

To the Editor: Diabetic nephropathy (DN) is the leading cause of chronic renal diseases and accounts for almost 50% of all end-stage renal diseases worldwide. The prevalence of DN increased significantly after 2010 in China.[1,2] DN is clinically characterized by proteinuria and morphological and ultrastructural changes in the kidney. The pathogenesis of DN is multifactorial and extremely complex, including hyperglycemia, transforming growth factor (TGF)-β1, angiotensin II, DNA methylation, chromatin histone modification, microRNAs, and long noncoding RNAs.
In the past 10 years, advances in high-throughput transcriptomics technology and analysis methods have provided the unprecedented opportunity to investigate the mechanisms of complex diseases with high resolution. A series of transcriptomics studies have been performed in DN patients and animal models, but most of these studies only focused on the differentially expressed genes (DEGs).
In addition to identifying the DEGs, identifying the regulators, which are relevant or even causative to the phenotypic changes of DN, is a challenging and worthwhile goal. Unfortunately, this goal cannot be achieved using differential expression analysis alone, not only because the causal genes are always buried within a large amount of DEGs but also because a causal regulator may not necessarily be a DEG. For example, if a posttranslational modification occurs to a transcription factor (TF) in a disease state, the TF, at its original expression level, can no longer activate its original target genes, instead activating a new group of target genes. In such a circumstance, the expression correlation between the TF and its targets can be affected; this TF might be captured using differential coexpression analysis (DCEA) and differential regulation analysis (DRA).[3] To further investigate the potential gene regulation patterns in the DN, DCEA and DRA were used in this study to identify molecular regulators, which play important roles in the pathogenesis of DN.
The protocol for the use of human samples was approved by the Human Subjects Committee of Jinling Hospital, Nanjing University (No. 2013KLY-013-01), and a signed consent form was obtained from each patient and control donor.
Based on the guidelines of NKF-K/DOQI (2007 edition) and the Expert Consensus on the Prevention and Treatment of Diabetic Nephropathy by the Chinese Medical Association (2014 edition), type 2 DN was diagnosed if the below criteria were met: (1) having a diagnosis of type 2 diabetes; (2) presence of a ratio of urinary albumin to urinary creatinine of at least 30 mg/g for a first morning specimen on two occasions or by a 24-h urinary protein excretion concentration ≥500 mg on two consecutive occasions; and (3) presence of diabetic retinopathy but absence of any clinical or laboratory evidence of other kidney or renal tract diseases.
A total of 41 DN patients confirmed by renal biopsy were enrolled in this study. Kidney samples were obtained from surgical nephrectomies and from leftover portions of diagnostic kidney biopsies. All of the patients were categorized based on the pathologic classification, which was described in our previous work.[4] The clinical and pathologic characteristics of DN patients are detailed in Supplementary Table 1. Control samples (Ctrl, n = 20) were obtained from biopsy samples of the unaffected portions of tumor nephrectomies. Controls met the criteria described below: estimated glomerular filtration rate (eGFR) more than 90 ml/min, the absence of proteinuria, and normal serum creatinine and blood urea nitrogen. Based on the clinical and pathologic characteristics, the DN patients were divided into the early-stage DN group (Early: n = 20, eGFR more than 60 ml/min, the glomerular classifications were Class I or Class IIa) and the late-stage DN group (Late: n = 21, eGFR between 15 ml/min and 60 ml/min, the glomerular classifications were Class III or Class IV).
Supplementary Table 1.
Clinical and pathologic characteristics of DN patients
| Characteristics | Early (n = 20) | Late (n = 21) | χ2 | P |
|---|---|---|---|---|
| Age (years) | 46.5 ± 8.7 | 47.7 ± 7.0 | 0.04 | 0.835 |
| Sex (female), n (%) | 9 (45.0) | 3 (14.3) | – | 0.043 |
| Ethnicity | Han | Han | – | – |
| BMI (kg/m2) | 25.2 ± 2.12 | 24.6 ± 1.22 | 0.43 | 0.514 |
| Serum creatinine (mg/dl) | 0.81 ± 0.19 | 1.95 ± 0.70 | 25.08 | <0.001 |
| eGFR (ml/min) | 99.16 ± 15.47 | 42.61 ± 13.47 | 30.00 | <0.001 |
| Proteinuria (g/24 h) | 0.60 ± 0.27 | 5.41 ± 3.88 | 22.78 | <0.001 |
| HbA1c (%) | 6.51 ± 1.12 | 6.91 ± 2.08 | 3.63 | 0.056 |
| Glomerular lesions, n | ||||
| Class I | 5 | 0 | – | <0.001 |
| Class II | 0 | 0 | ||
| IIa | 15 | 0 | ||
| IIb | 0 | 0 | ||
| Class III | 0 | 16 | ||
| Class IV | 0 | 5 | ||
| IFTA, n | ||||
| 0 | 6 | 0 | – | <0.001 |
| 1 | 14 | 2 | ||
| 2 | 0 | 7 | ||
| 3 | 0 | 12 | ||
| Interstitial inflammation, n | ||||
| 0 | 6 | 0 | – | <0.001 |
| 1 | 9 | 1 | ||
| 2 | 5 | 20 | ||
| Arteriolar hyalinosis, n | ||||
| 0 | 0 | 0 | – | 1 |
| 1 | 0 | 0 | ||
| 2 | 20 | 21 | ||
| Arteriosclerosis, n | ||||
| NA | 0 | 0 | – | 0.007 |
| 0 | 6 | 0 | ||
| 1 | 3 | 2 | ||
| 2 | 11 | 19 |
Values are presented as n or mean ± SD. P values were obtained using the Wilcoxon rank-sum test for continuous variables and Fisher's exact test for categorical variables. –: Not available. DN: Diabetic nephropathy; BMI: Body mass index; eGFR: Estimated glomerular filtration rate, calculated using the EPI-CKD (Chronic Kidney Disease Epidemiology Collaboration) formula; HbA1c: Glycated hemoglobin; SD: Standard deviation; IFTA: Interstitial fibrosis and tubular atrophy.
Tissue was placed into RNALater and manually microdissected at 4°C to obtain the glomerular compartment. Dissected glomeruli were homogenized, and RNA was prepared using RNAeasy mini kit (Qiagen Sciences Inc., Germantown, MD, USA) according to the manufacturer's instructions. Genome-wide gene expression profiling was performed using an Affymetrix GeneChip® Human Transcriptome Array 2.0 (Affymetrix, Santa Clara, California, USA).
Gene microarray data were received in the form of Affymetrix CEL files. The CEL files were submitted to the GEO database (accession number: GSE96804).[5] These files were then read into Expression Console (Affymetrix, Santa Clara, California, USA) and preprocessed using the gene level (RMA-Sketch) workflow. All of the samples were determined to be high quality during the quality control step of the workflow. Next, we exported the gene expression profile with annotation information into a TXT file to process it using R language (Free Software, https://www.r-project.org). To decrease computational constraints and to facilitate subsequent analysis, probe sets that were not associated with mRNA were omitted, and probe sets for the same mRNA were collapsed into one using the median value. We ultimately constructed an expression table, listing 61 samples (Ctrl: n = 20; Early: n = 20; Late: n = 21) and 18,418 genes.
TRANSFAC (geneXplain GmbH, Wolfenbüttel, Germany) is a unique knowledge database containing published data on eukaryotic TFs and miRNAs, their experimentally demonstrated binding sites, and regulated genes.[6] The human TFs and their regulated human genes were extracted from TRANSFAC (TFP_2013.4_data.tar.gz) and compiled as the human TF-to-target library, including 7569 relationships involving 573 human TFs and 2024 target genes.
Using the R package “limma”, we obtained two sets of DEGs (Benjamini-Hochberg adjusted P value <0.05 and |log fold change| >1.5) through pairwise comparisons (Early vs. Ctrl and Late vs. Early). We then utilized the R package “GeneAnswers” to find the enriched categories for the DEGs. The “GeneAnswers” package functionally categorizes the gene list based on Fisher's exact test with annotation libraries of gene ontology (GO), the Kyoto Encyclopedia of Genes and Genomes (KEGG), and the human TF-to-target library based on TRANSFAC.
In the transcriptome analysis domain, DCEA is emerging as a unique complement to traditional differential expression and coexpression analysis. The rationale behind DCEA is that changes in gene coexpression patterns between two contrasting phenotypes (e.g., healthy and disease) provide hints regarding the disrupted regulatory relationships or affected regulatory subnetworks specific to the phenotype of interest (in this case, the disease phenotype). DCGL (Differential Co-expression Analysis and Differential Regulation Analysis of Gene Expression Microarray Data) is an R package designed to identify DCGs (differentially coexpressed genes), DCLs (differentially coexpressed links), DRGs (differentially regulated genes), and DRLs (differentially regulated links).[3]
For our transcriptome data, we applied the R package “DCGL” to identify DCGs and DCLs through two comparisons (Early vs. Ctrl and Late vs. Early). The method “DCp” was used for identifying DCGs, while the method “DCe” was used for identifying both DCGs and DCLs, and the function “DCsum” was used for summarizing DCGs and DCLs.
In pairwise comparisons between two states, there are four types of DCLs. For example, a pair of genes is positively correlated in state A and is still positively correlated in state B; this pair is noted as “Positive-Positive”. By that analogy, the other three types of DCLs are “Positive-Negative”, “Negative-Positive”, and “Negative-Negative.” The “Positive-Positive” and “Negative-Negative” are defined as “Same type relationship”, while “Positive-Negative” and “Negative-Positive” are defined as “Different type relationship”. If the correlation coefficients of a pair of genes in two states both exceed the threshold, this DCL is defined as “Regulation-switch”, which is a special case of “Different type relationship”.
We incorporated the human TF-to-target library into the DCGs and DCLs to identify DRGs and DRLs respectively. If a DCG was a TF, this type of DCG was termed a DRG. It was intuitively speculated that its related differential coexpression might be attributed to the change in its transcriptional regulation activity. If a DCL coincided with a TF-to-target relation, this type of DCL was termed a TF2target_DCL, belonging to the DRLs. The rationale here was that the disruption of regulatory relations could affect the coexpression links between a regulator and its targets. With the function “DRplot”, TF2target_DCLs could be visualized as a DRG-highlighted, DRL-centered network. The algorithm DR rank, calculating Targets' Enrichment Density (TED) scores, was used to prioritize TFs that were putatively causative to DN occurrence and development.
In pairwise comparisons between Early and Ctrl, a total of 194 genes were determined to be DEGs, including 20 upregulated and 174 downregulated genes. The enriched GO terms and pathways were primarily involved in metabolic processes [Supplementary Figure 1 (531.1KB, tif) ]. In pairwise comparisons between Late and Early, a total of 374 genes were identified as DEGs, including 239 upregulated and 135 downregulated genes. The most enriched GO terms were “extracellular region”, “protein binding”, “extracellular structure organization”, “immune system process”, and “cell adhesion” among others. The most enriched KEGG pathways were “complement and coagulation cascades”, “Staphylococcus aureus infection”, “focal adhesion”, and “extracellular matrix (ECM)-receptor interaction” among others [Supplementary Figure 2 (538.2KB, tif) ].
GO categories and KEGG pathways enriched in the glomerular DEGs between the early stage of DN patients and the control group. (a) GO cellular component; (b) GO molecular function; (c) GO biological process; (d) KEGG pathways. GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; DEGs: Differentially expressed genes; DN: Diabetic nephropathy.
GO categories and KEGG pathways enriched in the glomerular DEGs between the late and early stage of DN patients. (a) GO cellular component; (b) GO molecular function; (c) GO biological process; (d) KEGG pathways. GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; DEGs: Differentially expressed genes; DN: Diabetic nephropathy.
The enriched TFs (Benjamini-Hochberg adjusted P value <0.2) of DEGs between Early and Ctrl were HNF1A, MAFK, NFIC, NR1H3, and RXRA [Figure 1a]. HNF1A, NR1H3, and RXRA were nuclear hormone receptors, and their target genes were all downregulated in Early versus Ctrl. The enriched TFs (Benjamini-Hochberg adjusted P value <0.2) of DEGs between Late and Early were SMAD2, SMAD3, SPI1, STAT1, GATA2, and FLI1 [Figure 1b]. The target genes of SMAD2, SMAD3, and STAT1 were all upregulated in Late versus Early.
Figure 1.
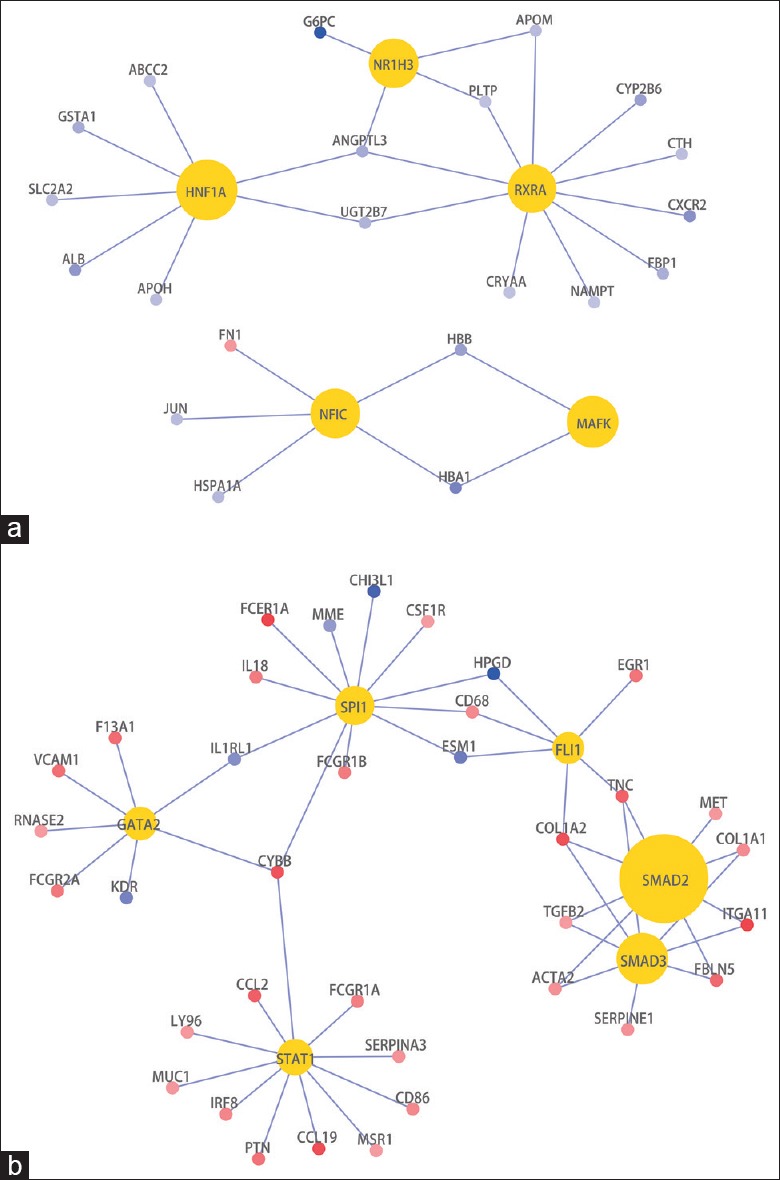
TFs enriched in the DEGs between the early-stage DN group (n = 20) and the control group (n = 20) (a). TFs enriched in the DEGs between the late-stage DN group (n = 21) and the early-stage DN group (n = 20) (b). Gold nodes represent the TFs; red nodes represent upregulated genes, while blue nodes represent downregulated genes. All gene symbols are approved HGNC symbols. TFs: Transcription factors; DEGs: Differentially expressed genes; DN: Diabetic nephropathy; HGNC: HUGO Gene Nomenclature Committee.
We applied the DCGL package to our DN transcriptome data to identify the DCGs and DCLs. Based on the criterion of P < 0.05, 872 DCGs were identified in pairwise comparisons between Early and Ctrl, 440 DCGs were identified between Late and Ctrl, and 317 DCGs were identified between Late and Early [Table 1]. The number of “Regulation-switch” DCLs may indicate the difference in gene coexpression relationships between different phenotypes. Therefore, the difference in gene coexpression relationships between Ctrl and Early was the largest (41,555 “Regulation-switch” DCLs), while that between Early and Late was the smallest (9384 “Regulation-switch” DCLs).
Table 1.
Statistics information of DCGs and DCLs
| Comparison | DCG | DCL | |||
|---|---|---|---|---|---|
| Total number, n | Same type relationship, n | Different type relationship, n | Regulation-switch, n | Total number, n | |
| Early versus Ctrl | 872 | 147,805 | 112,339 | 41,555 | 301,699 |
| Late versus Ctrl | 440 | 74,142 | 61,759 | 26,916 | 162,817 |
| Late versus Early | 317 | 61,495 | 36,742 | 9384 | 107,621 |
DCGs: Differentially coexpressed genes; DCLs: Differentially coexpressed links.
We compared the DCLs of the three comparisons in pairs to observe the similarities and differences in gene coexpression relationship changes [Supplementary Table 2]. The consistently related common DCLs between “Early versus Ctrl” and “Late versus Ctrl” had the highest proportion (4.22%), which indicated the highest consistency of differential coexpression relationships is between “Early versus Ctrl” and “Late versus Ctrl”.
Supplementary Table 2.
Comparison of DCLs between different comparisons
| Items (Comparison A vs. Comparison B) | Number of DCLs | Number of DCLs common to the double comparison (%) | Number of DCLs specifically found in Comparison A | Number of DCLs specifically found in Comparison B | |
|---|---|---|---|---|---|
| Consistently related | Inconsistently related | ||||
| (Early vs. Ctrl) versus (Late vs. Ctrl) | 443,820 | 18,713 (4.22) | 1983 (0.45) | 281,003 | 142,121 |
| (Early vs. Ctrl) versus (Late vs. Early) | 402,133 | 5114 (1.27) | 2073 (0.52) | 294,512 | 100,434 |
| (Late vs. Ctrl) versus (Late vs. Early) | 266,605 | 2464 (0.92) | 1369 (0.51) | 158,984 | 103,788 |
DCLs: Differentially coexpressed links.
We further matched the DCGs and DCLs to the known regulatory data between TFs and target genes and then obtained a number of disrupted regulatory relationships between TFs and target genes. To focus on the most meaningful information, we calculated the TED scores of each TF and selected the important TFs (Benjamini-Hochberg adjusted P value <0.05) in Early versus Ctrl and Late versus Early, respectively [Supplementary Table 3].
Supplementary Table 3.
Important TFs with high TED scores
| TF | TED score |
|---|---|
| Early versus Ctrl | |
| STAT2 | 5.249 |
| IRF9 | 4.397 |
| IRF5 | 1.920 |
| TAF12 | 1.919 |
| TAF6 | 1.919 |
| TAF9 | 1.919 |
| IRF1 | 1.353 |
| IRF3 | 1.207 |
| IRF7 | 1.207 |
| TFAP4 | 1.207 |
| Late versus Early | |
| IRF9 | 4.962 |
| STAT2 | 4.627 |
| NFKB2 | 1.704 |
TF: Transcription factor; TED: Targets’ Enrichment Density.
The pathogenesis of DN is complex and still not fully elucidated. Identifying the regulators that are relevant or even causative to the phenotypic changes of DN is a challenging and worthwhile goal. In our study, based on the gene expression profiles, we investigate the changes of transcription regulation in DN glomeruli, especially comparing the early-stage DN samples with the control and the late-stage DN samples, and tried to dissect the regulatory factors in the early stage of DN. These findings will improve the understanding of the molecular mechanisms of DN. In addition, our results will provide a novel resource for investigators focused on DN.
There were only 194 DEGs between Early and Ctrl, mainly involving metabolic processes. The DEGs between Late and Early were related to “extracellular structure organization”, “immune system process”, “cell adhesion”, “complement and coagulation cascades”, “focal adhesion”, and “ECM-receptor interaction” among others [Supplementary Figure 2 (538.2KB, tif) ]. These GO terms and KEGG pathways were widely related to the pathogenesis of DN and agreed well with our previous works.[4]
Three of the five enriched TFs in DEGs of the early stage of DN are nuclear hormone receptors, including HNF1A, NR1H3, and RXRA. Nuclear hormone receptors are TFs that regulate carbohydrate metabolism, lipid metabolism, the immune response, and inflammation. The nuclear hormone receptors, including peroxisome-proliferator-activated receptors (PPARs), estrogen receptors, the Vitamin D receptor (VDR), hepatocyte nuclear factor-1α (HNF-1α), hepatocyte nuclear factor-4α (HNF-4α), farnesoid X receptor (FXR), and liver X receptors (LXRs), were thought to play roles in the pathogenesis of DN, and many of them have been suggested to provide protection against DN.[7]
HNF1A, which encodes HNF-1α, is responsible for maturity-onset diabetes of the young type 3. Certain combinations of common variants in HNF1A are associated with decreased transcriptional activity in vitro and a decreased insulin response to oral glucose in vivo.[8] In the early stage of DN, the target genes of HNF-1α were downregulated, which indicated that the transcriptional activity of HNF-1α decreased in the glomeruli of DN patients.
NR1H3, which encodes LXR-α, is found in every segment along the nephron. In the glomeruli, this receptor seems to be present in all major renal cells, including mesangial cells, endothelial cells, and podocytes. Activation of LXR-α reduces the activation of RAS in mouse kidney.[9] In the early stage of DN, the target genes of LXR-α were downregulated, but the NR1H3 mRNA expression was unchanged. Collectively, these data suggest that reduced LXR-α activity may aggravate the progression of DN.
RXRA, which encodes retinoid X receptor-α (RXR-α), serves as a common heterodimeric partner for a number of nuclear receptors, including PPARs, LXRs, FXRs, and VDRs. RXR heterodimers act as ligand-dependent transcriptional regulators and increase the DNA-binding efficiency of its partner. In the early stage of DN, the target genes of RXR-α were all downregulated. Collectively, these data indicated that the transcriptional activities of PPARs, FXRs, and VDRs might also decrease in the glomeruli of DN patients.
SMAD2, SMAD3, STAT1 and other 3 TFs were enriched in DEGs between the late and early stage of DN. The target genes of these three TFs were all upregulated, which may indicate that the transcriptional activity of the three TFs increased during the progression of DN.
SMAD2 and SMAD3 are TFs activated by TGF-β receptor signaling, which is a leading candidate to mediate the progression of DN. SMAD2 and SMAD3 are phosphorylated by TGF-β type I receptor, homodimerize, and subsequently bind to SMAD4. The heteromeric complex of SMAD2 or SMAD3 and SMAD4 translocates to the nucleus, where it regulates the transcription of target genes.[10] The Janus kinase (JAK) family substrate STAT1 is tyrosine phosphorylated and translocates to the nucleus in response to angiotensin II.[11]
A number of DCGs and “Regulation-switch” DCLs in pairwise comparisons between Early and Ctrl were more than those between Late and Early, which indicated that the change of gene coexpression relationships in the early stage is more than that in the progression stage. In summary, we observed that the gene expression changes mainly occurred during the progression of DN, while the gene coexpression relationship changes mainly occurred in the early stage of DN.
After differential regulatory network construction and TED scoring, we found that the interferon regulatory factor (IRF) family members, including IRF1, IRF3, IRF5, IRF7, and IRF9, are important TFs in the early stage of DN. Among them, IRF1 and IRF9 are DRGs, which indicate that IRF1 and IRF9 might be posttranslationally modified in the early stage of DN.
The mammalian IRF family has nine members, IRF-1 through IRF-9. IRFs are critical regulators of immune responses and immune cell development, and abnormalities in IRF expression and/or function have been increasingly linked to numerous diseases. Interestingly, IRFs are also involved in the pathogenesis of metabolic diseases.[12,13] It is not surprising that a regulator originally considered to be involved in the immune system has been subsequently shown to play a role in metabolism, as numerous studies have reported that the immune and metabolic systems are intrinsically interconnected.[14]
The high glucose-induced proliferation of vascular smooth muscle cells (VSMCs) plays an important role in the development of diabetic angiopathy. IRF1 is a positive regulator of the high glucose-induced proliferation of VSMCs.[12] Under normal glucose conditions, IRF1 overexpression led to downregulation of cyclin D1/CDK4 and inhibited cell cycle progression in VSMCs. In high glucose conditions, IRF1 overexpression led to an upregulation of cyclin E/CDK2 and an acceleration of cell cycle progression.[15] The opposite effects of IRF1 under normal and high glucose conditions are consistent with our finding that IRF1 is a DRG.
IRF9 is primarily localized in the nucleus, and by promoting PPARα transactivation, it accelerates lipid catabolism and mitigates hepatic steatosis, suggesting a key role for IRF9 in metabolic functions that is independent of its role in immunity.[13]
Our results show that IRF9, STAT2, and NFKB2 are important TFs during the progression of DN. Among them, STAT2 is a DRG, which indicates that STAT2 might be posttranslationally modified during the progression of DN.
Excessive cellular growth is a major contributor to pathological changes associated with DN. In particular, high glucose-induced growth of glomerular mesangial cells is a characteristic feature of diabetes-induced renal complications. High glucose and ANG II activate intracellular signaling processes, including the polyol pathway and the generation of reactive oxygen species. These pathways activate the JAK/signal transducers and activators of transcription (STAT) signaling cascades in glomerular mesangial cells. Activation of the JAK/STAT signaling cascade can stimulate excessive proliferation and growth of glomerular mesangial cells, contributing to DN.[11]
Nuclear factor-κB (NF-κB) is the most important TF in the pathogenesis of DN. NF-κB1 or NF-κB2 is bound to REL (V-rel avian reticuloendotheliosis viral oncogene homolog), RELA, and RELB to form the NF-κB complex, which binds to the promoter regions of several genes, including those encoding TGF-β1, AKR1B1 (aldo-keto reductase family 1, member B1), CCL2 (CC chemokine ligand 2), and ICAM1 (intercellular adhesion molecule 1). NF-κB is also integrated in various biological pathways that are functionally involved in the pathogenesis of DN, such as PKCβ, RAS, AGE accumulation, and oxidative stress.
There were two strengths in our study. First, we enrolled patients at two stages of DN, especially including the early stage of DN, which provided us a great opportunity to investigate the special gene regulatory pattern in the initiation stage of DN. Second, we utilized the DCEA and DRA to explore the changes in transcription regulatory relationships in different stages of DN, which might lead to a greater understanding of the molecular mechanism of DN. One limitation of our studies was that the study was performed in Chinese Han patients; thus, the results might not be applied to other ethnic groups. Another limitation was that the results should be validated in cell culture and animal models.
In summary, this study utilized the gene expression profiles of glomeruli from DN patients to further investigate the potential transcriptional regulatory mechanisms in the early and late stages of the disease. We observed that the gene coexpression relationship changes occurred mainly in the early stage of DN, while the gene expression changes occurred mainly during the progression of DN. Although the results should be further validated, this study offered additional insights into the transcriptional regulatory mechanisms of DN.
Supplementary information is linked to the online version of the paper on the Chinese Medical Journal website.
Financial support and sponsorship
The work was supported by grants from the National Key Research and Development Program of China (2016YFC0904103), the National Natural Science Foundation of China (No. 81500556, No. 81500548, and No. 81500547), and the Innovation Capability Development Project of Jiangsu Province (No. BM2015004).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Ning-Ning Wang
REFERENCES
- 1.Zhang L, Long J, Jiang W, Shi Y, He X, Zhou Z, et al. Trends in chronic kidney disease in China. N Engl J Med. 2016;375:905–6. doi: 10.1056/NEJMc1602469. doi: 10.1056/NEJMc1602469. [DOI] [PubMed] [Google Scholar]
- 2.Yang YZ, Wang JW, Wang F, Wu YT, Zhao HY, Chen M, et al. Incidence, development, and prognosis of diabetic kidney disease in China: Design and methods. Chin Med J. 2017;130:199–202. doi: 10.4103/0366-6999.198002. doi: 10.4103/0366-6999.198002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu BH, Yu H, Tu K, Li C, Li YX, Li YY. DCGL: An R package for identifying differentially coexpressed genes and links from gene expression microarray data. Bioinformatics. 2010;26:2637–8. doi: 10.1093/bioinformatics/btq471. doi: 10.1093/bioinformatics/btq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi J, Jiang S, Qiu D, Le W, Wang X, Lu Y, et al. Rapid identification of potential drugs for diabetic nephropathy using whole-genome expression profiles of glomeruli. Biomed Res Int 2016. 2016:1634730. doi: 10.1155/2016/1634730. doi: 10.1155/2016/1634730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan Y, Jiang S, Hou Q, Qiu D, Shi J, Wang L, et al. Dissection of glomerular transcriptional profile in patients with diabetic nephropathy: SRGAP2a protects podocyte structure and function. Diabetes. 2018;67:717–30. doi: 10.2337/db17-0755. doi: 10.2337/db17-0755. [DOI] [PubMed] [Google Scholar]
- 6.Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, et al. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–8. doi: 10.1093/nar/gkg108. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang XX, Jiang T, Levi M. Nuclear hormone receptors in diabetic nephropathy. Nat Rev Nephrol. 2010;6:342–51. doi: 10.1038/nrneph.2010.56. doi: 10.1038/nrneph.2010.56. [DOI] [PubMed] [Google Scholar]
- 8.Holmkvist J, Cervin C, Lyssenko V, Winckler W, Anevski D, Cilio C, et al. Common variants in HNF-1 alpha and risk of type 2 diabetes. Diabetologia. 2006;49:2882–91. doi: 10.1007/s00125-006-0450-x. doi: 10.1007/s00125-006-0450-x. [DOI] [PubMed] [Google Scholar]
- 9.Kuipers I, van der Harst P, Kuipers F, van Genne L, Goris M, Lehtonen JY, et al. Activation of liver X receptor-alpha reduces activation of the renal and cardiac renin-angiotensin-aldosterone system. Lab Invest. 2010;90:630–6. doi: 10.1038/labinvest.2010.7. doi: 10.1038/labinvest.2010.7. [DOI] [PubMed] [Google Scholar]
- 10.Reeves WB, Andreoli TE. Transforming growth factor beta contributes to progressive diabetic nephropathy. Proc Natl Acad Sci U S A. 2000;97:7667–9. doi: 10.1073/pnas.97.14.7667. doi: 10.1073/pnas.97.14.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marrero MB, Banes-Berceli AK, Stern DM, Eaton DC. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am J Physiol Renal Physiol. 2006;290:F762–8. doi: 10.1152/ajprenal.00181.2005. doi: 10.1152/ajprenal.00181.2005. [DOI] [PubMed] [Google Scholar]
- 12.Yu S, Xi Z, Hai-Yan C, Ya-Li C, Shao-Hu X, Chuan-Sen Z, et al. Interferon regulatory factor-1 as a positive regulator for high glucose-induced proliferation of vascular smooth muscle cells. J Cell Biochem. 2012;113:2671–8. doi: 10.1002/jcb.24142. doi: 10.1002/jcb.24142. [DOI] [PubMed] [Google Scholar]
- 13.Wang XA, Zhang R, Jiang D, Deng W, Zhang S, Deng S, et al. Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology. 2013;58:603–16. doi: 10.1002/hep.26368. doi: 10.1002/hep.26368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8:1–26. doi: 10.1038/nri2449. doi: 10.1038/nri2449. Nutrient. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Liu L, Chen C, Chi YL, Yang XQ, Xu Y, et al. Interferon regulatory factor-1 together with reactive oxygen species promotes the acceleration of cell cycle progression by up-regulating the cyclin E and CDK2 genes during high glucose-induced proliferation of vascular smooth muscle cells. Cardiovasc Diabetol. 2013;12:147. doi: 10.1186/1475-2840-12-147. doi: 10.1186/1475-2840-12- [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GO categories and KEGG pathways enriched in the glomerular DEGs between the early stage of DN patients and the control group. (a) GO cellular component; (b) GO molecular function; (c) GO biological process; (d) KEGG pathways. GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; DEGs: Differentially expressed genes; DN: Diabetic nephropathy.
GO categories and KEGG pathways enriched in the glomerular DEGs between the late and early stage of DN patients. (a) GO cellular component; (b) GO molecular function; (c) GO biological process; (d) KEGG pathways. GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; DEGs: Differentially expressed genes; DN: Diabetic nephropathy.