

Abstract
Background
Previous evidence demonstrated that restoration of wild type VHL in human renal cancer cells decreased in vitro NK susceptibility. To investigate on the role of tumoral VHL status versus NK capability in renal cancer patients, 51 RCC patients were characterized for VHL mutational status and NK function.
Methods
VHL mutational status was determined by direct DNA sequencing on tumor tissue. NK cytotoxicity was measured against specific target cells K562, VHL-wild type (CAKI-1) and VHL-mutated (A498) human renal cancer cells through externalization of CD107a and IFN-γ production. Activating NK receptors, NKp30, NKp44, NKp46, NKG2D, DNAM-1, NCAM-1 and FcγRIIIa were evaluated through quantitative RT-PCR. RCC tumoral Tregs were characterized as CD4+CD25+CD127lowFoxp3+ and Treg function was evaluated as inhibition of T-effector proliferation.
Results
VHL mutations were detected in 26/55 (47%) RCC patients. IL-2 activated whole-blood samples (28 VHL-WT-RCC and 23 VHL-MUT-RCC) were evaluated for NK cytotoxicity toward human renal cancer cells A498, VHL-MUT and CAKI-1, VHL-WT. Efficient NK degranulation and increase in IFN-γ production was detected when IL-2 activated whole-blood from VHL-MUT-RCC patients were tested toward A498 as compared to CAKI-1 cells (CD107a+NK: 7 ± 2% vs 1 ± 0.41%, p = 0.015; IFN-γ+NK: 6.26 ± 3.4% vs 1.78 ± 0.9% respectively). In addition, IL-2 activated NKs induced higher CD107a exposure in the presence of RCC autologous tumor cells or A498 as compared to SN12C (average CD107a+NK: 4.7 and 2.7% vs 0.3% respectively at 10E:1 T ratio).
VHL-MUT-RCC tumors were NKp46+ cells infiltrated and expressed high NKp30 and NKp46 receptors as compared to VHL-WT-RCC tumors. A significant lower number of Tregs was detected in the tumor microenvironment of 13 VHL-MUT-RCC as compared to 13 VHL-WT-RCC tumors (1.84 ± 0.36% vs 3.79 ± 0.74% respectively, p = 0.04). Tregs isolated from VHL-MUT-RCC patients were less suppressive of patients T effector proliferation compared to Tregs from VHL-WT-RCC patients (Teff proliferation: 6.7 ± 3.9% vs 2.8 ± 1.1%).
Conclusions
VHL tumoral mutations improve NKs effectiveness in RCC patients and need to be considered in the evaluation of immune response. Moreover therapeutic strategies designed to target NK cells could be beneficial in VHL-mutated-RCCs alone or in association with immune checkpoints inhibitors.
Electronic supplementary material
The online version of this article (10.1186/s13046-018-0952-7) contains supplementary material, which is available to authorized users.
Keywords: Von Hippel-Lindau, Natural killer, Renal cell carcinoma, CD107a, Tumor microenvironment
Background
Renal cell carcinoma (RCC) is the most common kidney tumor in adults. Annually ~ 295,000 new kidney cancer cases are diagnosed and ~ 134,000 deaths are recorded worldwide [1, 2]. The most common histotype is clear cell-RCC (ccRCC) (70–80%) of which 50–70% carry Von Hippel-Lindau (VHL) gene mutations (VHL promoter hyper methylation and biallelic VHL inactivation) [3–6]. The VHL protein (pVHL) functions as an E3 ubiquitin ligase that targets the hypoxia-inducible factors (HIFs) for ubiquitin-mediated degradation via the proteasome [7–9]. Although VHL plays a key role in RCC pathogenesis, the prognostic meaning of VHL mutations is controversial. VHL mutations were reported not to affect prognosis [10, 11] or to improving it [12]. Natural killer cells (NKs) are effectors of the innate immune response [13] activated by a net between inhibitory and activating receptors: human activating receptors comprise the natural cytotoxicity receptors (NCRs: NKp46, NKp30, and NKp44), C-type lectin receptors (CD94/NKG2C, NKG2D, NKG2E/H, and NKG2F), and killer cell immunoglobulin-like receptors (KIRs) (KIR-2DS and KIR-3DS). Inhibitory receptors are C-type lectin receptors (CLRs) and KIRs (KIR-2DL and KIR-3DL) [14]. Certain VHL mutations correlate with low expression of classical HLA-I molecules and HLA-E expression that determine higher NK susceptibility reducing the engagement of KIR and NKG2A inhibitory receptors, respectively. Nevertheless multiple mechanisms concur in determining the NK sensitivity of cancer cells such as NK receptors surface ligands, LFA-1/ICAM-1 and HLA-I/NKR interactions, HLA-E molecules, soluble MICA A-B ligands, HLA-G, TGF beta secretion [15]. Moreover mechanisms affecting NK sensitivity may regulate gene transcription such as autophagy genes [16].
T regulatory cells (Tregs) partecipate in the control of tumor immunity attenuating antitumor immune response [17]. Increase in peripheral and tumoral Tregs were observed in several cancers and inversely correlated to NKs number and function [18]. Tregs mainly regulate NK activity in a transforming growth factor-beta (TGF-β)-dependent manner. Soluble and membrane-bound TGF-β on regulatory T cells impaired NK cytolytic activity through down-regulation of NKG2D receptor on the NK cells [19].
To investigate on the role of tumoral VHL status versus NK capability in renal cancer patients, 51 RCC patients were characterized for VHL mutational status and NK cytolytic activity toward A498-VHL-MUT and CAKI-1-VHL-WT human renal cancer cells.
Materials and methods
Patients and samples
Fifty five patients with primary RCC (41 clear cell; 4 papillary; 5 cromophobe; 5 unknown) and twelve healthy donors (HD) were enrolled in the study. The patients underwent surgery as part of their standard treatment at the Department of Urology, National Cancer Institute “G. Pascale” (Naples, Italy) and Genitourinary Oncology and Rare Cancer Center, Federico II University (Naples, Italy). RCC tumor and peritumoral tissues were obtained immediately after surgical resection, stabilized in RNA later (Qiagen) and stored at − 80 °C. The distance of 1 cm was the minimal distance between tumor and normal-appearing renal tissue sampled. Informed consent from each patient was sought. The research protocol was approved by Human Ethical Committee of Institute (n. CEI/423/13).
Cell culture
The human renal cancer cell lines CAKI-1 (HLA-A*23:01/A*24:02; HLA-B*35:02/B*44:03; HLA-C*04/C*04new) and SN12C (HLA-A*03/A*24new; HLA-B*07/B*44; HLA-C*05/C*07:02) VHL wild-type (VHL-WT), A498 (HLA-A*02:01; HLA-B*08:01; HLA-C*07) and 786-O (HLA-A*03:01; HLA-B*07/B*44, HLA-C*05/C*07:02) VHL mutated (VHL-MUT) and the human leukemic cell line K562 were obtained from the NCI 60 cancer cell line collection [20, 21]. All cells were cultured in the recommended growth medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% L-glutamine, 1% penicillin/streptomycin and maintained in 95% air 5% CO2 at 37 °C.
VHL somatic mutational status
VHL mutational status was determined by PCR amplification and direct sequencing. DNA was extracted from primary tumor tissues using Nucleon Genomic DNA extraction Kit (GE Healthcare). The three VHL coding exons, with exon-intron junctions, were PCR amplified using the following primers: exon 1, 5′-agcgcgttcc atcctctac-3′ (forward) and 5′-agttccccgtctgcaaaat-3′ (reverse); exon 2, 5′-aggacggtcttgatctcctg-3′ (forward) and 5′-gcccaaagtgcttttgagac-3′ (reverse); exon 3, 5′-ttgttggcaaagcctcttgt-3′ (forward) and 5′-tgcccctaaacatcacaatg-3′ (reverse). The thermal cycler (Applied Biosystems® 2720 Thermal Cycler) was programmed to first incubate the sample for 10 min for 95 °C followed by 35 cycles consisting of 95 °C for 45 s, 56 °C (exon1 and 3) and 60 °C (exon 2) for 45 s with final extension for 7 min at 72 °C. Purified PCR products were then sequenced by using the Big Dye terminators version 3.1 cycle sequencing kit (Applied Biosystems, Courtaboeuf, France) and the 3130 Genetic Analyzer (Applied Biosystems).
Flow cytometry
FITC-conjugated major histocompatibility complex (MHC) class I–specific antibody (IgG2a, W6/32, CBL139F, Cymbus Biotech, Hants, UK) and PE anti-human CD155/PVR (clone SKIL.4, Biolegend, Cat No 337609) were used for flow cytometry. FACSAria II Cell Sorter 8-colour flow cytometer with BD FACSDiva™ 8.0 Software (BD Biosciences, San Jose, CA) was daily calibrated with calibrite beads (Fitc, Pe, PerCP and APC) and compbeads (Pe-Cy7 and APC-Cy7; BD Biosciences). PerCP-Cy5.5 anti-CD3 (BD Biosciences Cat No 560835), FITC-conjugated anti-CD56 (BD Biosciences, Cat No 345811) and PE-conjugate anti-CD107a antibody (BD Bioscience, Cat No 555801) were used to identify degranulating NK-cells from peripheral blood. For intracellular staining PE-conjugated anti-IFN-γ antibody (BD Biosciences Cat No 554701) was used. Fluorochrome-labelled monoclonal antibodies for identification of tissue Treg cells were: FITC-anti-FOXP3 (BD Biosciences, Cat No 560047), Pe-Cy7-anti-CD25 (BD Biosciences, Cat No 557741) and APC-Cy7-anti-CD4 (BD Biosciences, Cat No 557871). Intracellular staining for FoxP3 was performed using a commercially available kit (BD Cytofix/Cytoperm; fixation and permeabilization kit; BD Pharmingen) according to the manufacturer’s instructions. A minimum of 100.000 events for each sample were collected.
NK cells and autologous tumor cells isolation
CD56+NK cells were isolated from PBMCs of 4 RCC patients (1 VHL-WT, 3 VHL-MUT) after Ficoll gradient centrifugation using the NK cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Purified NK cells were cultured in RPMI 1640 medium (GIBCO Invitrogen) in the presence of 100 U/ml recombinant interleukin 2 (IL-2) overnight at 37 °C, 5%CO2. Autologous tumor cells were isolated from tumor tissue with single cell sorting by FACS ARIAII post mechanical disruption and labelings with Antibody AbI RCC pure Mouse Dako (clone SPM314), 7AAD (Thermo Fischer) and PE anti-CD45 (BD Biosciences, cat No 555483) and AbII Goat anti-Mouse FITC IgG(H + L) (Caltag Laboratory) for indirect staining. Through the gate strategy 7AAD−/CD45−/RCC+ ~ 10,000 of tumor cells were isolate and cultured 24 h in 96-well plates with RPMI-1640 with 10% FBS medium.
Tregs dependent-CSFE T effector proliferation assay
Tumor CD4+CD25+ T regulatory cells (Tregs) and peripheral CD4+CD25− T effector (Teff) cells were isolated using the Dynabeads Regulatory CD4+CD25+ T cell kit as previously described [22]. All purification steps were performed according to the manufacturer’s instructions (Invitrogen by Life Technologies) and collected cells were found to be > 95% pure by flow cytometry. Tregs function was assessed by evaluating the CFSE-labeled Teff cells by FACS analysis. Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled autologous CD4+CD25− T cells from peripheral blood (CellTrace CFSE Cell Proliferation Kit, Molecular Probes, by Life Technologies) were cultured with tumor CD4+CD25+ Tregs in 1:1 ratio. Cells were cultured (5 × 103 cells/well) in U-bottom 96-well plates with RPMI medium (Thermo scientific, HyClone Laboratories, Inc) supplemented with 2-mM L-glutamine, 100 U/ml penicillin, 100 μg streptomycin, and 10% fetal bovine serum. Cells were stimulated for 5 days in the presence of Dynabeads Human T-Activator CD3/CD28 (Gibco by Life Technologies).
CD107a degranulation assay and IFN-γ production
Lysosome-associated membrane protein LAMP-1 (CD107a) is a marker of NK-cell degranulation. CD107a expression was measured by flow cytometry as previously described [23, 24]. Briefly, 1 ml whole blood was diluted with one volume RPMI-1640 containing 10% heat-inactivated fetal bovine serum and incubated with IL-2 (100 U/ml) overnight at 37 °C, 5% CO2. The cytotoxic activity of NK cells was tested against NK-sensitive cell line K562 (HLA class I-negative) [25], human renal cancer cell lines CAKI-1-VHL-WT, A498 and 786-OVHL-MUT. 200 μl of IL-2 activated blood was co-cultured with 2 × 105 target cell lines in presence of PE-anti-CD107a antibody for 3-h at 37 °C in 5% CO2. Alternatively, purified IL-2 activated NK cells from peripheral blood of RCC patients were used as effector cells. The autologous RCC tumor cells, A498-VHL-MUT and SN12C-VHL-WT cell lines were used as target cells. Assays were performed at the indicated effector-target ratio (10E:1 T; 5E:1 T). To detect spontaneous degranulation samples were incubated in the absence of target cells. Following 3-h culture, cells were stained with anti-CD56 and NK cells were identified as CD3−CD56+ in the lymphocyte gate. CD107a was analyzed on CD56+CD3− NK. For intracellular staining of IFN-γ, 200 μl of IL-2 preactivated blood samples were incubated with 2 × 105 K562 or RCC target cells for 6 h at 37 °C in a humidified 5% CO2 incubator. The secretion inhibitor Brefeldin A (Sigma-Aldrich, Seelze, Germany) was added to the assay medium at a concentration of 10 μg/ml during the 6 h coincubation. After incubation, samples were placed on ice and stained with anti-CD56 and anti-CD3 mAbs, followed by erythrocyte lysis of blood samples with FACS Lysing Solution (BD Biosciences). Thereafter, cells were permeabilized using FACS permeabilizing solution (Perm2, BD Biosciences) and stained for intracellular IFN-γ with a PE-anti-IFN-γ antibody. Analysis was performed on a FACSAria II Cell Sorter 8-colour flow cytometer, blinded to patients’ clinical characteristics.
Real-time PCR
RNA was isolated from tumoral and peritumoral RCC tissues using the RNeasy Mini kit (Qiagen, Hilden, Germany), according to manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA using SuperScript III Reverse Transciptase (Invitrogen). Quantitative real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) and data were collected and quantitatively analyzed on an ABI Prism 7000 System (Applied Biosystems). Primers sequences for NCAM-1, FCGR3a (CD16), DNAM-1, NKG2D, NKp46, NKp44, NKp30,KLRB1 and TGFB1 are detailed in Additional file 1: Table S1. Relative mRNA expression was normalized with GUSB gene expression.
Immunohistochemistry (IHC)
Paraffin-embedded RCC tissue blocks, after heat induced epitope retrieval with citrate buffer (pH 6), were incubated overnight at 4 °C with the mouse monoclonal IgG2B, anti-human NKp46 antibody, Clone 195,314 (MAB1850 R&D Systems) diluted 1:50, followed by incubation with peroxidase-conjugated secondary antibodies according to the manufacturer’s instructions (EnVision+System, Dako North America, Inc., Carpinteria, CA). The NKp46 receptor demonstrates a high degree of lineage-specificity, being expressed almost exclusively in natural killer cells.. Five randomly selected areas were identified across the entire section from the center to the invasive margin (100× magnification). NKp46+ cells were counted in 5 consecutive non-overlapping high-power fields (HPF) 400x magnification (0.237 mm2/field), using an Olympus BX51 microscope (Olympus, Tokyo, Japan). The results were expressed as the mean of NKp46+/mm2 out of 5 regions of interest. Immunostaining and scoring were evaluated by 4 independent observers (C.D., G.B., E.L. and A.A.) blinded to patients’ clinical characteristics.
Cytokine assay
IFN-γ was measured by ELISA on the culture supernatant collected on day 5 from suppression experiments. Cytokine concentration was assessed by Human IFN-gamma Instant ELISA (Bender MedSystems). Samples were acquired by LB 940 Multimode Reader Mithras (Berthold Technologies).
Statistical analysis
Statistical analysis were performed using the MedCalc 9.3.7.0 and Excel software. Unpaired Student t test or non-parametrical Mann-Whitney test was conducted. Statistical tests of hypotheses was two-sided with p < 0.05 taken as the criterion for statistical significance.
Results
RCC-VHL mutations were detected in 47% of RCC patients
Clinical pathological features of patients are summarized in Table 1. The VHL status was assessed in 55 primary renal cancers by direct sequencing. As shown in Tables 1 and 2, VHL was mutated in 26/55 (47%) cases, with missense mutations in 7/26 (27%), nonsense in 3/26 (12%), splice site in 2/26 (7%), frameshift deletions in 11/26 (42%) and frameshift insertions in 3/26 (12%) cases. The mutations exon prevalence was: exon 1, 50%; exon 2, 30.7%; and exon 3, 19.3%.
Table 1.
Patient characteristics (N = 55)
| Age, n (%) | |
| < 65 years | 26 (47) |
| ≥ 65 years | 29 (53) |
| Mean age ± SD, yr | 63 ± 11 |
| Gender, n | |
| Male | 33 |
| Female | 22 |
| Histological variant, n | |
| Clear Cell | 41 |
| Papillar | 4 |
| Cromophobe | 5 |
| Missing | 5 |
| VHL mutational status, n | |
| WT | 29 |
| MUT | 26 |
| Pathological stage, n | |
| T1 | 28 |
| T2 | 9 |
| T3 | 12 |
| Missing | 6 |
| Furhman grade, n | |
| I | 6 |
| II | 16 |
| III | 16 |
| IV | 2 |
| Missing | 15 |
Table 2.
VHL mutational status in RCC patients
| Exon | Mutation | Mutation type |
|---|---|---|
| 3 | c. 565C > T; p.E189X | Nonsense |
| 3 | c.481 C > T; p.R161X | Nonsense |
| 1 | c.203C > A; p.S68X | Nonsense |
| 2 | c.345_349delCCTTT; p.G114 fs*15 | Frameshift |
| 1 | c.162 insG; p. M54 fs*12 | Frameshift |
| 2 | c.450delT; p.N150 fs*9 | Frameshift |
| 1 | c. 203_209delCGCGCGA; p. S68 fs*89 | Frameshift |
| 1 | c.215_222delCCAGGTC; p. E70fs*57 | Frameshift |
| 3 | c.502_505delAGCC; p. S168 fs*1 | Frameshift |
| 2 | c.391_394delAACC; p.N131 fs*26 | Frameshift |
| 3 | c.638_639insT; p. D213fs*2 | Frameshift |
| 1 | c.199delA; p.N67Tfs*97 | Frameshift |
| 1 | c.306_314delGCCTGGCA p.P102Pfs*27 | Frameshift |
| 2 | c.411_413 insGT; p. V137Cfs*22 | Frameshift |
| 1 | c.339_340delAG; p.G114 fs*17 | Frameshift |
| 2 | c.444delT; p.F148 fs*11 | Frameshift |
| 1 | c.1101delA; p.V87 fs*47 | Frameshift |
| 1 | c.340G > T; p.G114C | Missense |
| 1 | c.234 T > G; p.N78 K | Missense |
| 3 | c.488 T > A; p.L163H | Missense |
| 1 | c.240 T > A; p.S80R | Missense |
| 1 | c.506C > T; p. L169P | Missense |
| 2 | c.602 T > C; p.L201R | Missense |
| 1 | c.194C > T p.S65 L | Missense |
| 2 | c.IVS2 + 2 T > C | Splicing |
| 2 | c.IVS1–1 G > C | Splicing |
VHL-MUT-RCC patients whole blood cytotoxicity was more efficient toward A498-VHL-MUT than CAKI-1-VHL-WT cells
IL-2 activated whole-blood-NK derived cytotoxicity (as CD107a externalization and intracellular IFNγ production) was evaluated in 51 available RCC patients (28 VHL-WT and 23 VHL-MUT) and 12 healthy donors (HD). IL-2 activated whole-blood samples were co-cultured with target cells K562, A498-VHL-MUT (containing a 4-nt deletion at nt 639–642 in VHL exon 2) [26] and CAKI-1-VHL-WT cell lines. As reported in Additional file 2: Figure S1 the target cells K562 are MHC-I negative, while A498 and CAKI-1 cell lines express high levels of MHC-I molecules (81 and 86.2% respectively). In Fig. 1 NKs from HD (A) and RCC patients, either VHL-WT (B) or –MUT (C), displayed comparable externalization of CD107a against K562 cells (average CD107a+NK cells: 15 ± 0.02% vs 15 ± 3% vs 17 ± 2.5% respectively); absence of CD107a+NK cells activity was detected evaluating HD blood samples after incubation with human RCC lines (Fig. 1A). Comparable CD107a+NK cells exposure was observed in VHL-WT-RCC patients after stimulation with human renal cells, A498 and CAKI-1 (average CD107a+NK cells: 3 ± 1.3% vs 3 ± 1% respectively, p = 0.84, Fig. 1B). Interestingly, high percentage of NK cells expressing CD107a was observed for blood derived from VHL-MUT-RCC patients against A498-VHL-MUT cells and not versus CAKI-1-VHL-WT cells (average CD107a+NK: 7 ± 2% vs 1 ± 0.41% respectively, p = 0.015, Fig. 1C). Similar results were obtained when IL-2 activated whole blood samples from VHL-MUT-RCC patients (n = 5) were evaluated toward another VHL-MUT RCC cell line, 786-O cells (MHC-I expressing cells, Additional file 2: Figures S1-S2A) carrying a single mutation in VHL exon 1 (1-nt deletion at nt 523) [26] different from A498 cells. Low expression of CD107a+NK cells was observed incubating RCC-VHL-WT blood samples (n = 9) with 786-O (Additional file 2: Figure S2B). The higher percent of CD107a+NK induced in VHL-MUT patients by the culture with A498 and 786-O VHL-MUT cell lines suggest that RCC-VHL mutated tumors displayed higher susceptibility to NK lysis .
Fig. 1.

VHL-MUT-RCC patients display higher NK cytotoxicity toward A498, VHL-MUT RCC cell line. NK cell function was evaluated in whole blood through CD107a cell-surface expression and production of IFN-γ in response to ex vivo stimulation with K562, A498 and CAKI-1 cells. Degranulation (CD107a; A, B, C) and cytokine production (IFN-γ; D, E, F) were evaluated after gating on CD3−CD56+ cells. Percent of degranulation was obtained subtracting the percent of degranulation occurring in the absence of target cells. CD107a+NK cells in 12 HD (A), 28 VHL-WT (B) and 23 VHL-MUT (C) RCC patients versus K562, CAKI-1 and A498 renal cell lines. Representative profiles of the percentages of CD107a+NK cells in HD (Ai) VHL-WT (Bi) and VHL-MUT (Ci) RCC patients. IFN-γ production was assayed by intracellular cytokine staining in 8 HD (D), 13 VHL-WT (E) and 14 VHL-MUT (F) RCC patients versus K562, CAKI-1 and A498 renal cell lines. Representative profiles of the percentages of IFN-γ+ NK cells in HD (Di) VHL-WT (Ei) and VHL-MUT (Fi) RCC patients. The results are presented mean ± SEM. Statistical significance was calculated by unpaired Student t test (p < 0.05)
Clinical-pathological features and the CD107a assay results from single patient were shown in Additional file 3: Table S2 (VHL-MUT-RCC patients) and Additional file 4: Table S3 (VHL-WT-RCC patients). NK derived-IFN-γ was evaluated in 8 HD (Fig. 1D) versus 13 VHL-WT (E) and 14 VHL-MUT (F) RCC patients in the presence of target cells (K562, A498 and CAKI-1). The highest IFN-γ production was registered with VHL-MUT blood versus A498 cells as compared to CAKI-1 cells (average IFN-γ+NK: 6.26 ± 3.4% vs 1.78 ± 0.9% respectively; p = 0.23 Fig. 1F). In addition autologous NK cell degranulation was evaluated in the presence of MUT-VHL tumors. Patients derived (1 VHL-WT, 3 VHL-MUT)-IL-2 activated NK cells were cultured in the presence of RCC cell lines or freshly isolated autologous tumor cells. As shown in Fig. 2, NKs derived from VHL-MUT patients (#24, #25, #26) displayed higher CD107a exposure versus RCC autologous tumor cells or A498-VHL-MUT cells as compared to SN12C-VHL-WT cells (average CD107a+NK: 4.7 and 2.7% vs 0.3% respectively at 10E:1 T ratio). Conversely, NKs derived from VHL-WT patient (#29) did not significantly increase CD107a exposure either toward RCC autologous tumor cells than toward SN12C or A498 cell lines. Thus we can speculate that NKs from VHL-MUT-RCC patients are more efficient toward human renal cancer VHL-MUT cells.
Fig. 2.
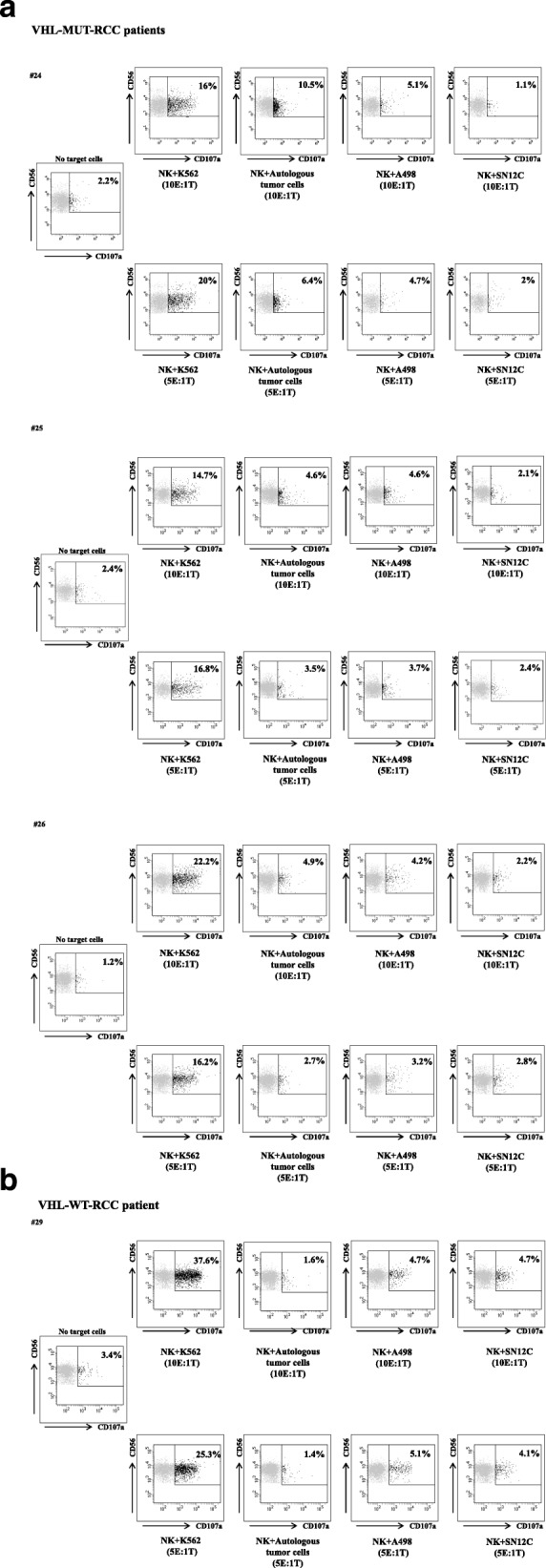
Isolated NKs from VHL-MUT-RCC patients show higher cytolytic capacity against autologous tumor cells. a-b Flow cytometry plots represent the percent NK cells isolated from 4 RCC patients (#24, #25, #26 VHL-MUT and #29 VHL-WT patients) that express CD107a following no stimulation (NK no target cells) and stimulation with K562, autologous tumor cells, A498 VHL-MUT and SN12C VHL-WT cell lines. Isolated NK cells were activated overnight with IL-2 (100 U/mL). Assays were performed at the indicated effector-target ratio (10E:1 T; 5E:1 T)
VHL-MUT-RCC are NKp46+cells infiltrated and express NK activator receptors
With the intent to correlate peripheral and tumoral NK status, NK cell infiltration was analyzed on paraffin-embedded primary RCC tissues by immunohistochemical staining using the NK cell–specific marker NKp46. 23 RCC-patients, 15/23 (65.2%) VHL-MUT and 8/23 (34.8%) VHL-WT were evaluated for the expression of NKp46+ cells. Although RCCs generally display low NK cell infiltration [27], NKp46 was expressed in the cytosol and on cell surface in lymphocytes (Fig. 3A). In VHL-WT-RCC the NKp46+ cells ranged from 0 to 5 per mm2 (median 0.5 cells/mm2), (4/8 VHL-WT-RCC were totally negative for NKp46). In 15 VHL-MUT-RCCs NKp46+ cells ranged from 1 to 12 per mm2 (median 6 cells/mm2); a significant higher number of NKp46+cells was detected in VHL-MUT-RCCs as compared to VHL-WT-RCCs (average NKp46+cells: 5.553 ± 0.78 vs 1.625 ± 0.78, p = 0.0041, Mann–Whitney test, Fig. 3B).
Fig. 3.

Higher NKp46+ cells infiltration and expression of NKp30 and NKp46 in VHL-MUT-RCCs. A Representative tissue staining in VHL-WT (Ai-Aii) and VHL-MUT-RCC patients (Aiii-Aiv) (Magnification 200X detail 400x). B NKp46+ cell densities [NKp46+ cells/mm2] in RCC patients grouped in VHL-WT (n = 8) and VHL-MUT (n = 15). The results are presented as mean ± SEM; p = 0.0041, Mann–Whitney test. C-D mRNA expression of NKp30 and NKp46 in 17 VHL-WT and 17 VHL-MUT RCC patients. Transcript levels are presented as mean ± SEM. Statistical significance was calculated by unpaired Student t test (p < 0.05)
NKs function is regulated by activating receptors other than NKp46 that include NKp30, NKp44, C-type lectin receptors NKG2D, CD94/NKG2C, FcγRIIIa (CD16), activator KIRs, DNAX accessory molecule-1 (DNAM-1, CD226), and costimulatory receptors such as NCAM-1, and the main NK cell-activating ligands on A498 and CAKI-1 cells was recently reported [27–29]. The expression of NKp30, NKp44, NKp46, NKG2D, DNAM-1, NCAM-1 and FcγRIIIa was evaluated by RT-PCR analysis in VHL-WT (n = 17) and VHL-MUT (n = 17) RCC samples. A significant high expression of NKp30 and NKp46 was detected in VHL-MUT-RCC as compared to VHL-WT-RCC samples (p = 0.04 and p = 0.017, respectively, Fig. 3C and D) and NKp44 and DNAM-1 were slightly overexpressed in VHL-MUT compared to VHL-WT patients. Conversely expression of NCAM-1, FcγRIIIa and NKG2D were comparable in the two groups (Additional file 2: Figure S3). In addition, NKp30, NKp44, NKp46 and FcγRIIIIa mRNA were highly expressed in VHL-MUT tumors as compared to surrounding unaffected tissue (Additional file 2: Figure S4A) while in the VHL-WT tumors no significant difference was retrieved (Additional file 2: Figure S4B). Thus VHL-MUT tumors were characterized by high density of NKp46+ cells infiltration and high expression of NK activating genes such as NKp30, NKp46, NKp44 and DNAM-1.
VHL-MUT-RCC tumors display less functional Tregs
Since Tregs are supposed to suppress NK activity tumoral, Tregs (tTreg) identified as CD4+CD25hiFoxp3+ cells, were evaluated in 26 RCC patients (13 VHL-WT and 13 VHL-MUT). tTregs were significantly lower in VHL-MUT-RCC as compared to VHL-WT-RCC tumors (1.84 ± 0.36% vs 3.79 ± 0.74%; p = 0.04, Fig. 4A). Treg function was evaluated ex vivo on patients T effectors (Teff) proliferation. Tregs isolated from VHL-MUT tumors were less efficient in inhibiting T-effectors proliferation (VHL-MUT: 6.7 ± 3.9% vs VHL-WT: 2.8 ± 1.1%, p = 0.14 Fig. 4B) as confirmed by increased IFN-γ production in co-colture of Tregs:Teff isolated from VHL-MUT-RCC patients (22.8 ± 2.8 vs 12.36 ± 3.9 pg/mL, p = 0.09 Fig. 4C). Few patients were also evaluated for TGF-β1 mRNA (6 VHL-WT/6 VHL-MUT peritumoral and tumoral tissues). Consistent with a reduction of Tregs function, TGF-β1 expression, though not significant, was lower in VHL-MUT RCC as compared to VHL-WT-RCC (2.5 ± 1.0 vs 4.9 ± 1.15, p = 0.16 Fig. 4D).
Fig. 4.

Tumor-Tregs from VHL-WT-RCC patients are more suppressive than tumor-Tregs from VHL-MUT-RCC patients. A Percentage of tumor CD4+CD25hiFoxp3+cells (13 VHL-WT-RCCs vs 13 VHL-MUT-RCCs, p = 0.04). (Ai) Representative plots of Tregs from 1 VHL-WT-RCCs vs 1 VHL-MUT-RCCs. B Autologous CFSE-labeled CD4+CD25− T cells were co-cultured with CD4+CD25+ isolated from tumor of 6 VHL-WT and 4 VHL-MUT RCC patients. After 5 days of stimulation with Dynabeads Human T-Activator CD3/CD28, CFSE+CD4+ T cells were analyzed for their proliferation by CFSE dilution. C IFN-γ-tumor-Treg dependent ELISA from 3 VHL-WT vs 3 VHL-MUT RCC patients. D Measurement of TGF-β1 mRNA expression in 6 VHL-WT vs 6 VHL-MUT RCC patients by real-time PCR in tumor and corresponding peritumoral tissues. Relative mRNA expression was calculated according to 2-ΔΔCt method. The results are presented mean ± SEM. Statistical significance was calculated by unpaired Student t test (p < 0.05)
Discussion
In the present study RCC-VHL mutational status was correlated to patients NK cytotoxicity through ex vivo evaluation toward human renal cancer cell lines A498 VHL-MUT and CAKI-1-VHL-WT. In 51 RCC patients (23 VHL-MUT and 28 VHL-WT) enhanced NK cell cytotoxicity, as CD107a expression in whole blood, was reported versus VHL-MUT A498 cells in VHL-MUT RCC patients. Moreover, in an autologous setting, IL-2 activated NKs isolated from RCC-VHL-MUT patients more efficiently degranulated toward RCC autologous tumor cells or A498 VHL-MUT cells as compared to SN12C (RCC-VHL-WT) cell line. Higher NK susceptibility was previously reported to correlate with decreased MHC Class I expression in cancer cells [30]. Herein it was shown that A498 (containing a 4-nt deletion at nt 639–642 in VHL exon 2) and 786-O (containing a 1-nt deletion at nt 523 in VHL exon 1), express high level of classical MHC-I molecules not significantly different from CAKI-1, VHL-WT (A498: 81%; 786-O: 92.7% and CAKI-1: 86.2%) suggesting that tested MHC-I molecules are not mainly involved in the different NK sensitivity. It is noteworthy that A498 and 786–0 cell lines, although carrying a MUT-VHL, display phenotypical and molecular differences. 786–0 appearing poorly differentiated with sarcomatoid features while A498 displays epithelial cells with clear cytoplasm. Moreover 786-O carries (VHL−/− and PTEN−/−), A498 (VHL−/− and PTEN+/+) and CAKI-1 carries (VHL+/+ and PTEN+/+) [31, 32]. These differences might accomplish for different phenotypes affecting NK sensitivity [15].
Nowadays the effects of VHL mutations on NK activity is controversial. Previous evidence demonstrated that restoration of wild type VHL in human renal cancer cells decreased their NK susceptibility [33] while VHL mutations increased resistance to NK-mediated lysis through EPAS1/HIF-2α stabilization [16, 34]. NK cells induced a contact-dependent autophagy in ccRCC cells that was mediated by the HIF-2a targeted, inositol triphosphate receptor1 (ITPR1) in tumor cells. Blocking ITPR1 expression in ccRCC cells inhibited NK cell-induced autophagy and suppressed ccRCC resistance to NK cells [35].
In our study blood derived from patients carrying RCC-VHL-MUT tumors display better NK susceptibility toward human renal cancer cells VHL-MUT suggesting an improvement in the innate immune response toward RCC.
To further correlate peripheral and tumoral NK status, NK cell infiltration was analyzed through the evaluation of NKp46+ cells and the NKs activating receptors NKp30, NKp44, NKp46, NKG2D, DNAM-1, NCAM-1 and FcγRIIIa. NK cell-activating ligands expression on A498 and CAKI-1 cells was recently reported [27–29]. Although a significant increase in NKG2D+ cells was not detected in our tested cells, A498 and CAKI-1 express high levels of DNAM-1 ligand PVR (94.8 and 99.1% respectively, Additional file 2: Figure S5). Previous authors stated that VHL-mutational status may not affect the expression of ligands for NKG2D and DNAM-1 activating NK receptors [33]. Higher infiltration of NKp46+ cells and increased expression of NKp30 and NKp46 [36–38] were detected in VHL-MUT-RCC compared to VHL-WT-RCC. Although the NKp46 receptor demonstrates a high degree of lineage-specificity, being expressed almost exclusively in natural killer cells [39] the presence of a rare NKp46+CD3+ population was reported. These include rare subsets of T cells (αβ and δγ) in humans and mice and a subset of group 3 innate lymphoid cells (ILCs) (NCR+ ILC3) [36, 40]. Also DNAM1 or NKG2D may be expressed by T cells [36, 40, 41]. This is a limit of the present study. However the concomitant expression of NKp46 plus NKp30, NKp44 and DNAM-1 strongly suggest the hypothesis that NKs are involved.
To better define the role of NK cells in RCC tumors, Tregs and NKs are currently evaluated within the study REVOLUTION aimed at identify biomarkers of the checkpoint inhibitor nivolumab efficacy in metastatic renal cancer patients. Preliminary evidence showed that while the absolute value of CD3−CD56dim was not affected by nivolumab treatment NK cytotoxicity correlated with the responsiveness to treatment (Proceedings AACR 2017: Abstract #580).
Tregs suppress NK cell effector functions [42] impairing TGF-β pathway which, among other effects, induces downregulation of the activating NK cell receptor NKG2D [43]. Herein we report high infiltration of NKp46+ cells and reduced CD4+CD25hiFoxp3+ T cells in VHL-MUT-RCC tumors, although the reduction of TGF-β levels did not correspond to increased expression of NKG2D gene in VHL-MUT-RCC tumors. As in our results, Shen showed no significant correlation between peripheral Tregs and NKG2D expression in colorectal cancer patients NK cells [44]. Since VHL mutations induces PD-L1 expression in RCC, NK function may be a crucial element in nivolumab sensitivity [45]. NKs isolated from renal carcinoma patients expressed PD-1 on their surface and engagement of PD-1 signaling reduced their cytolytic potential. Treatment of patient-derived PD-1+ NK cells with an anti-PD-1 antibody (pidilizumab, CT-011) increased NK killing of autologous cancer cells in vitro [46]. NK cells also engage antibody-dependent cell mediated cytotoxicity (ADCC) and ex vivo treatment with the anti-CTLA4 ipilimumab enhances cetuximab-mediated ADCC targeting CTLA-4+ Treg in head and neck squamous cell cancer (SCCHN)[47, 48]. Nowadays the immune checkpoint targeting agents nivolumab and ipilimumab represent a clinical standard for mRCC. Nevertheless tyrosine kinase inhibitors (TKI) still play a central role in metastatic RCC treament [49] and modify the tumor immune sensitivity. TKIs affect the frequency and composition of tumor immune cell subpopulations including Tregs, myeloid-derived suppressor cells as well as T and NK cells [50–52]. Sorafenib significantly reduce NKs activity against the RCC cell lines A498, ACHN and CAKI-2 [53] while axitinib promotes NK cell recognition and degranulation toward A498 RCC cells in a ROS-dependent manner [28].
Conclusions
VHL tumoral mutations improve the NKs effectiveness in RCC patients and thus need to be considered in the evaluation of TKIs or immune based therapies. Moreover therapeutic strategies designed to target NK cells could be beneficial in VHL-mutated-RCCs alone or in association with immune checkpoints inhibitors.
Additional files
Table S1: Primer sequences for SYBR Green RT-qPCR. (PPTX 68 kb)
Figure S1. MHC Class-I profile in K562, CAKI-1, SN12C, A498 and 786-O cells. The expression of MHC-I was evaluated in RCC target cells by flow cytometry (FITC-conjugated major histocompatibility complex (MHC) class I–specific antibody (IgG2a, W6/32, CBL139F, Cymbus Biotech, Hants, UK). Figure S2. NKs from VHL-MUT-RCC patients display higher cytotoxicity toward human renal cancer cells VHL mutated A498 and 786-O. NK cell function was evaluated through CD107a cell-surface expression in response to ex vivo stimulation with K562, CAKI-1 (VHL-WT), A498 and 786-O (VHL-MUT) cells. Degranulation (CD107a) was evaluated after gating on CD3-CD56+ cells. CD107a+NK cells in 5 VHL-MUT (A) and 9 VHL-WT (B) RCC patients versus K562, CAKI-1, A498 and 786-O renal cell lines. Figure S3. NCAM-1, DNAM-1, FcγRIIIa, NKp44 and NKG2D are slightly overexpressed in VHL-MUT RCC tumors. RNA from 34 RCC tumors (17 VHL-WT and 17 VHL-MUT). Transcript levels are presented as mean±SEM. Statistical significance was calculated by unpaired Student t test (p < 0.05 ). Figure S4. NCAM-1, DNAM-1, FcγRIIIa, NKp30, NKp46, NKp44, NKG2D expression are upregulated in VHL-MUT tumors as compared to VHL-WT tumors. 17 VHL-MUT (A) and 17 VHL-WT (B). RT-PCR was performed on total RNA isolated from 34 tumors and relative peritumoral tissues (17 VHL-WT and 17 VHL-MUT). Relative gene expression levels were normalized to GUSB. Statistical significance was calculated by unpaired Student t test (p < 0.05). Figure S5. Expression of DNAM-1 ligand (PVR) in CAKI-1 and A498 cell lines. The expression of PVR (CD155) was evaluated in RCC target cells by flow cytometry (PE anti-human CD155/PVR, ( clone SKIL.4, Biolegend, Cat No 337609). (ZIP 561 kb)
Table S2. Detailed characteristics of 23 VHL-MUT-RCC patients. (PPTX 71 kb)
Table S3. Detailed characteristics of 28 VHL-WT-RCC patients. (PPTX 71 kb)
Acknowledgements
We acknowledge our collaborator, Sonia Desicato for her contributions in recruitment of patients.
Funding
This work was supported by from Italian Ministry of Health: Grants Ricerca Corrente TITOLO.
Availability of data and materials
All data and materials can be provided upon request.
Abbreviations
- ADCC
Antibody-dependent cell mediated cytotoxicity
- ccRCC
Clear cell renal cell carcinoma
- CFSE
Carboxyfluorescein diacetate succinimidyl ester
- CLRs
C-type lectin receptors
- HD
Healthy donors
- HIFs
Hypoxia-inducible factors
- ITRI1
Inositol triphosphate receptor1
- KIRs
Killer cell immunoglobulin-like receptors
- LAMP-1 (CD107a)
Lysosome-associated membrane protein
- NCRs
Natural cytotoxicity receptors
- NKs
Natural killer cells
- PB
Peripheral blood
- PT
Peritumoral tissue
- RCCs
Renal cell carcinomas
- SCCHN
Head and neck squamous cell cancer
- Teff
T effector cells
- TGF-β
Transforming growth factor-beta
- TKI
Tyrosine kinase inhibitor
- Tregs
T regulatory cells
- TT
Tumor tissue
- VHL
Von Hippel-Lindau
- VHL-MUT
Von Hippel-Lindau mutated
- VHL-WT
Von Hippel-Lindau wild-type
Author’s contributions
AMT designed, performed, and analyzed data, and wrote the manuscript. SS SZ, CD, MN, GR, RC, GB. contributed to experimental design and data interpretation. FE organized the recruitment of patients. EL and AA provided clinical-pathological features of patients. GB contributed to data interpretation and editing the manuscript. NL, SP and SP provided patients sample, patients information, provided intellectual input and critically reviewed the manuscript. SS conceived of, designed, supervised the project and experimental plan, interpreted experiments, and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Primary RCC samples were from the Department of Urology, National Cancer Institute “G. Pascale” (Naples, Italy) and Genitourinary Oncology and Rare Cancer Center, Federico II University (Naples, Italy). The research protocol was approved by Human Ethical Committee of Institute (n. CEI/423/13).
Consent for publication
The author agree for publication.
Competing interests
The authors have declared that no competing interest exists.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Anna Maria Trotta, Email: amt78@libero.it.
Sara Santagata, Email: sara.santagata@yahoo.it.
Serena Zanotta, Email: serena.zanotta@alice.it.
Crescenzo D’Alterio, Email: crescenzodalterio76@gmail.com.
Maria Napolitano, Email: maria_napolitano@yahoo.it.
Giuseppina Rea, Email: pina.rea@hotmail.it.
Rosa Camerlingo, Email: camerlingorosa@gmail.com.
Fabio Esposito, Email: fabioesposito025@gmail.com.
Elvira Lamantia, Email: doctorelvy.lm@gmail.com.
Annamaria Anniciello, Email: a.anniciello@istitutotumori.na.it.
Giovanni Botti, Email: bottigiovanni88@gmail.com.
Nicola Longo, Email: nicolalongo20@yahoo.it.
Gerardo Botti, Email: g.botti@istitutotumori.na.it.
Sandro Pignata, Email: s.pignata@istitutotumori.na.it.
Sisto Perdonà, Email: s.perdona@istitutotumori.na.it.
Stefania Scala, Phone: +39-081-5903678, Email: scalaste@gmail.com, Email: s.scala@istitutotumori.na.it.
References
- 1.Fitzmaurice C, Dicker D, Pain A, et al. The global burden of Cancer 2013. JAMA Oncol. 2015;1(4):505–527. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Massari F, Bria E, Maines F, et al. Adjuvant treatment for resected renal cell carcinoma: are all strategies equally negative? Potential implications for trial design with targeted agents. Clin Genitourin Cancer. 2013;11(4):471–476. doi: 10.1016/j.clgc.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 4.Herrmann E, Weishaupt C, Poppelmann B, et al. New tools for assessing the individual risk of metastasis in renal cell carcinoma. Clin Exp Metastasis. 2013;30(2):215–224. doi: 10.1007/s10585-012-9529-6. [DOI] [PubMed] [Google Scholar]
- 5.Schraml P, Struckmann K, Hatz F, et al. VHL mutations and their correlation with tumour cell proliferation, microvessel density, and patient prognosis in clear cell renal cell carcinoma. J Pathol. 2002;196(2):186–193. doi: 10.1002/path.1034. [DOI] [PubMed] [Google Scholar]
- 6.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22(24):4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 7.Young AC, Craven RA, Cohen D, et al. Analysis of VHL gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clin Cancer Res. 2009;15(24):7582–7592. doi: 10.1158/1078-0432.CCR-09-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015;15(1):55–64. doi: 10.1038/nrc3844. [DOI] [PubMed] [Google Scholar]
- 9.Kaelin WG., Jr The von Hippel-Lindau tumor suppressor gene and kidney cancer. Clin Cancer Res. 2004;10(18 Pt 2):6290s–6295s. doi: 10.1158/1078-0432.CCR-sup-040025. [DOI] [PubMed] [Google Scholar]
- 10.Kondo K, Yao M, Yoshida M, et al. Comprehensive mutational analysis of the VHL gene in sporadic renal cell carcinoma: relationship to clinicopathological parameters. Genes Chromosomes Cancer. 2002;34(1):58–68. doi: 10.1002/gcc.10054. [DOI] [PubMed] [Google Scholar]
- 11.Smits KM, Schouten LJ, van Dijk BA, et al. Genetic and epigenetic alterations in the von hippel-Lindau gene: the influence on renal cancer prognosis. Clin Cancer Res. 2008;14(3):782–787. doi: 10.1158/1078-0432.CCR-07-1753. [DOI] [PubMed] [Google Scholar]
- 12.Patard JJ, Fergelot P, Karakiewicz PI, et al. Low CAIX expression and absence of VHL gene mutation are associated with tumor aggressiveness and poor survival of clear cell renal cell carcinoma. Int J Cancer. 2008;123(2):395–400. doi: 10.1002/ijc.23496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moretta L, Bottino C, Pende D, et al. Human natural killer cells: their origin, receptors and function. Eur J Immunol. 2002;32(5):1205–1211. doi: 10.1002/1521-4141(200205)32:5<1205::AID-IMMU1205>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 14.Cheng M, Chen Y, Xiao W, et al. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10(3):230–252. doi: 10.1038/cmi.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fregni G, Perier A, Avril MF, et al. NK cells sense tumors, course of disease and treatments: consequences for NK-based therapies. Oncoimmunology. 2012;1(1):38–47. doi: 10.4161/onci.1.1.18312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messai Y, Noman MZ, Hasmim M, et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. 2014;74(23):6820–6832. doi: 10.1158/0008-5472.CAN-14-0303. [DOI] [PubMed] [Google Scholar]
- 17.Chang WC, Li CH, Chu LH, et al. Regulatory T cells suppress natural killer cell immunity in patients with human cervical carcinoma. Int J Gynecol Cancer. 2016;26(1):156–162. doi: 10.1097/IGC.0000000000000578. [DOI] [PubMed] [Google Scholar]
- 18.Orentas RJ, Kohler ME, Johnson BD. Suppression of anti-cancer immunity by regulatory T cells: back to the future. Semin Cancer Biol. 2006;16(2):137–149. doi: 10.1016/j.semcancer.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Ghiringhelli F, Menard C, Terme M, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. 2005;202(8):1075–1085. doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6(10):813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 21.Adams S, Robbins FM, Chen D, et al. HLA class I and II genotype of the NCI-60 cell lines. J Transl Med. 2005;3(1):11. doi: 10.1186/1479-5876-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santagata S, Napolitano M, D'Alterio C, et al. Targeting CXCR4 reverts the suppressive activity of T-regulatory cells in renal cancer. Oncotarget. 2017;8(44):77110–77120. doi: 10.18632/oncotarget.20363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294(1–2):15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Claus M, Watzl C. Evaluation of human natural killer cell activities in whole blood. Curr Protoc Immunol. 2010;Chapter 7:Unit7.39. doi: 10.1002/0471142735.im0739s91. [DOI] [PubMed] [Google Scholar]
- 25.Fauriat C, Long EO, Ljunggren HG, et al. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115(11):2167–2176. doi: 10.1182/blood-2009-08-238469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whaley JM, Naglich J, Gelbert L, et al. Germ-line mutations in the von Hippel-Lindau tumor-suppressor gene are similar to somatic von Hippel-Lindau aberrations in sporadic renal cell carcinoma. Am J Hum Genet. 1994;55(6):1092–1102. [PMC free article] [PubMed] [Google Scholar]
- 27.Sconocchia G, Spagnoli GC, Del Principe D, et al. Defective infiltration of natural killer cells in MICA/B-positive renal cell carcinoma involves beta(2)-integrin-mediated interaction. Neoplasia. 2009;11(7):662–671. doi: 10.1593/neo.09296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morelli MB, Amantini C, Santoni M, et al. Axitinib induces DNA damage response leading to senescence, mitotic catastrophe, and increased NK cell recognition in human renal carcinoma cells. Oncotarget. 2015;6(34):36245–36259. doi: 10.18632/oncotarget.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luan Y, Liu J, Liu X, et al. Tetramethypyrazine inhibits renal cell carcinoma cells through inhibition of NKG2D signaling pathways. Int J Oncol. 2016;49(4):1704–1712. doi: 10.3892/ijo.2016.3670. [DOI] [PubMed] [Google Scholar]
- 30.Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in Cancer immunotherapy. Front Immunol. 2017;8:1124. doi: 10.3389/fimmu.2017.01124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha R, Winer AG, Chevinsky M, et al. Analysis of renal cancer cell lines from two major resources enables genomics-guided cell line selection. Nat Commun. 2017;8:15165. doi: 10.1038/ncomms15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell L, Al-Jayyoussi G, Gutteridge R, et al. Caveolin-1 in renal cell carcinoma promotes tumour cell invasion, and in co-operation with pERK predicts metastases in patients with clinically confined disease. J Transl Med. 2013;11:255. doi: 10.1186/1479-5876-11-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perier A, Fregni G, Wittnebel S, et al. Mutations of the von Hippel-Lindau gene confer increased susceptibility to natural killer cells of clear-cell renal cell carcinoma. Oncogene. 2011;30(23):2622–2632. doi: 10.1038/onc.2010.638. [DOI] [PubMed] [Google Scholar]
- 34.Messai Y, Noman MZ, Hasmim M, et al. HIF-2alpha/ITPR1 axis: a new saboteur of NK-mediated lysis. Oncoimmunology. 2015;4(2):e985951. doi: 10.4161/2162402X.2014.985951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasmim M, Messai Y, Ziani L, et al. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol. 2015;6:482. doi: 10.3389/fimmu.2015.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spits H, Artis D, Colonna M, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 37.Schleypen JS, Von Geldern M, Weiss E, et al. Renal cell carcinoma-infiltrating natural killer cells express differential repertoires of activating and inhibitory receptors and are inhibited by specific HLA class I allotypes. Int J Cancer. 2003;106(6):905–912. doi: 10.1002/ijc.11321. [DOI] [PubMed] [Google Scholar]
- 38.Schleypen JS, Baur N, Kammerer R, et al. Cytotoxic markers and frequency predict functional capacity of natural killer cells infiltrating renal cell carcinoma. Clin Cancer Res. 2006;12(3 Pt 1):718–725. doi: 10.1158/1078-0432.CCR-05-0857. [DOI] [PubMed] [Google Scholar]
- 39.Connelley TK, Longhi C, Burrells A, et al. NKp46+ CD3+ cells: a novel nonconventional T cell subset in cattle exhibiting both NK cell and T cell features. J Immunol. 2014;192(8):3868–3880. doi: 10.4049/jimmunol.1302464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudspeth K, Silva-Santos B, Mavilio D, et al. Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front Immunol. 2013;4:69. doi: 10.3389/fimmu.2013.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu J, Mitsui T, Wei M, et al. NKp46 identifies an NKT cell subset susceptible to leukemic transformation in mouse and human. J Clin Invest. 2011;121:1456–1470. doi: 10.1172/JCI43242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pedroza-Pacheco I, Madrigal A, Saudemont A. Interaction between natural killer cells and regulatory T cells: perspectives for immunotherapy. Cell Mol Immunol. 2013;10(3):222–229. doi: 10.1038/cmi.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zimmer J, Andres E, Hentges F. NK cells and Treg cells: a fascinating dance cheek to cheek. Eur J Immunol. 2008;38(11):2942–2945. doi: 10.1002/eji.200838813. [DOI] [PubMed] [Google Scholar]
- 44.Shen Y, Wang Q, Qi Y, et al. Peripheral Foxp3+ regulatory T cells and natural killer group 2, member D expression levels in natural killer cells of patients with colorectal cancer. Mol Med Rep. 2014;10(2):977–982. doi: 10.3892/mmr.2014.2229. [DOI] [PubMed] [Google Scholar]
- 45.Messai Y, Gad S, Noman MZ, et al. Renal cell carcinoma programmed death-ligand 1, a new direct target of hypoxia-inducible Factor-2 alpha, is regulated by von Hippel-Lindau gene mutation status. Eur Urol. 2016;70(4):623–632. doi: 10.1016/j.eururo.2015.11.029. [DOI] [PubMed] [Google Scholar]
- 46.Benson DM, Jr, Bakan CE, Mishra A, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116(13):2286–2294. doi: 10.1182/blood-2010-02-271874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jie HB, Schuler PJ, Lee SC, et al. CTLA-4(+) regulatory T cells increased in Cetuximab-treated head and neck Cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75(11):2200–2210. doi: 10.1158/0008-5472.CAN-14-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferris RL, Lenz HJ, Trotta AM, et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev. 2018;63:48–60. doi: 10.1016/j.ctrv.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zarrabi K, Fang C, Wu S. New treatment options for metastatic renal cell carcinoma with prior anti-angiogenesis therapy. J Hematol Oncol. 2017;10:38. doi: 10.1186/s13045-016-0374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stehle F, Schulz K, Fahldieck C, et al. Reduced immunosuppressive properties of axitinib in comparison with other tyrosine kinase inhibitors. J Biol Chem. 2013;288(23):16334–16347. doi: 10.1074/jbc.M112.437962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Geisler K, Reischer A, Kroeger I, et al. Nilotinib combined with interleukin-2 mediates antitumor and immunological effects in a B16 melanoma model. Oncol Rep. 2014;31:2015–2020. doi: 10.3892/or.2014.3070. [DOI] [PubMed] [Google Scholar]
- 52.Krieg S, Ullrich E. Novel immune modulators used in hematology: impact on NK cells. Front Immunol. 2013;3:388. doi: 10.3389/fimmu.2012.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krusch M, Salih J, Schlicke M, et al. The kinase inhibitors sunitinib and sorafenib differentially affect NK cell antitumor reactivity in vitro. J Immunol. 2009;183:8286–8294. doi: 10.4049/jimmunol.0902404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Primer sequences for SYBR Green RT-qPCR. (PPTX 68 kb)
Figure S1. MHC Class-I profile in K562, CAKI-1, SN12C, A498 and 786-O cells. The expression of MHC-I was evaluated in RCC target cells by flow cytometry (FITC-conjugated major histocompatibility complex (MHC) class I–specific antibody (IgG2a, W6/32, CBL139F, Cymbus Biotech, Hants, UK). Figure S2. NKs from VHL-MUT-RCC patients display higher cytotoxicity toward human renal cancer cells VHL mutated A498 and 786-O. NK cell function was evaluated through CD107a cell-surface expression in response to ex vivo stimulation with K562, CAKI-1 (VHL-WT), A498 and 786-O (VHL-MUT) cells. Degranulation (CD107a) was evaluated after gating on CD3-CD56+ cells. CD107a+NK cells in 5 VHL-MUT (A) and 9 VHL-WT (B) RCC patients versus K562, CAKI-1, A498 and 786-O renal cell lines. Figure S3. NCAM-1, DNAM-1, FcγRIIIa, NKp44 and NKG2D are slightly overexpressed in VHL-MUT RCC tumors. RNA from 34 RCC tumors (17 VHL-WT and 17 VHL-MUT). Transcript levels are presented as mean±SEM. Statistical significance was calculated by unpaired Student t test (p < 0.05 ). Figure S4. NCAM-1, DNAM-1, FcγRIIIa, NKp30, NKp46, NKp44, NKG2D expression are upregulated in VHL-MUT tumors as compared to VHL-WT tumors. 17 VHL-MUT (A) and 17 VHL-WT (B). RT-PCR was performed on total RNA isolated from 34 tumors and relative peritumoral tissues (17 VHL-WT and 17 VHL-MUT). Relative gene expression levels were normalized to GUSB. Statistical significance was calculated by unpaired Student t test (p < 0.05). Figure S5. Expression of DNAM-1 ligand (PVR) in CAKI-1 and A498 cell lines. The expression of PVR (CD155) was evaluated in RCC target cells by flow cytometry (PE anti-human CD155/PVR, ( clone SKIL.4, Biolegend, Cat No 337609). (ZIP 561 kb)
Table S2. Detailed characteristics of 23 VHL-MUT-RCC patients. (PPTX 71 kb)
Table S3. Detailed characteristics of 28 VHL-WT-RCC patients. (PPTX 71 kb)
Data Availability Statement
All data and materials can be provided upon request.