

Abstract
Quinazolin-4(3H)-ones have been prepared in one step from 2-aminobenzamides and orthoesters in the presence of acetic acid. Simple 2-aminobenzamides were easily converted to the heterocycles by refluxing in absolute ethanol with 1.5 equivalents of the orthoester and 2 equivalents of acetic acid for 12–24 h. Ring-substituted and hindered 2-aminobenzamides as well as cases incorporating an additional basic nitrogen required pressure tube conditions with 3 equivalents each of the orthoester and acetic acid in ethanol at 110 °C for 12–72 h. The reaction was tolerant towards functionality on the benzamide and a range of structures was accessible. Workup involved removal of the solvent under vacuum and either recrystallization from ethanol or trituration with ether-pentane. Several 5,6-dihydropyrimidin-4(3H)-ones were also prepared from 3-amino-2,2-dimethylpropionamide. All products were characterized by melting point, FT-IR, 1H-NMR, 13C-NMR, and HRMS.
Keywords: quinazolin-4(3H)-ones; 5,6-dihydropyrimidin-4(3H)-ones; heterocycle synthesis; 2-aminobenzamides; orthoesters
1. Introduction
Quinazolin-4(3H)-ones are valuable scaffolds in pharmaceutical chemistry due to their diverse biological activities. Drugs possessing the quinazolinone unit are used as anti-anxiety, antibacterial anticonvulsant, antifungal, anti-inflammatory, and antitumor agents [1]. Some examples of 2-alkyl- and 2-arylquinazolin-4(3H)-ones currently on the market include the sedative-hypnotics methaqualone (quaalude, 1) [2] and afloqualone (2) [3,4] as well as the antifungal albaconazole (3) [5]. Additionally, compound 4 [6] is currently under evaluation as an antibacterial agent against both Gram-positive and Gram-negative organisms.
In this study, we report the synthesis of 2-alkyl- and 2-arylquinazolin-4(3H)-ones from inexpensive 2-aminobenzamide and orthoester substrates without the need for metal catalysts or Lewis acids. The process is similar to earlier acid-catalyzed processes reported by our group for the preparation of 2-substituted 2,3-dihydroquinazolin-4(1H)-ones [7] and benzimidazoles [8] as well as for a synthesis of benzimidazo[1,2-c]quinazolines described by others [9]. Using the conditions disclosed herein, a small library of substituted quinazolin-4(3H)-ones has been generated with high yields (Figure 1). We also report the synthesis of a modest selection of 5,6-dihydropyrimidin-4(3H)-ones from 3-amino-2,2-dimethyl-propionamide under identical conditions.
Figure 1.

Bioactive quinazolin-4(3H)-ones.
To date, orthoesters have received minimal use for the preparation of these systems. Syntheses that did employ these building blocks focused almost exclusively on the use of triethyl orthoformate to prepare derivatives without C2 substitution [5,10]. Although the current method is somewhat limited by the sparse number of available orthoesters, the simplicity of our approach and its ability to provide a wide range of targets makes it a valuable addition to the field.
The current reaction is a variant of the classical Niementowski quinazoline synthesis, which involved the cyclocondensation of an N-alkylamide with anthranilic acid [11]. The original conditions for this transformation, however, proved somewhat limiting as they required extended heating at >200 °C and resulted in significant decarboxylation of the acid. Further work showed that methyl anthranilate was more stable at 200 °C and afforded higher yields with less degradation of the substrate [12]. The decarboxylation problem was also minimized through the application of microwave conditions, which considerably reduced the reaction time [13,14,15], and the use of thioamides, which permitted lower reaction temperatures [16]. Later efforts found that isatoic anhydride with amines and orthoesters under neat conditions gave clean cyclization to the quinazolinones at temperatures of 120 °C [17]. In a related procedure, heating isatoic anhydride with benzylamines at 120 °C in ionic liquids led to oxidation of the benzylic amine to the imine, followed by cyclization [18]. More recently, phosphoric acid was employed to promote the synthesis of 2,3-disubstituted quinazolin-4(3H)-ones from 2-amino-N- substituted benzamides and β-ketoesters [19]. Likewise, n-propanephosphonic anhydride was used as a condensing agent to assemble 2-fluoroalkyl-substituted derivatives from fluoroalkanoic acids and anthranilic acid [20]. Along a different line, two separate reports detailed the synthesis of quinazolinones by oxidative cyclization of aldehydes with 2-aminobenzamide under aerobic conditions [6] and in the presence of tert-butyl hydroperoxide (TBHP)-potassium iodide [21]. A similar oxidative condensation of primary alcohols with 2-aminobenzamide to generate quinazolinones was promoted by TBHP alone [22] and together with iodine and dimethyl sulfoxide [23,24]. Numerous metals such as manganese [25], iron [26], nickel [27], zinc [28], ruthenium [29], iridium [30], and platinum [31] in conjunction with TBHP were also reported to promote this reaction. The option of using primary alcohols for this transformation dramatically increased the scope of the synthesis due to the vast array of available substrates. Finally, other methods involved the reaction of lithium 2-(diethylaminocarbonyl)anilide with aryl- and alkylnitriles [32] as well as various copper [33,34,35,36,37,38,39,40] and palladium [2,41,42,43,44] catalyzed heterocyclizations.
2. Results and Discussion
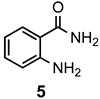
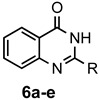
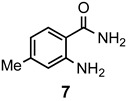
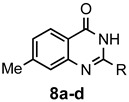
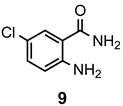
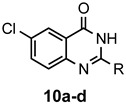
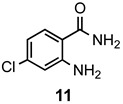
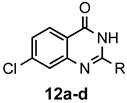
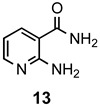
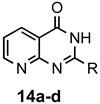
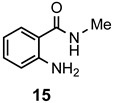
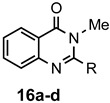
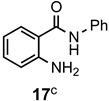
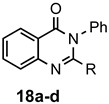
The results of our study are summarized in Table 1. The products listed illustrate the breadth of systems accessible using our procedure. Several acid catalysts were evaluated, but the most convenient and inexpensive was acetic acid. For 2-aminobenzamide (5), the reaction required heating 1 equivalent of the amide with 1.5–2 equivalents of the orthoester and 2 equivalents of acetic acid in absolute ethanol at reflux (78 °C) for 12–24 h (Method 1). Less reactive ring-substituted derivatives (7, 9, and 11), more hindered 2-amino-N-substituted benzamides (15 and 17) and substrates incorporating a second basic nitrogen (13 and 19) required pressure tube conditions with 3 equivalents of the orthoester and 3 equivalents of acetic acid in ethanol at 110 °C for 12–72 h (Method 2). Comparisons of Methods 1 and 2 are shown in the Table for several substrates and clearly demonstrate the advantage of using higher temperatures. Beyond these few examples, however, additional comparisons were not possible as pure material was often difficult to isolate from reactions run at 78 °C. Two examples that did permit comparison were the chloro-substituted 2-aminobenzamides 9 and 11 with triethyl orthobenzoate. Using Method 1, this reaction gave low conversions, and except for a small amount of product that initially crystallized from the cooling reaction mixture, the quinazolinones were contaminated with unreacted starting material and inseparable by-products. Increasing the stoichiometry of the orthoester under these conditions failed to improve the conversion to a satisfactory level. Alternatively, increasing the amount of acid to 6 equivalents, improved the yields of 10d and 12d to 56% and 77%, respectively, but repeated crystallizations were required for purification. Reacting these substrates according to Method 2, with 2–3 equivalents of the orthoester and 3 equivalents of acetic acid, however, gave nearly quantitative conversions to the products. Under this protocol, trituration of the crude products with 5% ether in pentane was sufficient to obtain spectroscopically pure material. Clearly, Method 2 proved superior for the broadest range of substrates. All reported yields were isolated, but not all were fully optimized. Procedures and copies of the 1H and 13C-NMR spectra are given in the Supplemental Material.
Table 1.
Synthesis of quinazolin-4(3H)-ones.

| Substrate | Product | R | Method a | Yield (%) |
|---|---|---|---|---|
|
|
a: Me b: Et c: Pr d: P he: H |
1 1 1 1 1 |
92 89 93 81 87 |
|
|
a: Me b: Et c: Pr d: Ph |
2 2 2 2 |
90 91 ND b 84 |
|
|
a: Me b: Et c: Pr d: Ph |
2 2 2 1(2) |
92 92 88 33(97) |
|
|
a: Me b: Et c: Pr d: Ph |
2 2 2 1(2) |
94 93 89 7(98) |
|
|
a: Me b: Et c: Pr d: Ph |
1(2) 1(2) 1(2) 1(2) |
71(86) 70(92) 55(87) 68(80) |
|
|
a: Me b: Et c: Pr d: Ph |
2 2 2 2 |
87 83 ND b 89 |
|
|
a: Me b: Et c: Pr d: Ph |
2 2 2 2 |
95 92 ND b 95 |
|
|
a: Me b: Et c: Pr d: Ph |
1(2) 2 2 2 |
21(82) 80 ND b 95 |
a For 1 equiv of the 2-aminobenzamide: Method 1: 1.5 equiv of orthoester, 2 equiv of AcOH, 3 mL of EtOH, 78 °C, 12–24 h; Method 2: 2–3 equiv of orthoester, 3 equiv of AcOH, 3 mL of EtOH, 110 °C, pressure tube, 12–72 h; b ND = not done; c Prepared from 2-nitrobenzoic acid as described in the experimental section.
The yields using Method 2 were generally high for all of the substrates evaluated. While reactions were followed by thin layer chromatography, attempts at preparative purifications using silica gel often led to decomposition of the products, even when the eluting solvent contained 1% triethylamine. Sublimation also afforded limited success. Thus, product purification was restricted to crystallization from ethanol or trituration with 5% ether in hexanes. Fortunately, all of the derivatives prepared were highly crystalline solids, which facilitated purification using these techniques.
Finally, we were able to extend the cyclization process to the preparation of a small family of 5,6-dihydropyrimidin-4(3H)-ones from 3-amino-2,2-dimethylpropionamide (21) (see Scheme 1). These compounds were prepared by Method 3 using 1.5 equivalents of the orthoester and 2 equivalents of acetic acid in ethanol at reflux for 24 h. Cyclizations to produce 22a–d were assisted by the Thorpe–Ingold (gem-dialkyl) effect [45,46], but the yields were modest due to difficulties in crystallizing the final products. Since the pyrimidinones were water-soluble and hygroscopic, it was necessary to utilize dry solvents for the crystallization. Best results were achieved by adding dry chloroform to the crude product and allowing the mixture to sit under nitrogen for four to seven days. The use of higher temperatures (Method 2) for these substrates had little effect on the yields.
Scheme 1.

Synthesis 5,6-dihydropyrimidin-4(3H)-ones.
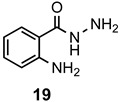
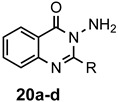
There are a number of very positive features in this synthesis. The reaction was free of metals and corrosive Lewis acids, requiring only a 2–3-fold excess of acetic acid to promote the reaction. This simplified the purification of the final products and eliminated the possibility of residual contaminants. Both sets of conditions were tolerant of alkyl, halide and basic functional groups present on the 2-aminobenzamide reacting partner and a small library of substituted derivatives was prepared. Employing Method 2, it was possible to obtain high yields of quinazolinones from ring-substituted 2-aminobenzamides 7, 9, and 11. This procedure also promoted excellent conversions of relatively hindered 2-amino-N-methyl- and 2-amino-N-phenylbenzamides 15 and 17, respectively, to 2,3-disubstituted quinazolinones 16 and 18. These hindered ring closures were likely facilitated by the rigid aromatic scaffold, which held the ortho-disposed reactive centers in close proximity. Finally, 13 and 19, which both possessed basic nitrogen groups, gave superb yields as well. In ring closures of hydrazide 19, it was noted that the less reactive amide nitrogen cyclized in preference to the more nucleophilic terminal nitrogen of the hydrazide to give exclusively the six-membered cyclic product, with none of the seven-membered ring observed. The preparation of the 5,6-dihydropyrimidin-4(3H)-ones was more limited due to challenges during the purification process. Additional experiments attempted to cyclize 3-aminopropionamide, which was commercially available as the hydrochloride salt. This substrate, however, lacked the gem-dimethyl moiety and did not undergo significant ring closure.
A plausible mechanism for quinazolinone formation is depicted for the reaction of 2-aminobenzamide with triethyl orthoacetate (see Figure 2). The process involves initial protonation of the orthoester and loss of ethanol to afford the stabilized carbocation A. Subsequent attack on A by the aniline amino group, proton exchange, and loss of a second molecule of ethanol would then yield the iminium ion B. Finally, closure of the amide nitrogen on the iminium carbon, proton exchange, and loss of ethanol would generate the quinazolinone product. A similar mechanism is likely operating for the closure of the 5,6-dihydropyrimidin-4(3H)-ones.
Figure 2.

Mechanism for quinazolin-4(3H)-one formation.
3. Experimental Section
3.1. General Methods
Most commercial chemicals and solvents were used as received, except for 2-amino-4-methylbenzamide, which required recrystallization from ethanol. 2-Amino-N-phenylbenzamide was prepared from 2-nitrobenzoic acid, according to the following sequence: (i) SOCl2 (2.0 equiv), benzene, reflux, 2 h; (ii) PhNH2 (1.1 equiv), CH2Cl2, Et3N (2.0 equiv), 0 °C to r.t., 4 h; (iii) Fe (3.2 equiv)/NH4Cl (1.0 equiv), aq EtOH, 85 °C, 2 h [47]; 82% overall, white solid, m.p. 123–125 °C (lit [48] m.p. 128–129.5 °C). The spectral data matched those reported [48].
Unless otherwise indicated, all reactions were carried out under dry N2 in oven-dried glassware. Reactions were monitored by thin layer chromatography on silica gel GF plates (Analtech no. 21521, Newark, DE, USA). Band elution was monitored using a hand-held UV lamp (Fisher Scientific, Pittsburgh, PA, USA). Melting points were obtained using a MEL-TEMP apparatus (Cambridge, MA, USA) and are uncorrected. FT-IR spectra were run using a Varian Scimitar FTS 800 spectrophotometer (Randolph, MA, USA) as thin films or nujol mulls on NaCl disks. 1H- and 13C-NMR spectra were measured using a Bruker Avance 400 system (Billerica, MA, USA) in the indicated solvents at 400 MHz and 101 MHz, respectively, with (CH3)4Si as the internal standard; coupling constants (J) are given in Hz. High-resolution mass spectra (HRMS-ESI) were obtained using a Thermo LTQ-Orbitrap XL mass spectrometer (Thermo Scientific, Waltham, MA, USA).
3.2. General Procedures to Prepare Quinazolin-4(3H)-ones
3.2.1. Method 1
The orthoester (1.5 equiv) was added to a mixture of the 2-aminobenzamide (1.0 equiv) in absolute ethanol (3 mL). Glacial acetic acid (2 equiv) was added and the reaction was heated at reflux for 12–24 h. The reaction mixture was cooled and concentrated under vacuum. If the crude product was pure by 1H-NMR, it was triturated with 5% ether in pentane. If it was not pure, it was recrystallized from ethanol. In some cases, it was necessary to remove excess orthoester under high vacuum at 50 °C prior to purification. The following compounds were prepared:
2-Methylquinazolin-4(3H)-one (6a). This compound was prepared from 2-aminobenzamide (125 mg, 0.92 mmol), triethyl orthoacetate (224 mg, 253 µL, 1.38 mmol) and acetic acid (110 mg, 105 µL, 1.84 mmol) in refluxing absolute ethanol (3 mL). Yield: 135 mg (92%) as an off-white solid, m.p. 236–238 °C (lit [40] m.p. 242–244 °C); IR (CHCl3): 3176, 1674 cm−1; 1H-NMR (400 MHz, CDCl3): δ 11.2 (br s, 1H), 8.28 (dd, J = 8.0, 1.0 Hz, 1H), 7.78 (td, J = 8.1, 1.5 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.48 (td, J = 8.0, 1.0 Hz, 1H), 2.58 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 163.9, 153.1, 149.6, 135.1, 127.2, 126.7, 126.4, 120.5, 22.4; HRMS (ESI): m/z for C9H8N2O [M + 1]+: calcd.: 161.0715; found: 161.0714.
2-Ethylquinazolin-4(3H)-one (6b). This compound was prepared from 2-aminobenzamide (125 mg, 0.92 mmol), triethyl orthopropionate (244 mg, 278 µL, 1.38 mmol) and acetic acid (110 mg, 105 µL, 1.84 mmol) in refluxing absolute ethanol (3 mL). Yield: 155 mg (89%) as an off-white solid, m.p. 235–237 °C (lit [15] m.p. 238–240 °C); IR (CHCl3): 3169, 1681 cm−1; 1H-NMR (400 MHz, CDCl3): δ 11.2 (br s, 1H), 8.29 (dd, J = 8.0, 1.2 Hz, 1H), 7.78 (ddd, J = 8.3, 7.0, 1.6 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.47 (ddd, J = 8.1, 7.1, 1.2 Hz, 1H), 2.82 (q, J = 7.3 Hz, 2H), 1.45 (t, J = 7.4 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 164.0, 157.5, 149.5, 134.9, 127.4, 126.6, 126.4, 120.8, 29.3, 11.6; HRMS (ESI): m/z for C10H10N2O [M + 1]+: calcd.: 175.0871; found: 175.0873.
2-Propylquinazolin-4(3H)-one (6c). This compound was prepared from 2-aminobenzamide (125 mg, 0.92 mmol), triethyl orthobutyrate (262 mg, 298 µL, 1.38 mmol) and acetic acid (110 mg, 105 µL, 1.84 mmol) in refluxing absolute ethanol (3 mL). Yield: 160 mg (93%) as an off-white solid, m.p. 212–214 °C (lit [15] m.p. 210–212 °C); IR (CHCl3): 3165, 1675 cm−1; 1H-NMR (400 MHz, CDCl3): δ 11.2 (br s, 1H), 8.29 (d, J = 7.8 Hz, 1H), 7.77 (t, J = 7.8 Hz, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 2.76 (t, J = 7.4 Hz, 2H), 1.92 (sextet, J = 7.4 Hz, 2H), 1.08 (t, J = 7.3 Hz, 3H) ; 13C-NMR (101 MHz, CDCl3): δ 169.3, 156.6, 149.5, 134.9, 127.4, 126.6, 126.4, 120.7, 38.0, 21.1, 13.9; HRMS (ESI): m/z for C11H12N2O [M + 1]+: calcd.: 189.1028; found: 189.1025.
2-Phenylquinazolin-4(3H)-one (6d). This compound was prepared from 2-aminobenzamide (125 mg, 0.92 mmol), triethyl orthobenzoate (309 mg, 312 µL, 1.38 mmol) and acetic acid (110 mg, 105 µL, 1.84 mmol) in refluxing absolute ethanol (3 mL). Yield: 134 mg (81%) as an off-white solid, m.p. 234–236 °C (lit [40] m.p. 233–235 °C); IR (CHCl3): 3196, 1668 cm−1; 1H-NMR (400 MHz, CDCl3): δ 11.1 (br s, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.21 (m, 2H), 7.86–7.78 (complex, 2H), 7.61–7.57 (complex, 3H), 7.52 (ddd, J = 7.9, 6.8, 1.2 Hz, 1H); 13C-NMR (101 MHz, CDCl3): δ 169.9, 151.5, 149.4, 134.9, 132.8, 131.7, 129.2, 128.1, 127.1, 126.9, 126.5, 121.0; HRMS (ESI): m/z for C14H10N2O [M + 1]+: calcd.: 223.0871; found: 223.0870.
Quinazolin-4(3H)-one (6e). This compound was prepared from 2-aminobenzamide (125 mg, 0.92 mmol), triethyl orthoformate (204 mg, 229 µL, 1.38 mmol) and acetic acid (110 mg, 105 µL, 1.84 mmol) in refluxing absolute ethanol (3 mL). Yield: 117 mg (87%) as an off-white solid, m.p. 212–214 °C (lit [16] m.p. 215.5–216.5 °C); IR (CHCl3): 3202, 1664 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.31 (dd, J = 8.0, 1.0 Hz, 1H), 8.08 (s, 1H), 7.28 (td, J = 8.1, 1.4 Hz, 1H), 7.77 (dd, J = 8.1, 1.0 Hz, 1H), 7.55 (td, J = 7.4, 1.4 Hz, 1H), NH not observed; 13C-NMR (101 MHz, CDCl3): δ 162.8, 149.0, 143.5, 135.1, 128.0, 127.6, 126.6, 122.7; HRMS (ESI): m/z for C8H6N2O [M + 1]+: calcd.: 147.0558; found: 147.0557.
3.2.2. Method 2
The 2-aminobenzamide (1 equiv), the orthoester (2–3 equiv) and absolute ethanol (2–3 mL) were placed in a 15-mL Chemglass screw-cap pressure tube (No. CG-1880-01, Chemglass, Vineland, NJ, USA). Glacial acetic acid (3 equiv) was added, N2 was introduced to the vessel and the cap was tightened. The vessel was heated at 110 °C for 12–72 h, then cooled and concentrated to give the quinazolinone, which was purified by crystallization from absolute ethanol or trituration from 5% ether in pentane. The following compounds were prepared:
2,7-Dimethylquinazolin-4(3H)-one (8a). This compound was prepared from 2-amino-4-methylbenzamide (100 mg, 0.67 mmol), triethyl orthoacetate (217 mg, 245 µL, 1.34 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h to give 105 mg (90%) as a white solid, m.p. 260–262 °C (lit [49] m.p. 263–264 °C); IR (CHCl3): 3169, 1677 cm−1; 1H-NMR (400 MHz, CDCl3): δ 11.6 (br s, 1H), 8.16 (d, J = 8.1 Hz, 1H), 7.47 (br s, 1H), 7.29 (dd, J = 8.1, 1.0 Hz, 1H), 2.57 (s, 3H), 2.51 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 163.9, 153.2, 149.5, 146.0, 128.0, 126.8, 126.1, 117.9, 22.1, 22.0; HRMS (ESI): m/z for C10H10N2O [M + 1]+: calcd.: 175.0871; found: 175.0867.
2-Ethyl-7-methylquinazolin-4(3H)-one (8b). This compound was prepared from 2-amino-4-methylbenzamide (100 mg, 0.67 mmol), triethyl orthopropionate (236 mg, 269 µL, 1.34 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h. Yield: 114 mg (91%) as a white solid, m.p. 244–245 °C; IR (CHCl3): 3178, 1674 cm−1; 1H-NMR (400 MHz, CDCl3): δ 11.2 (br s, 1H), 8.17 (d, J = 8.1 Hz, 1H), 7.50 (br s, 1H), 7.30 (dd, J = 8.1, 0.9 Hz, 1H), 2.80 (q, J = 7.6 Hz, 2H), 2.51 (s, 3H), 1.43 (t, J = 7.6 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 163.8, 157.4, 149.5, 145.8, 128.0, 127.0, 126.1, 118.2, 29.2, 22.0, 11.5; HRMS (ESI): m/z for C11H12N2O [M + 1]+: calcd.: 189.1028; found: 189.1031.
7-Methyl-2-phenylquinazolin-4(3H)-one (8d). This compound was prepared from 2-amino-4-methylbenzamide (100 mg, 0.67 mmol), triethyl orthobenzoate (300 mg, 303 µL, 1.34 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 48 h. Yield: 133 mg (84%) as a light yellow solid, m.p. 239–241 °C (lit [40] m.p. 240–241 °C); IR (CHCl3): 3156, 1672 cm−1; 1H-NMR (400 MHz, CDCl3): δ 10.9 (br s, 1H), 8.21 (d, J = 8.1 Hz, 1H), 8.18 (m, 2H), 7.64 (br s, 1H), 7.61–7.56 (complex, 3H), 7.33 (dd, J = 8.2, 0.9 Hz, 1H), 2.54 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 163.3, 151.6, 149.6, 146.0, 132.9, 131.6, 1291, 128.5, 127.8, 127.2, 126.2, 118.5, 22.0; HRMS (ESI): m/z for C15H12N2O [M + 1]+: calcd.: 237.1028; found: 237.1023.
6-Chloro-2-methylquinazolin-4(3H)-one (10a). This compound was prepared from 2-amino-5-chlorobenzamide (100 mg, 0.59 mmol) triethyl orthoacetate (191 mg, 216 µL, 1.18 mmol) and acetic acid (106 mg, 101 µL, 1.77 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 106 mg (92%) as a light tan powder, m.p. 288–290 °C (lit [49] m.p. 289–291 °C); IR (CHCl3): 3166, 1693, 1631 cm−1; 1H-NMR (400 MHz, CDCl3): δ 12.4 (br s, 1H), 8.00 (d, J = 2.5 Hz, 1H), 7.79 (dd, J = 8.7, 2.5 Hz, 1H), 7.59 (d, J = 8.7 Hz, 1H), 2.35 (s, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 160.7, 154.9, 147.7, 134.4, 130.1, 128.9, 124.7, 121.9, 21.5; HRMS (ESI): m/z for C9H735ClN2O [M + 1]+: calcd.: 195.0325; found: 195.0323.
6-Chloro-2-ethylquinazolin-4(3H)-one (10b). This compound was prepared from 2-amino-5-chlorobenzamide (100 mg, 0.59 mmol), triethyl orthopropionate (208 mg, 237 µL, 1.18 mmol) and acetic acid (106 mg, 101 µL, 1.77 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 114 mg (92%) as a light tan powder, m.p. 262–264 °C; IR (nujol): 3169, 1681, 1627 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.3 (br s, 1H), 8.01 (d, J = 2.5 Hz, 1H), 7.79 (dd, J = 8.7, 2.5 Hz, 1H), 7.62 (d, J = 8.7 Hz, 1H), 2.62 (q, J = 7.5 Hz, 2H), 1.24 (t, J = 7.5 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 160.8, 159.0, 147.7, 134.3, 130.1, 129.1, 124.7, 122.1, 27.8, 11.2; HRMS (ESI): m/z for C10H935ClN2O [M + 1]+: calcd.: 209.0482; found: 209.0484.
6-Chloro-2-propylquinazolin-4(3H)-one (10c). This compound was prepared from 2-amino-5-chlorobenzamide (100 mg, 0.59 mmol), triethyl orthobutyrate (224 mg, 255 µL, 1.18 mmol) and acetic acid (106 mg, 101 µL, 1.77 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 116 mg (88%) as a light tan powder, m.p. 252–254 °C (lit [33] m.p. 257–259 °C); IR (nujol): 3165, 1681, 1618 cm−1; 1H-NMR (400 MHz, CDCl3): δ 12.4 (br s, 1H), 8.01 (d, J = 2.4 Hz, 1H), 7.80 (dd, J = 8.7, 2.4 Hz, 1H), 7.62 (d, J = 8.7 Hz, 1H), 2.57 (t, J = 7.4 Hz, 2H), 1.74 (sextet, J = 7.4 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 160.8, 158.0, 147.7, 134.4, 130.1, 129.1, 124.7, 122.1, 36.3, 20.1, 13.5; HRMS (ESI): m/z for C11H1135ClN2O [M + 1]+: calcd.: 223.0638; found: 223.0637.
6-Chloro-2-phenylquinazolin-4(3H)-one (10d). This compound was prepared from 2-amino-5-chlorobenzamide (100 mg, 0.59 mmol), triethyl orthobenzoate (264 mg, 267 µL, 1.18 mmol) and acetic acid (106 mg, 101 µL, 1.77 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 147 mg (97%) as white crystals, m.p. 292–293 °C (lit [37] m.p. 295–296 °C); IR (nujol): 3151, 1679 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.7 (br s, 1H), 8.18 (dm, J = 6.8 Hz, 2H), 8.10 (d, J = 2.5 Hz, 1H), 7.87 (dd, J = 8.7, 2.5 Hz, 1H), 7.77 (d, J = 8.7 Hz, 1H), 7.64–7.53 (complex, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 161.8, 153.3, 148.0, 135.2, 132.9, 132.1, 131.2, 130.2, 129.1, 128.3, 125.3, 122.7; HRMS (ESI): m/z for C14H935ClN2O [M + 1]+: calcd.: 257.0482; found: 257.0477.
7-Chloro-2-methylquinazolin-4(3H)-one (12a). This compound was prepared from 2-amino-4-chlorobenzamide (100 mg, 0.59 mmol) triethyl orthoacetate (191 mg, 216 µL, 1.18 mmol) and acetic acid (106 mg, 101 µL, 1.77 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 108 mg (94%) as a light tan powder, m.p. 293–295 °C (lit [38] m.p. 295–297 °C); IR (nujol): 3163, 1674, 1636, 1609 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.3 (br s, 1H), 8.06 (d, J = 8.5 Hz, 1H), 7.62 (dd, J = 1.6 Hz, 1H), 7.48 (d, J = 8.5, 1.7 Hz, 1H), 2.35 (s, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 161.1, 156.0, 150.1, 138.8, 127.8, 126.1, 125.7, 119.5, 21.5; HRMS (ESI): m/z for C9H735ClN2O [M + 1]+: calcd.: 195.0325; found: 195.0322.
7-Chloro-2-ethylquinazolin-4(3H)-one (12b). This compound was prepared 2-amino-4-chlorobenzamide (100 mg, 0.59 mmol), triethyl orthopropionate (208 mg, 237 µL, 1.18 mmol) and acetic acid (106 mg, 101 µL, 1.77 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 115 mg (93%) as a light tan powder, m.p. 235–237 °C (lit [29] m.p. 205–207 °C); IR (nujol): 3172, 1681, 1620, 1607 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.3 (br s, 1H), 8.07 (d, J = 8.5 Hz, 1H), 7.65 (dd, J = 2.0 Hz, 1H), 7.49 (dd, J = 8.5, 2.0 Hz, 1H), 2.62 (q, J = 7.5 Hz, 2H), 1.23 (t, J = 7.5 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 161.2, 160.1, 150.1, 138.9, 127.8, 126.2, 126.0, 119.7, 27.9, 11.2; HRMS (ESI): m/z for C10H935ClN2O [M + 1]+: calcd.: 209.0482; found: 209.0481.
7-Chloro-2-propylquinazolin-4(3H)-one (12c). This compound was prepared from 2-amino-4-chlorobenzamide (100 mg, 0.59 mmol), triethyl orthobutyrate (224 mg, 255 µL, 1.18 mmol) and acetic acid (106 mg, 101 µL, 1.77 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 117 mg (89%) as a light tan powder, m.p. 214–216 °C; IR (nujol): 3169, 1681, 1613, 1603 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.3 (br s, 1H), 8.06 (d, J = 8.5 Hz, 1H), 7.65 (dd, J = 1.8 Hz, 1H), 7.49 (dd, J = 8.5, 1.9 Hz, 1H), 2.57 (t, J = 7.4 Hz, 2H), 1.73 (sextet, J = 7.4 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 161.2, 159.1, 150.1, 138.9, 127.8, 126.2, 125.9, 119.6, 36.4, 20.2, 13.5; HRMS (ESI): m/z for C11H1135ClN2O [M + 1]+: calcd.: 223.0638; found: 223.0635.
7-Chloro-2-phenylquinazolin-4(3H)-one (12d). This compound was prepared from 2-amino-4-chlorobenzamide (100 mg, 0.59 mmol), triethyl orthobenzoate (264 mg, 267 µL, 1.18 mmol) and acetic acid (106 mg, 101 µL, 1.77 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 148 mg (98%) as light yellow crystals, m.p. 276–278 °C (lit [43] m.p. > 300 °C); IR (nujol): 3151, 1679 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.7 (br s, 1H), 8.18 (dm, J = 6.9 Hz, 2H), 8.15 (d, J = 8.5 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.64–7.53 (complex, 4H); 13C-NMR (101 MHz, DMSO-d6): δ 162.1, 154.3, 150.4, 139.6, 132.8, 132.2, 129.1, 128.42, 128.38, 127.3, 127.1, 120.3; HRMS (ESI): m/z for C14H935ClN2O [M + 1]+: calcd.: 257.0482; found: 257.0480.
2-Methylpyrido[2,3-d]pyrimidin-4(3H)-one (14a). This compound was prepared from 2-aminonicotinamide (126 mg, 0.92 mmol), triethyl orthoacetate (224 mg, 253 µL, 1.38 mmol) and acetic acid (165 mg, 158 µL, 2.76 mmol) in absolute ethanol (3 mL) at 110 °C for 12 h. Yield: 127 mg (86%) as an off-white solid, m.p. 261–263 °C (lit [14] m.p. 262–264 °C); IR (CHCl3): 3207, 1698 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.5 (br s, 1H), 8.90 (dd, J = 4.6, 1.9 Hz, 1H), 8.45 (dd, J = 7.8, 1.9 Hz, 1H), 7.48 (td, J = 7.8, 4.6 Hz, 1H), 2.40 (s, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 162.3, 158.9, 157.9, 155.7, 135.3, 121.7, 115.6, 21.8; HRMS (ESI): m/z for C8H7N3O [M + 1]+: calcd.: 162.0667; found: 162.0671.
2-Ethylpyrido[2,3-d]pyrimidin-4(3H)-one (14b). This compound was prepared from 2-aminonicotinamide (126 mg, 0.92 mmol) and triethyl orthopropionate (244 mg, 278 µL, 1.38 mmol) and acetic acid (165 mg, 158 µL, 2.76 mmol) in absolute ethanol (3 mL) at 110 °C for 12 h. Yield: 148 mg (92%) as an off-white solid, m.p. 197–198 °C; IR (CHCl3): 3198, 1674 cm−1; 1H-NMR (400 MHz, CDCl3): δ 11.6 (br s, 1H), 9.02 (dd, J = 4.6, 2.1 Hz, 1H), 8.62 (dd, J = 7.9, 2.1 Hz, 1H), 7.44 (dd, J = 7.9, 4.6 Hz, 1H), 2.91 (q, J = 7.6 Hz, 2H), 1.48 (t, J = 7.6 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 164.3, 161.2, 159.4, 156.5, 135.9, 122.0, 115.8, 29.2, 11.0; HRMS (ESI): m/z for C9H9N3O [M + 1]+: calcd.: 176.0824; found: 176.0819.
2-Propylpyrido[2,3-d]pyrimidin-4(3H)-one (14c). This compound was prepared from 2-aminonicotinamide (126 mg, 0.92 mmol), triethyl orthobutyrate (262 mg, 298 µL, 1.38 mmol) and acetic acid (165 mg, 158 µL, 2.76 mmol) in absolute ethanol (3 mL) at 110 °C for 12 h. Yield: 164 mg (87%) as a tan solid, m.p. 168–170 °C; IR (CHCl3): 3205, 1674 cm−1; 1H-NMR (400 MHz, CDCl3): δ 11.4 (br s, 1H), 9.00 (dd, J = 4.6, 2.1 Hz, 1H), 8.60 (dd, J = 7.9, 2.1 Hz, 1H), 7.43 (dd, J = 7.9, 4.6 Hz, 1H), 2.81 (t, J = 7.5 Hz, 2H), 1.97 (sextet, J = 7.5 Hz, 2H), 1.07 (t, J = 7.5 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 164.5, 160.5, 159.4, 156.5, 135.9, 122.0, 115.7, 37.7, 20.5, 13.7; HRMS (ESI): m/z for C10H11N3O [M + 1]+: calcd.: 190.0980; found: 190.0977.
2-Phenylpyrido[2,3-d]pyrimidin-4(3H)-one (14d). This compound was prepared from 2-aminonicotinamide (126 mg, 0.92 mmol) and triethyl orthobenzoate (309 mg, 312 µL, 1.38 mmol) and acetic acid (165 mg, 158 µL, 2.76 mmol) in absolute ethanol (3 mL) at 110 °C for 12 h. Yield: 164 mg (80%) as an off-white solid, m.p. 181–182 °C (lit [35] m.p. 178–180 °C); IR (CHCl3): 3210, 1672 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.8 (br s, 1H), 8.99 (dd, J = 4.5, 2.0 Hz, 1H), 8.54 (dd, J = 7.8, 2.0 Hz, 1H), 8.25 (d, J = 8.1 Hz, 2H), 7.67–7.56 (complex, 3H), 7.55 (dd, J = 7.8, 4.5 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6): δ 163.4, 159.2, 156.6, 155.9, 136.0, 132.9, 132.4, 129.1, 128.5, 122.7, 116.6; HRMS (ESI): m/z for C13H9N3O [M + 1]+: calcd.: 224.0824; found: 224.0820.
2,3-Dimethylquinazolin-4(3H)-one (16a). This compound was prepared from 2-amino-N-methylbenzamide (100 mg, 0.67 mmol), triethyl orthoacetate (224 mg, 253 µL, 1.38 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 12 h. Yield: 102 mg (87%) as a light tan solid, m.p. 108–109 °C (lit [19] m.p. 112–114 °C); IR (CHCl3): 1661 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.27 (dd, J = 8.0, 1.2 Hz, 1H), 7.72 (ddd, J = 8.3, 7.2, 1.5 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.44 (td, J = 8.0, 1.0 Hz, 1H), 3.63 (s, 3H), 2.63 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 162.5, 154.6, 147.4, 134.3, 126.9, 126.8, 126.5, 120.4, 31.2, 23.8; HRMS (ESI): m/z for C10H10N2O [M + 1]+: calcd.: 175.0871; found: 175.0874.
2-Ethyl-3-methylquinazolin-4(3H)-one (16b). This compound was prepared from 2-amino-N-methylbenzamide (100 mg, 0.67 mmol), orthopropionate (236 mg, 269 µL, 1.34 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h. Yield: 105 mg (83%) as a light tan solid, m.p. 118–120 °C (lit [50] m.p. 120–121 °C); IR (CHCl3): 1684 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.26 (dd, J = 8.1, 1.2 Hz, 1H), 7.72 (ddd, (J = 8.2, 7.0, 1.5 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.43 (ddd, J = 8.2, 7.0, 1.3 Hz, 1H), 3.64 (s, 3H), 2.87 (q, J = 7.4 Hz, 2H), 1.41 (t, J = 7.4 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 162.6, 157.8, 147.3, 134.0, 126.9, 126.7, 126.3, 120.2, 30.3, 28.9, 11.1; HRMS (ESI): m/z for C11H12N2O [M + 1]+; calcd.: 189.1028; found: 189.1025.
2-Phenyl-3-methylquinazolin-4(3H)-one (16d). This compound was prepared from 2-amino-N-methylbenzamide (100 mg, 0.67 mmol), triethyl orthobenzoate (300 mg, 303 µL, 1.34 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 48 h. Yield: 140 mg (89%) as a light tan solid, m.p. 133–135 °C (lit [19] m.p. 134–138 °C); IR (CHCl3): 1674 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.34 (d, J = 7.8 Hz, 1H), 7.79 (complex, 2H), 7.60–7.49 (complex, 6H), 3.50 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 162.7, 156.1, 147.3, 135.4, 134.3, 130.1, 128.9, 128.0, 127.5, 127.0, 126.7, 120.6, 34.3; HRMS (ESI): m/z for C15H12N2O [M + 1]+: calcd.: 237.1028; found: 237.1024.
2-Methyl-3-phenylquinazolin-4(3H)-one (18a). This compound was prepared from 2-amino-N-phenylbenzamide (100 mg, 0.47 mmol), triethyl orthoacetate (228 mg, 258 µL, 1.41 mmol) and acetic acid (85 mg, 81 µL, 1.41 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h. Yield: 105 mg (95%) as a white solid, m.p. 142–143 °C (lit [21] m.p. 142–144 °C); IR (CHCl3): 1684 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.27 (dd, J = 7.9, 0.7 Hz, 1H), 7.77 (td, J = 8.1, 1.3 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.60–7.44 (complex, 4H), 7.27 (d, J = 7.1 Hz, 2H), 2.25 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 162.3, 154.2, 147.5, 137.8, 134.6, 130.0, 129.3, 128.0, 127.1, 126.8, 126.6, 120.8, 24.4; HRMS (ESI): m/z for C15H12N2O [M + 1]+: calcd.: 237.1028; found: 237.1023.
2-Ethyl-3-phenylquinazolin-4(3H)-one (18b). This compound was prepared from 2-amino-N-phenylbenzamide (100 mg, 0.47 mmol), triethyl orthopropionate (248 mg, 283 µL, 1.41 mmol) and acetic acid (85 mg, 81 µL, 1.41 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h. Yield: 108 mg (92%) as an off-white solid, m.p. 120–122 °C (lit [21] m.p. 124–125 °C); IR (CHCl3): 1684 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.28 (dd, J = 8.0, 1.4 Hz, 1H), 7.77 (ddd, J = 8.2, 6.8, 1.5 Hz, 1H), 7.73 (dd, J = 8.1, 1.5 Hz, 1H), 7.59–7.47 (complex, 4H), 7.27 (m, 2H), 2.45 (q, J = 7.4 Hz, 2H), 1.22 (t, J = 7.4 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 162.5, 157.8, 147.6, 137.4, 134.5, 129.9, 129.2, 128.3, 127.1, 127.0, 126.6, 120.8, 29.3, 11.7; HRMS (ESI): m/z for C16H14N2O [M + 1]+: calcd.: 251.1184; found: 251.1179.
2,3-Diphenylquinazolin-4(3H)-one (18d). This compound was prepared from 2-amino-N-phenylbenzamide (100 mg, 0.47 mmol), triethyl orthobenzoate (316 mg, 319 µL, 1.41 mmol) and acetic acid (85 mg, 81 µL, 1.41 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h. Yield: 132 mg (95%) as an off-white solid, m.p. 154–156 °C (lit [21] m.p. 154–155 °C); IR (CHCl3): 1692, 1664 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.37 (d, J = 8.0 Hz, 1H), 7.85–7.78 (complex, 2H), 7.54 (ddd, J = 8.1, 5.6, 2.8 Hz, 1H), 7.36–7.13 (complex, 10H); 13C-NMR (101 MHz, CDCl3): δ 162.3, 155.2, 147.5, 137.7, 135.5, 134.7, 129.3, 129.1, 129.0 (2C), 128.4, 128.0, 127.8, 127.3, 127.2, 121.0; HRMS (ESI): m/z for C20H14N2O [M + 1]: calcd.: 299.1184; found: 299.1180.
3-Amino-2-methylquinazolin-4(3H)-one (20a). This compound was prepared from 2-aminobenzhydrazide (100 mg, 0.67 mmol), triethyl orthoacetate (217 mg, 245 µL, 1.34 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h. Yield: 97 mg (82%) a tan solid, m.p. 147–149 °C (lit [51] m.p. 146–148 °C); IR (CHCl3): 3455, 3413, 1635 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.72 (dd, J = 8.0, 1.2 Hz, 1H), 7.26 (td, J = 8.1, 1.2 Hz, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.75 (td, J = 8.1, 0.9 Hz, 1H), 5.81 (br s, 2H), 2.61 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 165.0, 161.9, 147.0, 132.4, 127.9, 116.9, 116.3, 106.0, 11.1; HRMS (ESI): m/z for C9H9N3O [M + 1]+: calcd.: 176.0824; found: 177.0898.
3-Amino-2-ethylquinazolin-4(3H)-one (20b). This compound was prepared from 2-aminobenzhydrazide (100 mg, 0.67 mmol), triethyl orthopropionate (235 mg, 269 µL, 1.34 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h. Yield: 100 mg (80%) as a tan solid, m.p. 116–117 °C (lit [52] m.p. 121.5–122.5 °C); IR (CHCl3): 3421, 3320, 1627 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J = 7.9, 1.5 Hz, 1H), 7.25 (td, J = 8.1, 1.6 Hz, 1H), 6.79 (d, J = 8.2 Hz, 1H), 6.74 (t, J = 8.1 Hz, 1H), 5.82 (br s, 2H), 2.95 (q, J = 7.6 Hz, 2H), 1.44 (t, J = 7.6 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 166.0, 164.7, 146.8, 132.2, 127.7, 116.7, 116.1, 106.0, 19.0, 10.9; HRMS (ESI): m/z for C10H11N3O [M + 1]+: calcd.: 190.0980; found: 190.0978.
3-Amino-2-phenylquinazolin-4(3H)-one (20d). This compound was prepared from 2-aminobenzhydrazide (100 mg, 0.67 mmol), triethyl orthobenzoate (300 mg, 303 µL, 1.34 mmol) and acetic acid (121 mg, 115 µL, 2.01 mmol) in absolute ethanol (2 mL) at 110 °C for 24 h. Yield: 151 mg (95%) as a tan solid, m.p. 160–161 °C (lit [53] m.p. 181–182 °C); IR (CHCl3): 3435, 3316, 1612 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.15 (m, 2H), 7.87 (dd, J = 8.0, 1.5 Hz, 1H), 7.55 (m, 3H), 7.31 (ddd, J = 8.4, 7.2, 1.5 Hz, 1H), 6.82 (d, J = 8.4 Hz, 1H), 6.80 (td, J = 7.9, 1.1 Hz, 1H), 5.90 (br s, 2H); 13C-NMR (101 MHz, CDCl3): δ 164.6, 162.8, 147.1, 132.5, 131.7, 129.1, 127.8, 126.9, 123.9, 116.8, 116.2, 105.8; HRMS (ESI): m/z for C14H11N3O [M + 1]+: calcd.: 238.0980; found: 238.0974.
3.2.3. Method 3: General Procedure to Prepare 5,6-dihydropyrimidin-4(3H)-ones
The orthoester (1.5 equiv) was added to a mixture of the 3-amino-2,2-dimethylpropionamide (1.0 equiv) in anhydrous ethanol (3 mL). Glacial acetic acid (2 equiv) was added and the reaction was heated at reflux for 24 h. The reaction mixture was cooled and concentrated under vacuum. The product was hygroscopic and was best crystallized by adding purified chloroform and allowing it to sit under nitrogen for 4–7 days. Filtration and drying yielded the pure product.
2,5,5-Trimethyl-5,6-dihydropyrimidin-4(3H)-one (22a). This compound was prepared from 3-amino-2,2-dimethylpropionamide (100 mg, 0.86 mmol), triethyl orthoacetate (209 mg, 236 µL, 1.29 mmol) and acetic acid (103 mg, 99 µL, 1.72 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 60 mg (50%) as a white solid, m.p. 196–198 °C; IR (nujol): 3223, 1669 cm−1; 1H-NMR (400 MHz, D2O): δ 3.35 (s, 2H), 2.23 (s, 3H), 1.16 (s, 6H), NH exchanged; 13C-NMR (101 MHz, D2O): δ 184.4, 165.3, 43.0, 23.3 (2C), 18.4; HRMS (ESI): m/z for C7H12N2O [M + 1]+: calcd.: 141.1028; found: 141.1028.
2-Ethyl-5,5-dimethyl-5,6-dihydropyrimidin-4(3H)-one (22b). This compound was prepared from 3-amino-2,2-dimethylpropionamide (100 mg, 0.86 mmol), triethyl orthopropionate (227 mg, 259 µL, 1.29 mmol) and acetic acid (103 mg, 99 µL, 1.72 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 59 mg (44%) as a white solid, m.p. 185–187 °C; IR (nujol): 3184, 1642 cm−1; 1H-NMR (400 MHz, D2O): δ 3.36 (s, 2H), 2.50 (q, J = 7.7 Hz, 2H), 1.24 (t, J = 7.7 Hz, 3H), 1.15 (s, 6H), NH exchanged; 13C-NMR (101 MHz, D2O): δ 184.5, 169.7, 51.0, 43.8, 26.5, 23.3 (2C), 10.7; HRMS (ESI): m/z for C8H14N2O [M + 1]+: calcd.: 155.1184; found: 155.1187.
5,5-Dimethyl-2-propyl-5,6-dihydropyrimidin-4(3H)-one (22c). This compound was prepared from 3-amino-2,2-dimethylpropionamide (100 mg, 0.86 mmol), triethyl orthobutyrate (245 mg, 279 µL, 1.29 mmol) and acetic acid (103 mg, 99 µL, 1.72 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 52 mg (36%) as a white solid, m.p. 118–120 °C; IR (nujol): 3199, 1652 cm−1; 1H-NMR (400 MHz, D2O): δ 3.37 (s, 2H), 2.46 (t, J = 7.4 Hz, 2H), 1.69 (sextet, J = 7.4 Hz, 2H), 1.16 (s, 6H), 0.96 (t, J = 7.4 Hz, 3H), NH exchanged; 13C-NMR (101 MHz, D2O): δ 184.4, 168.5, 51.0, 43.8, 34.5, 23.3 (2C), 20.2, 12.3; HRMS (ESI): m/z for C9H16N2O [M + 1]+: calcd.: 169.1341; found: 169.1339.
5,5-Dimethyl-2-phenyl-5,6-dihydropyrimidin-4(3H)-one (22d). This compound was prepared from 3-amino-2,2-dimethylpropionamide (100 mg, 0.86 mmol), triethyl orthobenzoate (289 mg, 292 µL, 1.29 mmol) and acetic acid (103 mg, 99 µL, 1.72 mmol) in absolute ethanol (3 mL) at 110 °C for 24 h. Yield: 126 mg (72%) as a white solid, m.p. 153–155 °C; IR (nujol): 3231, 1700, 1651 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.46 (br s, 1H), 7.78 (d, J = 7.6 Hz, 2H), 7.53–7.43 (complex, 2H), 3.62 (s, 2H), 1.21 (s, 6H), NH exchanged; 13C-NMR (101 MHz, CDCl3): δ 177.2, 151.5, 133.3, 131.4, 129.0, 126.4, 57.7, 36.5, 23.4 (2C); HRMS (ESI): m/z for C12H14N2O [M + 1]+: calcd.: 203.1184; found: 203.1180.
4. Conclusions
We have developed an efficient strategy for the synthesis of 2-alkyl- and 2-arylquinazolin-4(3H)-ones. The procedure is straightforward, and gives the desired heterocycles in yields ≥80% without contamination by residual metals or Lewis acids. Although almost all of the compounds have been reported previously, the current work provides a unified approach that allows the convenient preparation of a broad range of targets derived from 2-aminobenzamides; ring-substituted 2-aminobenzamides, 2-aminonicotinamides, 2-amino-N-methyl (and N-phenyl) benzamides; as well as 2-aminobenzhydrazides. The conditions are mild and purification of the final products is easily accomplished. Additionally, we have extended the method to the synthesis of 5,6-dihydropyrimidin-4(3H)-ones, although these compounds proved difficult to purify due to their hygroscopic nature. The only limitation to our approach is that relatively few substituted orthoesters are available commercially. Nevertheless, the simplicity and high yields provided by the current procedure, as well as its ability to provide the target heterocycles free of contaminants, makes this method an important contribution to the field.
Acknowledgments
J.T.G. wishes to thank the 2018 NSF-supported REU Program at Oklahoma State University (CHE-1559874) for a summer appointment. The authors also wish to acknowledge the NSF (BIR-9512269), the Oklahoma State Regents for Higher Education, the W. M. Keck Foundation, and Conoco, Inc. for funds to establish our Oklahoma State-Wide NMR Laboratory. The OSU College of Arts and Sciences is also acknowledged for funds to purchase a new 400 MHz NMR for this facility.
Supplementary Materials
The following are available online. Electronic Supplementary Information (ESI) available: Copies of 1H and 13C-NMR spectra of the final products.
Author Contributions
J.T.G. and J.K.A. did the experimental work. R.A.B. optimized several of the procedures and wrote the paper. All of the authors read and approved the final version of the manuscript before submission.
Funding
This research was funded by the NSF REU Program (NSF CHE–1559874) and the NSF MRI Program (NSF BIR-9512269).
Conflicts of Interest
The authors declare no conflicts of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Michael J.P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 2005;22:627–646. doi: 10.1039/b413750g. [DOI] [PubMed] [Google Scholar]
- 2.Peng J.-B., Geng H.-Q., Wang W., Qi X., Ying J., Wu X.-F. Palladium-catalyzed four-component carbonylative synthesis of 2,3-disubstituted quinazolin-4(3H)-ones: Convenient methaqualone preparation. J. Catal. 2018;365:10–13. doi: 10.1016/j.jcat.2018.06.007. [DOI] [Google Scholar]
- 3.Inoue I., Oine T., Yamada Y., Tani J., Ishida R., Ochiai T. 2-Fluoromethyl-3-(o-tolyl)- 6-amino-4(3H)-quinazolinone. DE 2,449,113 A1. German Patent. 1975 Apr 24;
- 4.Ochiai T., Ishida R. Pharmacological studies on 6-amino-2-fluoromethyl-3-(o-tolyl)- 4(3H)-quinazolinone (afloqualone), a new centrally acting muscle relaxant. (II). Effects on the spinal reflex potential and the rigidity. Jpn. J. Pharmacol. 1982;32:427–438. doi: 10.1254/jjp.32.427. [DOI] [PubMed] [Google Scholar]
- 5.Bartroli J., Turmo E., Agueró M., Boncompte E., Vericat M.L., Conte L., Ramis J., Merlos M., García-Rafanell J., Forn J. New azole antifungals. 3. Synthesis and antifungal activity of 3-substituted-4(3H)-quinazolinones. J. Med. Chem. 1998;41:1869–1882. doi: 10.1021/jm9707277. [DOI] [PubMed] [Google Scholar]
- 6.Mabkhot Y.N., Al-Har M.S., Barakat A., Aldawsari F.D., Aldelbahi A., Ul-Haq Z. Synthesis, anti-microbial and molecular docking studies of quinazolin-4(3H)-ones derivatives. Molecules. 2014;19:8725–8739. doi: 10.3390/molecules19078725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunce R.A., Nammalwar B. New conditions for the synthesis of (±)-2-monosubstituted and (±)-2,2-disubstituted 2,3-dihydro-4(1H)-quinazolinones from 2-nitro- and 2-aminobenzamide. J. Heterocycl. Chem. 2011;48:991–997. doi: 10.1002/jhet.672. [DOI] [Google Scholar]
- 8.Fortenberry C., Nammalwar B., Bunce R.A. Ammonium chloride-catalyzed synthesis of benzo-fused heterocycles from o-substituted anilines and orthoesters. Org Prep. Proced. Int. 2013;45:57–65. doi: 10.1080/00304948.2013.743751. [DOI] [Google Scholar]
- 9.Khajavi M.S., Rad-Moghadam K., Hazarkhani H. A facile synthesis of 6-substituted benzimidazo[1,2-c]quinazolines under microwave irradiation. Synth. Commun. 1999;29:2617–2624. doi: 10.1080/00397919908086422. [DOI] [Google Scholar]
- 10.Helali A.Y.H., Sarg M.T.M., Koraa M.M.S., El-Zoghbi M.S.F. Utility of 2-methyl-quinazolin-4(3H)-one in the synthesis of heterocyclic compounds with anticancer activity. Open J. Med. Chem. 2014;4:12–37. doi: 10.4236/ojmc.2014.41002. [DOI] [Google Scholar]
- 11.Von Niementowski S. Synthesen von Chinazolinverbindungen. J. Prakt. Chem. 1895;51:564–572. doi: 10.1002/prac.18950510150. [DOI] [Google Scholar]
- 12.Meyer J.F., Wagner E.C. The Niementowski reaction. The use of methyl anthranilate or isatoic anhydride with substituted amides or amidines in the formation of 3-substituted-4-keto-3,4-dihydroquinazolines. The course of the reaction. J. Org. Chem. 1943;8:239–252. doi: 10.1021/jo01191a005. [DOI] [Google Scholar]
- 13.Li F., Feng Y., Meng Q., Li W., Li A., Wang Q., Tao Z. An efficient construction of quinazolin-4(3H)-ones under microwave irradiation. Arkivoc. 2007;2007:40–50. [Google Scholar]
- 14.Baghbanzadeh M., Molnar M., Damm M., Reidlinger C., Dabiri M., Kappe C.O. Parallel microwave synthesis of 2-styrylquinazolin-4(3H)-ones in a high throughput platform using HPLC/GC vials as reaction vessels. J. Comb. Chem. 2009;11:676–684. doi: 10.1021/cc900036a. [DOI] [PubMed] [Google Scholar]
- 15.Wang W.-L., Liu X.-X., Zhang T., Zhang J.-M., Zhou J.-H. New Facile and solvent-free method for the one-pot synthesis of quinazolin-4(3H)-ones catalyzed by SbCl3 under microwave irradiation. J. Chem. Soc. Pak. 2016;38:1196–1202. [Google Scholar]
- 16.Endicott M.M., Wick E., Mercury M.L., Sherrill M.L. Quinazoline derivatives. I. The synthesis of 4-(4’-diethylamino-1’-methylbutylamino)-quinazoline (SN 11,534) and the corresponding 2-phenylquinazoline (SN 11,535) J. Am. Chem. Soc. 1946;68:1299–1301. doi: 10.1021/ja01211a055. [DOI] [PubMed] [Google Scholar]
- 17.Kumar D., Jadhavar P.S., Nautiyal M., Sharma H., Meena P.K., Adane L., Pancholia S., Chakraborti A.K. Convenient synthesis of 2,3-disubstituted quinazolin-4(3H)-ones and 2-styryl-3-substituted quinazolin-4(3H)-ones: Applications towards the synthesis of drugs. RSC Adv. 2015;5:30819–30825. doi: 10.1039/C5RA03888J. [DOI] [Google Scholar]
- 18.Sharma R., Abdullaha M., Bharate S.B. Metal-free ionic liquid-mediated synthesis of benzimidazoles and quinazolin-4(3H)-ones from benzylamines. Asian J. Org. Chem. 2017;6:1370–1374. doi: 10.1002/ajoc.201700214. [DOI] [Google Scholar]
- 19.Li Z., Dong J., Chen X., Li Q., Zhou Y., Yin S.-F. Metal- and oxidant-free synthesis of quinazolinones from -ketoesters with o-aminobenzamides via phosphorous acid-catalyzed cyclocondensation and selective C–C bond cleavage. J. Org. Chem. 2015;80:9392–9400. doi: 10.1021/acs.joc.5b00937. [DOI] [PubMed] [Google Scholar]
- 20.Almeida S., Marti R., Vanoli E., Abele S., Tortoioli S. One-pot synthesis of trifluoromethylated quinazolin-4(3H)-ones with trifluoroacetic acid as a CF3 source. J. Org. Chem. 2018;83:514–5113. doi: 10.1021/acs.joc.8b00389. [DOI] [PubMed] [Google Scholar]
- 21.Kumar R.A., Uma-Maheswari C., Ghantasala S., Jyothi C., Reddy K.R. Synthesis of 3H-quinazolin-4-ones and 4H-3,1-benzoxazin-4-ones via benzylic oxidation and oxidative dehydrogenation using potassium iodide-tert-butyl hydroperoxide. Adv. Synth. Catal. 2011;353:401–410. doi: 10.1002/adsc.201000580. [DOI] [Google Scholar]
- 22.Sun J., Tao T., Xu D., Cao H., Kong Q., Wang X., Liu Y., Zhao J., Wang Y., Pan Y. Metal-free oxidative cyclization of 2-aminobenzamides, 2-aminobenzenesulfonamide or 2-(aminomethyl)-anilines with primary alcohols for the synthesis of quinazolines and their analogues. Tetrahedron Lett. 2018;59:2099–2102. doi: 10.1016/j.tetlet.2018.04.054. [DOI] [Google Scholar]
- 23.Ge W., Zhu X., Wei Y. Iodine-catalyzed oxidative system for the cyclization of primary alcohols with o-aminobenzamides to quinazolinones using DMSO as the oxidant in dimethyl carbonate. RSC Adv. 2013;3:10817–10822. doi: 10.1039/c3ra40872h. [DOI] [Google Scholar]
- 24.Mohammed S., Vishwakarma R.A., Bharate S.B. Iodine catalyzed oxidative synthesis of quinazolin-4(3H)-ones and pyrazolo[4.3-d]pyrimidin-7(6H)-ones via amination of sp3 C–H bind. J. Org. Chem. 2015;80:6915–6921. doi: 10.1021/acs.joc.5b00989. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z., Wang M., Zhang C., Zhang Z., Lu J., Wang F. The cascade synthesis of quinazolinones and quinazolines using α-MnO2 catalyst and tert-butyl hydroperoxide (TBHP) as an oxidant. Chem. Commun. 2015;51:9205–9207. doi: 10.1039/C5CC02785C. [DOI] [PubMed] [Google Scholar]
- 26.Zhou D., Zhou Y.-R., Shen Q., Li J.-X. Iron-catalyzed oxidative synthesis of N-heterocycles from primary alcohols. RSC Adv. 2014;4:6486–6489. doi: 10.1039/c3ra46363j. [DOI] [Google Scholar]
- 27.Parua S., Das S., Sikari R., Sinha S., Paul N.D. One-pot cascade synthesis of quinazolin-4(3H)-ones via nickel-catalyzed dehydrogenative coupling of o-aminobenzamides with alcohols. J. Org. Chem. 2017;82:7165–7175. doi: 10.1021/acs.joc.7b00643. [DOI] [PubMed] [Google Scholar]
- 28.Sharif M., Opalach J., Langer P., Beller M., Wu X.-F. Oxidative synthesis of quinazolinones and benzothiadiazine 1,1-dioxides from 2-aminobenzamide and 2-aminobenzenesufonamide with benzyl alcohols and aldehydes. RSC Adv. 2014;4:8–17. doi: 10.1039/C3RA45765F. [DOI] [Google Scholar]
- 29.Zhang W., Meng C., Liu Y., Tang Y., Li F. Auto-tandem catalysis with ruthenium: From o-aminobenzamides and allylic alcohols to quinazolinones via redox isomerization/acceptorless dehydrogenation. Adv. Synth. Catal. 2018;360:1–10. doi: 10.1002/adsc.201800660. [DOI] [Google Scholar]
- 30.Zhou J., Fang J. One-pot synthesis of quinazolinones via iridium-catalyzed hydrogen transfers. J. Org. Chem. 2011;76:7730–7736. doi: 10.1021/jo201054k. [DOI] [PubMed] [Google Scholar]
- 31.Hakim Siddiki S.M.A., Kon K., Touchy A.S., Shimizu K. Direct synthesis of quinazolinones by acceptorless dehydrogenative coupling of o-aminobenzamide and alcohols by heterogeneous Pt catalysts. Catal. Sci. Technol. 2014;4:1716–1719. doi: 10.1039/C4CY00092G. [DOI] [Google Scholar]
- 32.Couture A., Cornet H., Grandclaudon P. An expeditious synthesis of 2-aryl- and 2-alkylquinazolin-4(3H)-ones. Synthesis. 1991:1009–1010. doi: 10.1055/s-1991-26632. [DOI] [Google Scholar]
- 33.Liu X., Fu H., Jiang Y., Zhao Y. A simple efficient approach to quinazolinones under mild copper-catalyzed conditions. Angew. Chem. Int. Ed. 2009;48:348–351. doi: 10.1002/anie.200804675. [DOI] [PubMed] [Google Scholar]
- 34.Xu W., Liu H., Jiang Y., Fu H. Copper-catalyzed domino synthesis of quinazolinones via Ullmann-type coupling and aerobic oxidative C–H amidation. Org. Lett. 2011;13:1274–1277. doi: 10.1021/ol1030266. [DOI] [PubMed] [Google Scholar]
- 35.Li T., Chen M., Yang L., Xiong Z., Wang Y., Li F., Chen D. Copper-catalyzed consecutive reaction to construct quinazolin-4(3H)-ones and pyrido[2,3-d]pyrimidin-4(3H)-ones. Tetrahedron. 2016;72:868–874. doi: 10.1016/j.tet.2015.12.059. [DOI] [Google Scholar]
- 36.Thakur M.S., Nayal O.S., Bhatt V., Sharma S., Kumar N. Rapid and efficient cascade synthesis of 4-amino-4(3H)-quinazolinones over an in situ-generated heterogeneous CuCO3-K2CO3 nanocomposite. Asian J. Org. Chem. 2016;5:750–754. doi: 10.1002/ajoc.201600113. [DOI] [Google Scholar]
- 37.Soheilizad M., Soroosh S., Pashazadeh R. Solvent-free copper-catalyzed three-component synthesis of 2-substituted quinazolin-4(3H)-ones. Monatsh. Chem. 2017;148:739–743. doi: 10.1007/s00706-016-1804-9. [DOI] [Google Scholar]
- 38.Abe T., Kida K., Yamada K. A copper-catalyzed Ritter-type cascade via iminoketene for the synthesis of quinazolin-4(3H)-ones and diazocines. Chem. Commun. 2017;53:4362–4365. doi: 10.1039/C7CC01406F. [DOI] [PubMed] [Google Scholar]
- 39.Nisha, Sharma M.C., Kumar R., Kumar Y. Regioselective copper(I) catalyzed Ullmann amination of halopyridyl carboxylates using sodium azide: A route for aminopyridyl carboxylates and their transformation to pyrido[2,3-d]pyrimidin-4(1H)-ones. Chem. Sel. 2018;3:4822–4826. doi: 10.1002/slct.201800907. [DOI] [Google Scholar]
- 40.Yu X., Gao L., Jia L., Yamamoto Y., Bao M. Synthesis of quinazolin-4(3H)-ones via the reaction of 2-halobenzamides with nitriles. J. Org. Chem. 2018;83:10352–10358. doi: 10.1021/acs.joc.8b01460. [DOI] [PubMed] [Google Scholar]
- 41.Hikawa H., Ino Y., Suzuki H., Yokoyama Y. Pd-catalyzed benzylic C–H amidation with benzyl alcohols in water: A strategy to construct quinazolinones. J. Org. Chem. 2012;77:7046–7051. doi: 10.1021/jo301282n. [DOI] [PubMed] [Google Scholar]
- 42.Hikawa H., Matsuda N., Suzuki H., Yokoyama Y., Azumaya I. N-Benzylation/benzylic C–H amidation cascade by the (η3-benzyl)palladium system in aqueous media: An effective pathway for the direct construction of 3-phenyl-3,4-dihydro-(2H)-1,2,4-benzothiadiazine 1,1-dioxides. Adv. Synth. Catal. 2013;355:2308–2320. doi: 10.1002/adsc.201300317. [DOI] [Google Scholar]
- 43.Jiang X., Tang T., Wang J.-M., Chen Z., Zhu Y.-M., Ji S.-J. Palladium-catalyzed one-pot synthesis of quinazolinones via tert-butyl isocyanide insertion. J. Org. Chem. 2014;79:5082–5087. doi: 10.1021/jo500636y. [DOI] [PubMed] [Google Scholar]
- 44.Qian C., Liu K., Tao S.-W., Zhang F.-L., Zhu Y.M., Yang S.-L. Palladium-catalyzed oxidative three-component coupling of anthranilamides with isocyanides and arylboronic acids: Access to 2,3-disubstituted quinazolinones. J. Org. Chem. 2018;83:9201–9209. doi: 10.1021/acs.joc.8b01218. [DOI] [PubMed] [Google Scholar]
- 45.Hammond G.S. In: Steric Effects in Organic Chemistry. Newman M.S., editor. Wiley; New York, NY, USA: 1956. p. 425. [Google Scholar]
- 46.Anslyn E.V., Dougherty D.A. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA, USA: 2006. p. 497. [Google Scholar]
- 47.Zhao G., Souers A.J., Voorbach M., Falls H.D., Droz B., Brodjian S., Lai Y.Y., Iyengar R.R., Gao J., Judd A.S., Wagaw S.H., et al. Validation of diacyl glycerolacyltransferase I as a novel target for the treatment of obesity and dyslipidemia using a potent and selective small molecule inhibitor. J. Med. Chem. 2008;51:380–383. doi: 10.1021/jm7013887. [DOI] [PubMed] [Google Scholar]
- 48.Kakuta K., Zheng X., Oda H., Harada S., Sugimoto Y., Sasaki K., Tai A. Cyclooxygenase-1-selective inhibitors are attractive candidates for analgesics that do not cause gastric damage. Design and in vitro/in vivo evaluation of a benzamide-type cyclooxygenase-1-selective inhibitor. J. Med. Chem. 2008;51:2400–2411. doi: 10.1021/jm701191z. [DOI] [PubMed] [Google Scholar]
- 49.Okano M., Mito J., Maruyama Y., Masuda H., Niwa T., Nakagawa S.-C., Nakamura Y., Matsuura A. Discovery and structure-activity relationships of 4-aminoquinazoline derivatives, a novel class of opioid receptor like-1 (ORL1) antagonists. Bioorg. Med. Chem. 2009;17:119–132. doi: 10.1016/j.bmc.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 50.Kato T., Takada A., Ueda T. Reaction of triethyloxonium fluoroborate with acid amide. III. Formation of quinazoline and 4H-3,1-benzooxazin-4-one derivatives. Chem. Pharm. Bull. 1976;24:431–436. doi: 10.1248/cpb.24.431. [DOI] [Google Scholar]
- 51.Youssef A.S.A., Hemdan M.M., El-Mariah F.A., Hashem H.E. Synthesis of some quinazolinone derivatives functionalized with N-3 heterocyclic side chain. J. Heterocycl. Chem. 2018;55:1626–1633. doi: 10.1002/jhet.3197. [DOI] [Google Scholar]
- 52.Koohang A., Stanchina C.L., Coates R.M. Regio- and stereoselective synthesis of N-H aziridines by N-N bond reduction of quinazolinyl aziridines. Tetrahedron. 1999;55:9669–9686. doi: 10.1016/S0040-4020(99)00522-0. [DOI] [Google Scholar]
- 53.Abdel-Rahman H.M., Abdel-Azziz M., Canzoneri J.C., Gary B.D. Novel quinazolin-4(3H)-one/Schiff base hybrids as antiproliferative and phosphodiesterase 4 inhibitors: Design, synthesis, and docking studies. Arch. Pharm. Chem. Life Sci. 2014;347:650–657. doi: 10.1002/ardp.201400083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.