

Abstract
Natural L-carvone was utilized as a starting material for an efficient synthesis of some terpenyl-derived 1,2,3-triazoles. Chlorination of carvone, followed by nucleophilic substitution with sodium azide resulted in the preparation of 10-azidocarvone. Subsequent CuAAC click reaction with propargylated derivatives provided an efficient synthetic route to a set of terpenyl-derived conjugates with increased solubility in water. All investigated compounds exhibit high antioxidant activity, which is comparable with that of vitamin C. It was also found that serum albumin and the terpenyl-1,2,3-triazoles hybrids spontaneously undergo reversible binding driven by hydrophobic interactions, suggesting that serum albumin can transport the target triazoles.
Keywords: carvone; 1,2,3-triazole; click chemistry; acetylene; antioxidant activity; bovine serum albumin
1. Introduction
Natural compounds play a significant role in the design of new drugs and prevention of various diseases [1]. Thus, more than 60% of current drugs for the treatment of cancer and infectious diseases are of natural origin [2]. Terpenoids are one of the largest classes of chiral natural compounds, which includes more than 23,000 compounds. Many terpene derivatives are widely used in perfumes, playing the role of cosmetic products and food additives [3]. However, medicine is not less important area of application of terpenes, since the majority of compounds of this class exhibits pronounced biological activity. For example, the antimalarial drug artemisinin and the anticancer drug paclitaxel (Taxol®) are the two most prominent members of the class of terpenes used in medicine (Figure 1).
Figure 1.

Structures of artemisinin and Taxol®.
Annual sales of terpenic derivatives in the world are estimated at about $20 billion [4]. The scope of terpenes is constantly expanding; therefore, terpenoids will play an increasingly important role as therapeutic and prophylactic agents for human diseases.
Monoterpenes–C10H16 compounds and their derivatives are the simplest type of terpenoids. They are the basis of essential oils, floral aromas and perform plant protective functions [5,6]. Despite the fact that these are small and fairly simple molecules, a number of monoterpenes have an antitumor effect, demonstrating not only the ability to prevent the formation or progression of cancer, but the ability to reduce existing malignant tumors [7]. Most abundant in nature are terpenoids having menthane skeleton (Figure 2). For example, limonene is the most common monocyclic monoterpene found in nature. It can be found in a variety of trees and grasses (for example, Mentha spp.). Limonene is an important component of the peel of oranges and lemons, as well as the essential oil of cumin. The limonene fragment can be found in the structure of many drugs [7,8,9,10,11]. Carvone, the main monoterpene of cumin seed. It has been shown that carvone oil prevents chemically induced carcinoma of the lung [12]. In addition, carveol has chemoprophylactic activity against breast cancer during the initiation phase [13]. Perillyl alcohol, a hydroxylated limonene analog, exhibits chemopreventive activity against chemically induced liver cancer [14] and tumor recurrences in animal models [15].
Figure 2.

Some menthane-derived monoterpenoids.
Nowadays, one of the most promising synthetic trends for expanding molecular complexity from a given scaffold have been the “click” reactions. “Click” reactions are a class of efficient, fast, universal, and selective reactions that are characterized by high yields and simple isolation of products. The standard “click” of chemistry for today is the formation of 1,2,3-triazoles catalyzed by copper salts (I) in the reaction of azides and terminal alkynes (CuAAC) [16,17,18,19,20]. Although 1,2,3-triazole fragment is generally absent in natural compounds, synthetic molecules containing 1,2,3-triazole cycles represent a wide class of physiologically active substances exhibiting various types of biological activity, for example, antibacterial, antiallergic, antiviral, antifungal properties [21,22]. Apparently, the pharmacological activity of compounds containing 1,2,3-triazole groups is due to the structural and electronic similarity of the 1,2,3-triazole fragment and the amide group. The 1,2,3-triazole fragment can be considered as a conformationally constrained bioisostere of the amide group. Greater stability of the 1,2,3-triazole scaffold under physiological conditions, led to the widespread use of the “click” reaction as an effective method for the synthesis of various biologically active compounds in drug design. The triazole fragment is an important pharmacophoric unit, and a large number of drugs containing this heterocycle are known (Figure 3). Due to the commercial success of some pharmaceutical preparations based on the triazole ring, many pharmaceutical companies and academic groups have shown interest in developing new methods for synthesizing triazole compounds and screening their biological activity [23,24]. For example, antifungal drugs containing triazole rings are known: itraconazole, fluconazole, voriconazole [25], antiviral drug ribavirin and murbritinib (used to treat breast, bladder, kidney and prostate cancer). Ribavirin is a drug for the treatment of viral infections such as herpes and hepatitis [26,27].
Figure 3.

Some triazole-derived drugs.
2. Results and Discussion
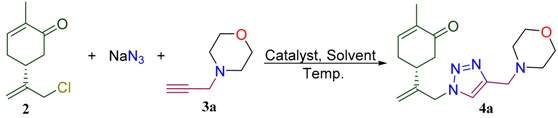
This study is devoted to the synthesis of carvone-derived 1,2,3-triazoles 4a–i prepared according to the strategy outlined in Scheme 1. We proposed that due to the presence of the enone fragment in the structure, such conjugates would behave as antioxidants. On the other hand, the triazole and amine moieties will play an auxiliary role of improving water solubility of these molecules. 10-Azido-carvone was chosen as a key building block to study copper-catalyzed azide alkyne cycloaddition (CuAAC). A number of propargylated amino derivatives 3 were studied as alkyne partners for this reaction. They can be prepared using standard alkylation of NH-derivatives with propargyl bromide (Scheme 1) [28].
Scheme 1.

Reagents and conditions: i–Ca(OCl)2, CO2, DCM/H2O, 0 °C; ii—NaN3, MeCN or DMSO; iii—3-bromoprop-1-yne, EtOH or MeCN, base; iv—Et3N, CuBr or CuI (5 mol%), MeCN or DMSO, 60 °C.
To achieve this goal, L-carvone (1) was chlorinated using calcium hypochlorite−CO2 system to provide 10-chlorocarvone (2) [29]. Subsequent treatment of 2 with sodium azide resulted in synthesis of 10-azidocarvone. We decided to utilize synthetically attractive one-pot protocol to avoid isolation and purification of this intermediate product. For this aim, chloride 2 was treated with sodium azide in DMSO or acetonitrile to yield 10-azido-carvone, which was used without isolation in model CuAAC reaction with N-propargylmorpholine 3a. It was found that the in the case of DMSO as a solvent the yield of model conjugate 4a is moderate. However, with acetonitrile as a solvent and copper iodide as a catalyst, the isolated yield was improved significantly. For example, 4a was obtained in up to 78% yield within a reasonable reaction time (10 h) using only 5 mol % of copper iodide (Table 1). Probably, such observation can be explained by formation of the complex of copper iodide with final products.
Table 1.
Optimization of synthesis of 4а.
| |||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Time, h | Temp., °C | Isolated Yield a |
| 1. | DMSO | CuBr(10) | 5 | 65 | 39 |
| 2. | DMSO | CuI(10) | 5 | 65 | 48 |
| 3. | MeCN: DMSO—1:1 | CuI(10) | 8 | 60 | 56 |
| 4. | MeCN | CuI (10) | 8 | 60 | 69 |
| 5. | MeCN | CuI (10) | 10 | 60 | 71 |
| 6. | MeCN | CuI (5) | 10 | 60 | 78 |
a Conditions: 10-chlorocarvone (1 mmol), solvent (3 mL), NaN3 (1.2 mmol), 60 °C 10 h, Et3N (1.2 mmol), CuHal, N-propargylmorpholine.
Next, the reaction with a number of propargylated amines and amides was investigated. To our delight, the corresponding triazole derived carvone conjugates 4 were isolated in up to 84% yield. The efficient procedure provided 4 as crystalline compounds. Their structure was confirmed by combination of spectroscopic methods (see SI). As a result, this part of study gave as a family of carvone derivatives 4 with variable amino- (amido) substituents in the triazole ring (Table 2).
Table 2.
Synthesis of terpenyl-1,2,3-triazoles 4a–i.
| Terpenyl-1,2,3-triazoles | Yield, % | Terpenyl-1,2,3-triazoles | Yield, % | ||
|---|---|---|---|---|---|
| 4a |
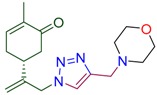
|
78 | 4f |
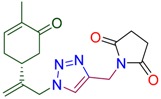
|
65 |
| 4b |
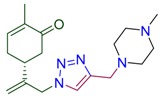
|
79 | 4g |
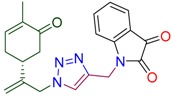
|
63 |
| 4c |
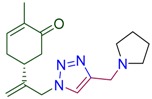
|
73 | 4h |
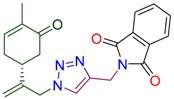
|
84 |
| 4d |
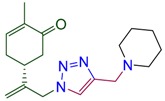
|
79 | 4i |
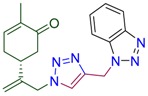
|
76 |
| 4e |
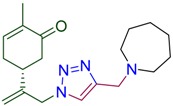
|
81 |
With these new compounds in hand, we decided to study the antioxidant properties of the synthesized terpenyl-1,2,3-triazoles 4. For this aim the interaction with HO• radicals in the presence of the competitive acceptor 4-nitroso-N,N-dimethylaniline (PNDMA) was studied [30,31,32]. The initiation of HO• radicals was carried out by photolysis of H2O2 (10−3 mol·L−1) under the action of UV radiation (λ = 313 nm with the use of a special filter). The rate of initiation of HO• radicals (Figure 4a–f) was determined by the change in PNDMA absorption (А440). It was found that compounds 4a–e have better water-solubility and their antioxidant properties were investigated. Below are given the kinetic data of the effect of 4а–e different concentrations on the optical density of PNDMA, depending on the time of irradiation (Figure 4a–e).
Figure 4.

(a) compound 4a; (b) compound 4b; (c) compound 4c; (d) compound 4d; (e) compound 4e. Dependence of optical density of PNDMA vs the time of UV irradiation (λ = 313 nm) at various concentrations of compounds 4a–e; (f) dependence of rate of PNDMA discoloring (in units) vs the concentration: compounds 4а–e.
The constants of the interaction rate of HO• radicals with 4a–e were calculated by the Equation (1) [31,32]:
| kOH + P = 1.25 × 1010([PNDMA]/[P]) × [(W1/W2) − 1], mol−1 s−1 L | (1) |
where 1.25 × 1010 mol−1 s−1 L is the constant of the interaction rate of HO• radicals with PNDMA, [P] is the concentration of 4a–e, W1 and W2 are rates of PNDMA discoloring in distilled water and in the presence of 4a–e respectively. The rate constants are shown in Table 3. As one can see from Table 3, all the compounds tested exhibit significant antioxidant activity. Moreover compound 4b is slightly inferior to the known antioxidant ascorbic acid used as a control compound.
Table 3.
The rate constants for the reaction.
| Tested Compound | Rate Constant HO• + 4a–e (M s−1) |
|---|---|
| 4a | 1.155 × 109 |
| 4b | 4.483 × 109 |
| 4c | 1.359 × 109 |
| 4d | 0.104 × 109 |
| 4e | 0.407 × 109 |
| Ascorbic acid (control) | 9.450 × 109 |
The transport of active aglycones of drugs is known to be carried out by various interactions with the proteins. The most abundant protein in the blood plasma is the transport protein serum albumin (up to 60%) which can reversibly bind various endogenous and exogenous compounds [33,34,35]. To reveal possibility of such binding, fluorescence spectroscopy was used for model compound 4e with the bovine serum albumin (BSA). The thermodynamic parameters (ΔH, ΔS and ΔG) can be used to propose the binding mode. For the typical hydrophobic interactions, both ΔH and ΔS are positive, while negative ΔH and ΔS result from the hydrogen bond formation and van der Waals forces, and electrostatic interactions are responsible for the cases when ΔH < 0 and ΔS > 0 [36]. The value of binding constant (Kb) for transport protein ligand interaction in the range of103–106 M−1 indicate the reversibility of binding [37]. The results obtained for BSA-4e system are summarized in Table 4. Negative values of ΔG show that the binding process proceeds spontaneously. The positive values of ΔH and ΔS show that the stability of the BSA-4e system is due mainly to hydrophobic interactions.
Table 4.
The values of thermodynamic parameters for BSA binding with 4e at 298 and 308 K.
| T, K | Kb, M−1 | N | ΔH, kJ mol−1 | ΔG, kJ mol−1 | ΔS, J mol−1 K−1 |
|---|---|---|---|---|---|
| 298 | 2.40·× 103 | 1.001 ± 0.079 | 67.989 | −19.283 | 292.86 |
| 308 | 5.85·× 103 | 1.061 ± 0.066 | −22.212 |
The distance between the protein and the ligand can be calculated using the theory of resonance energy transfer (Foerster theory) [38]. Average distance between BSA-4e decreases in the range of 2–8 nm and the energy transfer efficiency increases when the temperature was increased from 298 K to 308 K (Table 5). However, such interactions of BSA with 4e are not very strong. Thus, the transport of 4e can be performed by serum albumin-BSA.
Table 5.
The values of the overlap integral, the energy transfer efficiency, the Forster radius and the distance between the BSA and 4e.
| T, K | I, cm3 L × mol−1 | E | R0, nm | r, nm |
|---|---|---|---|---|
| 298 | 1.79 × 10−14 | 0.019 | 3.778 | 7.27 |
| 308 | 1.80 × 10−14 | 0.043 | 3.782 | 7.06 |
3. Materials and Methods
3.1. General Information
1H- and 13C-NMR spectra were recorded on a Mercury-300 MHz instrument (Varian, Palo Alto, CA, USA) in DMSO-CCl4 mixture (1:3) or on an AVANCE 400 MHz spectrometer (Bruker, Billerica, MA, USA) in CDCl3. Chemical shifts (δ) in ppm are reported as quoted relative to the residual signals of chloroform-d (7.26 for 1H-NMR and 77.16 for 13C-NMR) or DMSO-d6 (2.50 for 1H-NMR and 39.52 for 13C-NMR) as internal references. The coupling constants (J) are given in Hertz. ESI-MS spectra were measured with a MicroTof instrument (Bruker Daltonics, Billerica, MA, USA). TLC analysis was performed on Xtra SIL G/UV254 plates (Macherey-Nagel, Düren, German). All reagents were of reagent grade and were used as such or distilled prior to use. Compounds 2 and 3a–i were synthesized according to the procedure described in [28,29]. Melting points were determined on a Boetius micro-heating stage and a SMP10.
(5R)-5-(3-chloroprop-1-en-2-yl)-2-methylcyclohex-2-en-1-one. Yield 67%, b.p. 110–116 °C/1 Torr, 1.5285, −44.05 (C 2.17 DCM).
4-(Prop-2-yn-1-yl)morpholine (3a). Yield 53%, b.p. 71–72 °C/15 Torr, 1.4741. 1H-NMR (300 MHz, DMSO/CCl4—1/3) δ 3.63–3.56 (m, 4H, OCH2), 3.22 (d, J = 2.5 Hz, 2H, NCH2C≡CH), 2.51 (t, J = 2.5 Hz, 1H, ≡CH), 2.49–2.44 (m, 4H, NCH2). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 78.0(C≡CH), 73.9(C≡CH), 65.8 (OCH2), 51.3 (NCH2), 46.4(NCH2C≡).
1-Methyl-4-(prop-2-yn-1-yl)piperazine (3b). Yield 49%, b.p. 74 °C/12 Torr, 1.4799. 1H-NMR (300 MHz, DMSO/CCl4—1/3) δ 3.20 (d, J = 2.5 Hz, 2H, NCH2C≡CH), 2.55–2.42 (m, 5H, NCH2, ≡CH), 2.42–2.21 (m, 4H, NCH2), 2.18 (s, 3H, NCH3). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 78.4(C≡CH), 73.5(C≡CH), 54.2(NCH2), 50.8(NCH2), 46.1(NCH2C≡), 45.4(NCH3).
1-(Prop-2-yn-1-yl)pyrrolidine (3c). Yield 41%, b.p. 42–43 °C/17 Torr, 1.4590. 1H-NMR (300 MHz, DMSO/CCl4—1/3) δ 3.33 (d, J = 2.4 Hz, 2H, NCH2C≡CH), 2.58–2.50 (m, 4H, NCH2), 2.38 (t, J = 2.4 Hz, 1H, ≡CH), 1.82–1.70 (m, 4H, CH2). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 78.9(C≡CH), 72.7(C≡CH), 51.2(NCH2), 41.8(NCH2C≡), 23.2(CH2).
1-(Prop-2-yn-1-yl)piperidine (3d). Yield 50%, b.p. 55–57 °C/12 Torr, 1.4706. 1H-NMR (300 MHz, DMSO/CCl4—1/3) δ 3.18 (d, J = 2.4 Hz, 2H, NCH2C≡CH), 2.48–2.35 (m, 5H-NCH2, ≡CH), 1.62–1.50 (m, 4H, CH2), 1.47–1.33 (m, 2H, CH2). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 78.6(C≡CH), 73.2(C≡CH), 52.1(NCH2), 46.9(NCH2C≡), 25.2(CH2), 23.4(CH2).
1-(Prop-2-yn-1-yl)azepane (3e). Yield 55%, b.p. 71–72 °C/15 Torr, 1.4791. 1H-NMR (300 MHz, DMSO/CCl4—1/3) δ 3.28 (d, J = 2.4 Hz, 2H, NCH2C≡CH), 2.66–2.57 (m, 4H, NCH2), 2.35 (t, J = 2.4 Hz, 1H, ≡CH), 1.69–1.54 (m, 8H, (CH2)4). 13C-NMR (75 MHz, DMSO) δ 79.6(C≡CH), 72.3(C≡CH), 54.0(NCH2), 47.4(NCH2C≡), 27.8(CH2), 26.2(CH2).
1-(Prop-2-yn-1-yl)pyrrolidine-2,5-dione (3f). Yield 64%, b.p. 127 °C/2 Torr, 1.5124. 1H-NMR (300 MHz, DMSO/CCl4—1/3) δ 4.13 (d, J = 2.5 Hz, 2H, NCH2), 2.69 (s, 4H, CH2C=O), 2.60 (t, J = 2.5 Hz, 1H, ≡CH). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 175.0 (C=O), 77.1(C≡CH), 71.8(C≡CH), 27.7 CH2C=O), 26.8 (NCH2).
1-(Prop-2-yn-1-yl)-1H-indole-2,3-dione (3g). Yield 64%, m.p. 162–163 °C (EtOH/H2O—1/1). 1H-NMR (300 MHz, DMSO/CCl4—1/3) δ 7.66 (td, J = 7.8, 1.4 Hz, 1Harom), 7.57 (ddd, J = 7.4, 1.3, 0.4 Hz, 1Harom), 7.22–7.12 (m, 2Harom), 4.53 (d, J = 2.5 Hz, 2H, NCH2), 2.81 (t, J = 2.5 Hz, 1H, ≡CH). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 181.7(C=O), 156.3(NC=O), 149.3(Carom), 137.5(HCarom), 124.2(HCarom), 123.0(HCarom), 117.2(Carom), 110.8(HCarom), 76.1(C≡CH), 73.8(C≡CH), 28.7 (NCH2).
2-(Prop-2-yn-1-yl)-1H-isoindole-1,3(2H)-dione (3h). Yield 63%, m.p. 146–148 °C (EtOH). 1H-NMR (300 MHz, DMSO/CCl4—1/3) δ 7.89–7.84 (m, 2Harom), 7.83–7.78 (m, 2Harom), 4.37 (d, J = 2.5 Hz, 2H, NCH2), 2.64 (t, J = 2.5 Hz, 1H, ≡CH). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 165.7(C=O), 133.7(HCarom), 131.4(Carom), 122.9(HCarom), 77.2(C≡CH), 72.2(C≡CH), 26.2(NCH2).
1-(Prop-2-yn-1-yl)-1H-benzotriazole (3i). Yield 45%, m.p. 62–63 °C (PhH/Hex—1/1). 1H-NMR (300 MHz, DMSO) δ 8.00 (dt, J = 8.3, 1.0 Hz, 1Harom), 7.82 (dt, J = 8.3, 1.0 Hz, 1Harom), 7.52 (ddd, J = 8.2, 6.9, 1.0 Hz, 1Harom), 7.37 (ddd, J = 8.2, 6.9, 1.0 Hz, 1Harom), 5.58 (d, J = 2.6 Hz, 2H, NCH2), 3.04 (t, J = 2.6 Hz, 1H, ≡CH). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 145.3(Carom), 132.0(Carom), 126.7(HCarom), 123.2(HCarom), 119.1(HCarom), 109.9(HCarom), 75.9(C≡CH), 75.6(C≡CH), 37.1(NCH2).
(5R)-5-(3-azidoprop-1-en-2-yl)-2-methylcyclohex-2-en-1-one (10-N3-Car.). 369 mg (2 mmol) of (5R)-5-(3-chloroprop-1-en-2-yl)-2-methylcyclohex-2-en-1-one, 2 mL of acetonitrile, 156 mg (2.4 mmol) of sodium azide are placed in a round-bottomed 5 mL flask and heated with stirring for 10 h (60 °C). The mixture was poured into water (30 mL) and extracted with DCM (3 × 10 mL). The combined extracts were dried over Na2SO4, the volatiles were evaporated and the residue was purified via column chromatography on silica gel using DCM–Hex mixture (1:1) as an eluent. Yield 70%. 1H-NMR (300 MHz, DMSO/CCl4—1/.3) δ 6.74 (ddq, J = 5.5, 2.7, 1.3 Hz, 1H, HC=), 5.15 (s, J = 4.0 Hz, 1Ha, H2C=), 5.09 (s, 1Hb, H2C=), 3.87 (s, 2H, CH2N3), 2.93–2.66 (m, 1Ha, CH2C=O), 2.61–2.44 (m, 2H, CH, CH2 in carbocycle), 2.43–2.22 (m, 2H, CH2 in carbocycle), 1.73 (dt, J = 2.6, 1.4 Hz, 3H, CH3). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 196.5(C=O), 144.7, 142.8, 134.6, 113.4, 53.8, 42.2, 38.4, 30.6, 15.1(CH3).
3.2. General Method for the Preparation of Terpenyl-1,2,3-triazoles 4a–i
369 mg (2 mmol) of (5R)-5-(3-chloroprop-1-en-2-yl)-2-methylcyclohex-2-en-1-one, 2 mL of acetonitrile, 156 mg (2.4 mmol) of sodium azide are placed in a round-bottomed 5 mL flask and heated with stirring for 10 h (60 °C). After cooling, 242 mg (2.4 mmol) of triethylamine, 19 mg (0.1 mmol) of CuI and 2.4 mmol of the corresponding propargyl derivative are added to the resulting mixture. The whole is heated then for 10 h at 60 °C and the solvent is distilled off. The cooled mixture is dissolved in benzene, filtered and hexane is added to the filtrate. The precipitated crystals are filtered off, washed with hexane and dried.
(5R)-2-methyl-5-(3-{4-[(morpholin-4-yl)methyl]-1H-1,2,3-triazol-1-yl}prop-1-en-2-yl)cyclohex-2-en-1-one (4a). Yield 78%, m.p. 81–82 °C, −31.65 (C 1.15 DCM). 1H-NMR (400 MHz, CDCl3) δ 7.47 (s, 1H, NCH), 6.69 (ddd, J = 5.8, 2.6, 1.4 Hz, 1H, HC=CCO), 5.13 (s, 1Ha, H2C=), 5.06 (s, 1Hb, H2C=), 5.04 (d, J = 15.8 Hz, 1Ha, H2C=C-CH2N), 4.93 (d, J = 15.3 Hz, 1Ha, H2C=C-CH2N), 3.75–3.68 (m, 4H, O(CH2CH2)2NCH2), 3.67 (s, 2H, O(CH2CH2)2NCH2), 2.65–2.43 (m, 7H, CH2C=O, O(CH2CH2)2N, CH in carbocycle), 2.42–2.18 (m, 2H, CH2 in carbocycle), 1.76 (dt, J = 2.5, 1.3 Hz, 3H, CH3). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ: 196.5 (C=O), 145.6, 143.0, 142.9, 134.5, 123.1, 113.3, 65.9, 52.9, 52.6, 52.5, 42.1, 37.9, 30.4, 15.1. HRMS (ESI) m/z: [M + H]+ Calcd for C17H25N4O2+ 317.1978, Found 317.1972.
(5R)-2-methyl-5-(3-{4-[(4-methylpiperazin-1-yl)methyl]-1H-1,2,3-triazol-1-yl}prop-1-en-2-yl)cyclohex-2-en-1-one (4b). Yield 79%, m.p. 117 °C, −38.79 (C 1.044 DCM). 1H-NMR (400 MHz, CDCl3) δ 7.45 (s, 1H, NCH), 6.67 (ddd, J = 5.7, 2.7, 1.4 Hz, 1H, HC=CCO), 5.10 (s, 1Ha, =CH2), 5.04 (s, 1Hb, =CH2), 5.01 (d, J = 15.5 Hz, 1Ha, H2C=C-CH2N), 4.90 (d, J = 15.2 Hz, 1Hb, H2C=C-CH2N), 3.66 (s, 2H, CH2N(CH2CH2)2NCH3), 2.65–2.30 (m, 12H, CH and CH2 in carbocycle, CH2N), 2.25 (s, J = 7.6 Hz, 4H, CH2 in carbocycle, NCH3), 1.74 (dt, J = 2.5, 1.3 Hz, 3H, CH3). 13C-NMR (101 MHz, CDCl3) δ 198.5 (C=O), 145.2, 145.1, 143.8, 135.8, 122.6, 115.5, 55.1, 54.0, 53.4, 53.0, 46.1, 42.8, 38.3, 31.2, 15.7. HRMS (ESI) m/z: [M + H]+ Calcd for C18H28N5O+ 330.2294, Found 330.2293.
(5R)-2-methyl-5-(3-{4-[(pyrrolidin-1-yl)methyl]-1H-1,2,3-triazol-1-yl}prop-1-en-2-yl)cyclohex-2-en-1-one (4c). Yield 73%, m.p. 69–70 °C, −29.6 (C 1.066 DCM). 1H-NMR (400 MHz, CDCl3) δ 7.47 (s, 1H, NCH), 6.67 (ddd, J = 5.7, 2.6, 1.3 Hz, 1H, HC=CCO), 5.11 (s, 1Ha, =CH2), 5.05 (s, 1Hb, =CH2), 5.02 (d, J = 15.4 Hz, 1H, H2C=C-CH2N), 4.92 (d, J = 15.2 Hz, 1H, H2C=C-CH2N), 3.77 (s, 2H, CH2N(CH2)4), 2.64–2.52 (m, 2H, CH2C=O, 4H, N(CH2CH2)2), 2.47 (m, 1H, CH in carbocycle), 2.36 (m, 1H, CH2 in carbocycle), 2.29–2.17 (m, 1H, CH2 in carbocycle), 1.83–1.76 (m, 4H, N(CH2CH2)2), 1.76–1.71 (m, 3H, CH3). 13C-NMR (101 MHz, DMSO) δ 198.6 (C=O), 146.7, 145.2, 143.8, 135.8, 122.2, 115.5, 54.2, 54.0, 51.0, 42.8, 38.3, 31.3, 23.6, 15.7. HRMS (ESI) m/z: [M + H]+ Calcd for C17H25N4O+ 301.2028, Found 301.2026.
(5R)-2-methyl-5-(3-{4-[(piperidin-1-yl)methyl]-1H-1,2,3-triazol-1-yl}prop-1-en-2-yl)cyclohex-2-en-1-one (4d). Yield 79%, m.p. 80–81 °C, −35.04 (C 1.036 DCM). 1H-NMR (400 MHz, CDCl3) δ 7.46 (s, 1H, NCH), 6.67 (ddd, J = 5.8, 2.6, 1.4 Hz, 1H, HC=CCO), 5.12 (s, 1Ha, H2C=), 5.06 (s, 1Hb, H2C=), 5.02 (d, J = 15.4 Hz, 1Ha, H2C=C-CH2N), 4.92 (d, J = 15.2 Hz, 1Hb, H2C=C-CH2N), 3.64 (s, 2H, CH2N(CH2)5), 2.65–2.30 (m, 8H, CH2C=O, N(CH2CH2)2CH2, CH and CH2 in carbocycle), 2.29–2.16 (m, 1H CH2 in carbocycle), 1.75 (dt, J = 2.3, 1.2 Hz, 3H, CH3), 1.61–1.52 (m, 4H, N(CH2CH2)2CH2), 1.46–1.36 (m, 2H, N(CH2CH2)2CH2). 13C-NMR (75 MHz, DMSO/CCl4—1/3) δ 196.4 (C=O), 145.6, 143.8, 142.9, 134.4, 122.7, 113.3, 53.4, 52.5, 42.1, 37.9, 30.4, 25.3, 23.7, 15.1. HRMS (ESI) m/z: [M + H]+ Calcd for C18H27N4O+ 315.2185, Found 315.2182.
(5R)-2-methyl-5-(3-{4-[(azepan-1-yl)methyl]-1H-1,2,3-triazol-1-yl}prop-1-en-2-yl)cyclohex-2-en-1-one (4e). Yield 81%, m.p. 89–90 °C, −21.6 (C 1.000 DCM). 1H-NMR (400 MHz, CDCl3) δ 7.46 (s, 1H, NCH), 6.66 (ddd, J = 5.7, 2.6, 1.4 Hz, 1H, HC=CCO), 5.11 (s, 1Ha, H2C=), 5.05 (s, 1Hb, H2C=), 5.02 (d, J = 15.6 Hz, 1Ha, CH2=C-CH2N), 4.92 (d, J = 15.2 Hz, 1Hb, CH2=C-CH2N), 3.79 (s, 2H, CH2N(CH2)6), 2.68–2.60 (m, 4H, N(CH2CH2CH2)2), 2.60–2.29 (m, 4H, CH and CH2 in carbocycle), 2.29–2.04 (m, 1H, CH2 in carbocycle), 1.74 (dt, J = 2.4, 1.2 Hz, 3H, CH3), 1.68–1.51 (m, 8H, N(CH2CH2CH2)2). 13C-NMR (101 MHz, CDCl3) δ 198.6 (C=O), 146.9, 145.3, 143.8, 135.8, 122.4, 115.4, 55.6, 54.0, 53.6, 42.8, 38.3, 31.3, 28.1, 27.0, 15.7. HRMS (ESI) m/z: [M + H]+ Calcd for C19H29N4O+ 329.2341, Found 329.2339.
1-[(1-{2-[(1R)-4-methyl-5-oxocyclohex-3-en-1-yl]prop-2-en-1-yl}-1H-1,2,3-triazol-4-yl)methyl]pyrrolidine-2,5-dione (4f). Yield 65%, m.p. 127–128 °C, −23.2 (C 1.61 DCM). 1H-NMR (400 MHz, CDCl3) δ 7.56 (s, 1H, NCH), 6.68 (ddd, J = 5.6, 2.6, 1.4 Hz, 1H, HC=CCO), 5.13 (s, 1Ha, H2C=), 5.06 (s, 1Hb, H2C=), 5.00 (d, J = 15.2 Hz, 1Ha, CH2=C-CH2N), 4.90 (d, J = 15.2 Hz, 1Hb, CH2=C-CH2N), 4.79 (s, 2H, CH2N-C=O), 2.74 (s, 4H, CH2C(O)N), 2.61–2.43 (m, 3H, CH and CH2 in carbocycle), 2.37–2.18 (m, 2H, CH and CH2 in carbocycle), 1.76–1.74 (m, 3H, CH3). 13C-NMR (101 MHz, CDCl3) δ 198.5(C=O), 176.7(C=O), 144.9, 143.8, 142.6, 135.8, 123.2, 115.8, 54.1, 42.8, 38.2, 33.8, 31.2, 29.8, 28.3, 15.7. HRMS (ESI) m/z: [M + H]+ Calcd for C17H21N4O3+ 329.1614, Found 329.1610.
1-[(1-{2-[(1R)-4-methyl-5-oxocyclohex-3-en-1-yl]prop-2-en-1-yl}-1H-1,2,3-triazol-4-yl)methyl]-1H-indole-2,3-dione (4g). Yield 63%, m.p. 144–145 °C, −23.1 (C 1.6(6) DCM). 1H-NMR (400 MHz, CDCl3) δ 7.58 (s, 1H, NCH), 7.53–7.44 (m, 2Harom), 7.16 (d, J = 8.3 Hz, 1Harom), 7.02 (t, J = 7.5 Hz, 1Harom), 6.57 (ddd, J = 5.7, 2.6, 1.3 Hz, 1H, HC=CCO), 5.03 (s, 1Ha, H2C=), 4.95 (s, 1Hb, H2C=), 4.92 (s, 2H, CH2N-C=O), 4.91 (d, J = 15.6 Hz, 1Ha, CH2=C-CH2N), 4.85 (d, J = 15.2 Hz, 1Hb, CH2=C-CH2N), 2.54–2.47 (m, 1Ha, CH2 in carbocycle), 2.47–2.40 (m, 1Hb, CH2 in carbocycle), 2.40–2.30 (m, 1H, CH in carbocycle), 2.29–2.19 (m, 1Ha, CH2 in carbocycle), 2.19–2.07 (m, 1Hb, CH2 in carbocycle), 1.69–1.58 (m, 3H, CH3). 13C-NMR (101 MHz, CDCl3) δ 198.4(C=O), 183.1(C=O), 158.0, 150.2, 144.6, 143.7, 142.3, 138.7, 135.7, 125.4, 124.2, 123.1, 117.6, 115.9, 111.5, 54.1, 42.7, 38.2, 35.5, 31.1, 15.7. HRMS (ESI) m/z: [M + H]+ Calcd for C18H24N5O3+ 377.1614, Found 377.1609.
2-[(1-{2-[(1R)-4-methyl-5-oxocyclohex-3-en-1-yl]prop-2-en-1-yl}-1H-1,2,3-triazol-4-yl)methyl]-1H-isoindole-1,3(2H)-dione (4h). Yield 84%, m.p. 132 °C, –21.3 (C 1.6(6) DCM). 1H-NMR (400 MHz, CDCl3) δ 7.93–7.80 (m, 2Harom), 7.80–7.66 (m, 2Harom), 7.59 (s, 1H, NCH), 6.73–6.59 (m, 1H, HC=CCO), 5.11 (s, 1Ha, H2C=), 5.05 (s, 1Hb, H2C=), 5.00 (d, J = 14.6 Hz, 1Ha, CH2=C-CH2N), 4.99 (s, 1H, CH2N-C=O), 4.90 (d, J = 15.2 Hz, 1Hb, CH2=C-CH2N), 2.66-2.51 (m, 2H, CH2 in carbocycle), 2.47 (m, 1H, CH in carbocycle), 2.36 (m, 1H, CH2 in carbocycle), 2.28–2.12 (m, 1H, CH2 in carbocycle), 1.82–1.63 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ 198.6(C=O), 167.8(C=O), 144.9, 143.8, 135.8, 134.5, 134.3, 132.1, 123.6, 123.0, 115.7, 54.1, 42.7, 38.2, 33.2, 31.2, 15.7. HRMS (ESI) m/z: [M + H]+ Calcd for C21H21N4O3+ 377.1614, Found 377.1609.
(5R)-5-(3-{4-[(1H-benzotriazol-1-yl)methyl]-1H-1,2,3-triazol-1-yl}prop-1-en-2-yl)-2-methylcyclohex-2-en-1-one (4i). Yield 76%, m.p. 104–105 °C, −24.0 (C 1.67 DCM). 1H-NMR (400 MHz, CDCl3) δ 8.04 (dt, J = 8.4, 0.9 Hz, 1Harom), 7.68 (dt, J = 8.4, 0.9 Hz, 1Harom), 7.50 (s, J = 3.2 Hz, 1H, NCH), 7.47 (ddd, J = 8.3, 7.0, 1.0 Hz, 1Harom), 7.39–7.34 (m, 1Harom), 6.62 (ddd, J = 5.6, 2.7, 1.3 Hz, 1H, HC=CCO), 5.97 (s, 2H, CH2N), 5.09 (s, 1Ha, H2C=), 5.00 (s, 1Hb, H2C=), 4.96 (d, J = 15.4 Hz, 1Ha, CH2=C-CH2N), 4.88 (d, J = 15.2 Hz, 1Hb, CH2=C-CH2N), 2.59–2.47 (m, 2H, CH2 in carbocycle), 2.45–2.30 (m, 2H, CH2 in carbocycle), 2.30–2.11 (m, 1H, CH2 in carbocycle), 1.75–1.72 (m, 3H, CH3). 13C-NMR (101 MHz, CDCl3) δ 198.3(C=O), 144.6, 143.6, 142.8, 135.8, 127.9, 124.3, 123.0, 120.0, 116.0, 110.1, 54.3, 42.7, 38.2, 31.1, 29.8, 15.7. HRMS (ESI) m/z: [M + H]+ Calcd for C19H21N6O+ 349.1777, Found 349.1773.
The 1H-NMR and 13C-NMR for all the synthesized compounds as well as details of fluorescence experiments and BSA binding are available in Supplementary Material.
4. Conclusions
Efficient synthesis of 1,2,3-triazolyl conjugates of various amines with carvone was developed. Chlorination of carvone followed by the reaction with sodium azide open excess to 10-azidocarvone. Its CuAAC with propargylated amines can be performed as a one-pot procedure to give target products in up to 84% isolated yield. The prepared conjugates demonstrated high antioxidant activity and spontaneously undergo hydrophobic reversible interaction with serum albumin.
Supplementary Materials
The 1H-NMR and 13C-NMR for all the synthesized compounds as well as details of fluorescence experiments and BSA binding are available online.
Author Contributions
Conceptualization, A.S.G., T.V.G. and V.G.N. A.S.G., M.A.S. performed experiments on the synthesize and assay the data. A.I.M. developed and performed the antioxidant activity experiments. K.R.G. and A.G.G. developed experiments and analyzed the data and fluorescence spectroscopy.
Funding
This work was supported by the RA MES State Committee of Science and Russian Foundation for Basic Research (RF) in the frames of the joint research project SCS 18RF-020 and RFBR 18-53-05007 accordingly.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of all the compounds 4a–i are available from the authors.
References
- 1.Newman D.J., Cragg G.M., Snader K.M. The influence of natural products upon drug discovery. Nat. Prod. Rep. 2000;17:215–234. doi: 10.1039/a902202c. [DOI] [PubMed] [Google Scholar]
- 2.Newman D.J., Cragg G.M., Snader K.M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 3.Hill R.A. The Chemistry of Natural Products. Chapman & Hall. Blackie Academic & Professional; New York, NY, USA: 1993. Terpenoids. [Google Scholar]
- 4.Zhang L. Natural Products: Drug Discovery and Therapeutic Medicine. Humana Press; Totowa, NY, USA: 2005. [Google Scholar]
- 5.Loza-Tavera H. Monoterpenes in essential oils-biosynthesis and properties. Adv. Exp. Med. Biol. 1999;464:49–62. doi: 10.1007/978-1-4615-4729-7_5. [DOI] [PubMed] [Google Scholar]
- 6.Little D.B., Croteau R. Flavor Chemistry: Thirty Years of Progress. Kluwer Academic/Plenum; New York, NY, USA: 1999. Biochemistry of Essential Oil Plants: A Thirty-Year Overview. [Google Scholar]
- 7.Crowell P.L. Prevention and therapy of cancer by dietary monoterpenes. J. Nutr. 1999;129:775S–778S. doi: 10.1093/jn/129.3.775S. [DOI] [PubMed] [Google Scholar]
- 8.Griffin S.G., Wyllie S.G., Markham J.L. The role of structure and molecular properties of terpenoids in determining their antimicrobial activity. Flavour Fragr. J. 1999;5:322–332. doi: 10.1002/(SICI)1099-1026(199909/10)14:5<322::AID-FFJ837>3.0.CO;2-4. [DOI] [Google Scholar]
- 9.Fabian C.J. Breast cancer chemoprevention: Beyond tamoxifen. Breast Cancer Res. 2001;3:99–103. doi: 10.1186/bcr279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oldfield E., Lin F.Y. Terpene Biosynthesis: Modularity Rules. Angew. Chem. Int. Ed. 2012;51:1124–1137. doi: 10.1002/anie.201103110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.González-Burgos E., Gómez-Serranillos M.P. Terpene Compounds in Nature: A Review of Their Potential Antioxidant Activity. Curr. Med. Chem. 2012;19:5319–5341. doi: 10.2174/092986712803833335. [DOI] [PubMed] [Google Scholar]
- 12.Wattenberg L.W., Sparnins V.L., Barany G. Inhibition of N-nitrosodiethylamine carcinogenesis in mice by naturally occurring organosulfur compounds and monoterpenes. Cancer Res. 1989;49:2689–2692. [PubMed] [Google Scholar]
- 13.Crowell P.L., Kennan W.S., Vedejs E., Gould M.N. Chemoprevention of limonene carcinogenesis by hydroxylated metabolites; Proceedings of the 81st Annual Meeting of the American Association for Cancer Research; Washington, DC, USA. 23–26 May 1990. [Google Scholar]
- 14.Mills J.J., Chari R.S., Boyer I.J., Gould M.N., Jirtle R.L. Induction of apoptosis in liver tumors by the monoterpene perillyl alcohol. Cancer Res. 1995;55:979–983. [PubMed] [Google Scholar]
- 15.Haag J.D., Gould M.N. Mammary carcinoma regression induced by perillyl alcohol, a hydroxylated analog of limonene. Cancer Chemother. Pharmacol. 1994;34:477–483. doi: 10.1007/BF00685658. [DOI] [PubMed] [Google Scholar]
- 16.Rostovtsev V.V., Green L.G., Fokin V.V., Sharpless K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)—Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Tornøe C.W., Christensen C., Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002;67:3057–3062. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 18.Sokolova N., Nenajdenko V.G. Recent Advances in the CuI-catalyzed Alkyne-Azide Cycloaddition (CuAAC): Focus on Functionally Substituted Azides and Alkynes. RSC Adv. 2013;3:16212–16242. doi: 10.1039/c3ra42482k. [DOI] [Google Scholar]
- 19.Sokolova N.V., Latyshev G.V., Lukashev N.V., Nenajdenko V.G. Design and synthesis of bile acid-peptide conjugates linked via triazole moiety. Org. Biomol. Chem. 2011;9:4921–4926. doi: 10.1039/c0ob01188f. [DOI] [PubMed] [Google Scholar]
- 20.Nenajdenko V.G., Gulevich A.V., Sokolova N.V., Mironov A.V., Balenkova E.S. Chiral Isocyanoazides: Efficient Bifunctional Reagents for Bioconjugation. Eur. J. Org. Chem. 2010:1445–1449. doi: 10.1002/ejoc.200901326. [DOI] [Google Scholar]
- 21.Vasilevsky S.F., Govdi A.I., Sorokina I.V., Tolstikova T.G., Baev D.S., Tolstikov G.A., Mamatuyk V.I., Alabugin I.V. Rapid Access to New Bioconjugates of Betulonic Acid via Click Chemistry. Bioorg. Med. Chem. Lett. 2011;21:62–65. doi: 10.1016/j.bmcl.2010.11.072. [DOI] [PubMed] [Google Scholar]
- 22.Vasilevsky S.F., Govdi A.I., Shults E.E., Shakirov M.M., Tolstikov G.A., Alabugin I.V. Efficient Synthesis of the Betulonic Acid -Acetylene Hybrids and Their Hepatoprotective and Anti-Inflammatory Activity. Biorgan. Med. Chem. 2009;17:5164–5169. doi: 10.1016/j.bmc.2009.05.059. [DOI] [PubMed] [Google Scholar]
- 23.Ferreira V.F., da Rocha D.R., da Silva F.C., Ferreira P.G., Boechat N.A., Magalhães J.L. Novel 1H-1,2,3-, 2H-1,2,3-, 1H-1,2,4- and 4H-1,2,4-triazole derivatives: A patent review (2008–2011) Expert Opin. Ther. Pat. 2013;23:319–331. doi: 10.1517/13543776.2013.749862. [DOI] [PubMed] [Google Scholar]
- 24.Mandal S.K., Saha D., Jain V.K., Jain B. Synthesis and antitubercular activity of some triazole derivatives of propyl gallate. Int. J. Pharm. Sci. Res. 2010;1:465–472. [Google Scholar]
- 25.Tan S.L., Pause A., Shi Y., Sonenberg N. Hepatitis C therapeutics: Current status and emerging strategies. Nat. Rev. Drug. Discov. 2002;1:867–881. doi: 10.1038/nrd937. [DOI] [PubMed] [Google Scholar]
- 26.Prusiner P., Sundaralingam M. A New Class of Synthetic Nucleoside Analogues with Broad-spectrum Antiviral Properties. Nat. New Biol. 1973;244:116–118. doi: 10.1038/newbio244116a0. [DOI] [PubMed] [Google Scholar]
- 27.Prusiner P., Sundaralingam M. The crystal and molecular structures of two polymorphic crystalline forms of virazole (1-β-d-ribo furanosyl-1,2,4-triazole-3-carboxamide). A new synthetic broad sprectrum antiviral agent. Acta Cryst. Sect. B. 1976;32:419–426. doi: 10.1107/S0567740876003154. [DOI] [Google Scholar]
- 28.Kushwaha K., Kaushik N., Jain S.C. Design and synthesis of novel 2H-chromen-2-one derivatives bearing 1,2,3-triazole moiety as lead antimicrobials. Bioorg. Med. Chem. Lett. 2014;24:1795–1801. doi: 10.1016/j.bmcl.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Weinges K., Schwarz G. Radikalische Cyclisierung von Dienen, VI. Substratkontrollierte asymmetrische Synthese von (3aS,6aR)-(+)-3,3a,6,6a-Tetrahydro-2H-cyclopenta[b]furan-2-on. Liebigs Ann. Chem. 1993;1993:811–814. doi: 10.1002/jlac.1993199301128. [DOI] [Google Scholar]
- 30.Bielski B.H.J., Cabelli D.E., Arudi R.L., Ross A.B. Reactivity of HO2/O2− Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data. 1985;14:1041–1100. doi: 10.1063/1.555739. [DOI] [Google Scholar]
- 31.Ernestova L.S., Semenova I.V., Vlasova G.V., Lee Wolf N. Hydrological, Chemical and Biological Processes of Transformation and Transport of Contaminants in Aquatic Environments. Volume 219. IAHS Publisher; New York, NY, USA: 1994. Redox transformation of pollutants in natural waters; pp. 67–74. [Google Scholar]
- 32.Simonsen M.E., Muff J., Bennedsen L.R., Kowalski K.P., Søgaard E.G. Photocatalytic bleaching of p-nitrosodimethylaniline and a comparison to the performance of other AOP technologies. J. Photochem. Photobiol. A Chem. 2010;216:244–249. doi: 10.1016/j.jphotochem.2010.07.008. [DOI] [Google Scholar]
- 33.Urquiza N.M., Naso L.G., Manca S.G., Lezama L., Rojo T., Williams P.A.M., Ferrer E.G. Antioxidant activity of methimazole–copper (II) bioactive species and spectroscopic investigations on the mechanism of its interaction with Bovine Serum Albumin. Polyhedron. 2012;31:530–538. doi: 10.1016/j.poly.2011.10.008. [DOI] [Google Scholar]
- 34.Li Q., Yang W., Wu L., Qi H.Y., Huang Y., Zhang Z. Interaction of Warfarin with Human Serum Albumin and Effect of Ferulic Acid on the Binding. J. Spectrosc. 2014 doi: 10.1155/2014/834501. [DOI] [Google Scholar]
- 35.Manjunath D.M., Sharanappa T.N., Shrinivas D.J., Uttam A.M., Shivamurti A.C. Multi-spectroscopic investigation of the binding interaction of fosfomycin with bovine serum albumin. J. Pharm. Anal. 2015;5:249–255. doi: 10.1016/j.jpha.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross D.P., Subramanian S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry. 1981;20:3096–3102. doi: 10.1021/bi00514a017. [DOI] [PubMed] [Google Scholar]
- 37.Kragh-Hansen U. Structure and ligand binding properties of human serum albumin. Dan. Med. Bull. 1990;37:57–84. [PubMed] [Google Scholar]
- 38.Lakowicz J. Principles of Fluorescence Spectroscopy. Springer; New York, NY, USA: 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.