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.