

Abstract
Patients with neuropsychiatric and neurological disorders often express limbic circuit abnormalities and deficits in information processing. While these disorders appear to have diverse etiology, their common features suggest neurodevelopmental origins. Neurodevelopment is a prolonged process of diverse events including neurogenesis/apoptosis, axon pathfinding, synaptogenesis, and pruning, to name a few. The precise timing of the neurodevelopmental insult to these processes likely determines the resulting functional outcome. We used the epilepsy and schizophrenia-related gestational day 17 methylazoxymethanol acetate (GD17-MAM) model to examine the impact of this timed neurodevelopmental insult on principal cell morphology and synaptic network function of the dorsal hippocampus (dHPC) circuit. Our observed structural and functional alterations in dHPC are compartment-specific, indicating that adverse global exposure during gestation can produce specific alterations and distort information processing in neural circuits that underlie cognitive abilities.
Keywords: neurodevelopment, hippocampus, MAM, schizophrenia, morphology, physiology
Introduction
Neurodevelopmentally altered brain circuits are thought to underlie schizophrenia (Insel 2010; Lewis and Levitt 2002; Weinberger 1987, 1996; Jaaro-Peled et al. 2009), a multifaceted and heterogeneous disease that has had few promising treatment advances over the last decades (Lieberman et al. 2005). Neurodevelopmental abnormalities in schizophrenia are proposed to arise from genetic susceptibility, neurodevelopmental insults during gestation, such as maternal illness, malnutrition, or exposure to environmental toxins, and complications during labor (Lewis and Levitt 2002), the timing of which is anticipated to be important in determining which systems and processes are impacted. If this neurodevelopmental hypothesis is correct, developmental consequences are predicted to be present in affected individuals at multiple levels of biological function and cognitive behavior.
Studies utilizing the epilepsy and schizophrenia-related gestational day 17 (GD17) methylazoxymethanol acetate (MAM) model have shown that exposure to the mitotoxic DNA methylating agent results in abnormalities at various neurobiological levels investigated, including structural, physiological and behavioral abnormalities during adulthood (Hernan et al. 2018). Most studies have focused on the abnormal interactions between the medial prefrontal cortex and ventral HPC that results in abnormal dopamine function (Lodge and Grace 2007), thought to be related to behavioral abnormalities such as sensorimotor gating deficits, measured as impaired prepulse inhibition of startle (Moore et al. 2006), hyperlocomotion (Moore et al. 2006; O’Reilly et al. 2016; Ratajczak et al. 2015) and working memory deficits (Gourevitch et al. 2004).
The dHPC has been understudied in GD17-MAM rats, in spite of reported reductions in dHPC area and altered dHPC morphology (Matricon et al. 2010; O’Reilly et al. 2016; Moore et al. 2006) and in spite of reported behavioral abnormalities typically associated with dHPC function, including deficits in spatial learning (Gastambide et al. 2015) and cognitive ability in epilepsy (Lucas et al. 2011; Hernan et al. 2018). We previously showed that GD17-MAM exposed rats have minor memory impairments with intact cognitive control in the dorsal hippocampus (dHPC) dependent two-frame active place avoidance task (O’Reilly et al. 2016), indicating that some, but not all, components of the dHPC circuit will be abnormal. To investigate the aspects of dHPC circuitry abnormalities that may underlie these observed memory deficits, we examined cellular morphology and functionality of input and output within dHPC. We find that there are not global abnormalities in circuit structure and function, but that sub-circuit input, output, and input-output relationships are altered.
Methods
Animals
Pregnant Long-Evans rats arrived at the New York University animal facilities on GD10 and were housed individually. On GD17, rats were given either 26 mg/kg MAM (in 500 μL saline) or saline intraperitoneally (i.p.). Male pups were weaned at P24 and group-housed until P42–56, after which they were individually housed. Rats were maintained on a 12:12 light:dark cycle and had free access to food and water.
Golgi Studies
Tissue preparation
Adult rats aged P70–75 were anesthetized with isoflurane and immediately decapitated. Their brains were quickly extracted and the front of the brain was removed by creating a coronal cut just posterior to the optic chiasm. The cerebellum was removed from the posterior portion of the brain, which was then cut into two hemispheres. The frontal piece and the left and right hemispheres were Golgi impregnated using the FD rapid Golgi Stain kit (FD NeuroTechnologies, Inc., Columbia, MD) according to the manufacturer’s instructions. Briefly, the brains were allowed to incubate at room temperature in solutions A+B for 14 days and then were stored at 4°C in solution C for 4–5 days, at which point they were cut into 250 μm thick coronal sections on a vibratome in a bath of solution C. The sections were placed on gelatin coated slides and allowed to air dry for up to one week before being processed for the Golgi stain.
Layer Measurements
Sections were selected from each animal that were at approximately the same anterior-posterior location. Three measurements were taken for each layer (stratum oriens, stratum pyramidale, stratum radiatum (str. rad.), stratum lacunosum moleculare (slm), molecular layer, and granule cell layer) using the quick measure tool of Neurolucida morphometry software (version 11.11.2, Microbrightfield, Inc., Cholchester, VT) at 4× magnification using an Olympus BX5T microscope. These three measurements were averaged to give a value for each animal.
Neuronal tracing
Neurons were traced using the same morphometry software at 20× or 40× magnification. Neurons were selected for tracing on the criteria that they were well isolated from other neurons and were located in the dorsal hippocampus. Dentate gyrus neurons were selected from the upper blade of the granule cell layer. The Sholl analysis was performed using Neurolucida Explorer 11 Software with an increment of 20 μm set for the increasing diameter of the concentric circles. The branch order was determined on the traced neurons by manually coding each branch based on the first branch which protrudes directly from the soma (first order) and increasing the order at each node. The thickness of the granule cell and molecular layers were measured close to the location of each traced neuron and recorded using the same software. The molecular layer can be divided into thirds, each of which receives projections predominantly from the return projections of the hilus, the medial entorhinal cortex, and the lateral entorhinal cortex, respectively. The thickness of the molecular layer was used on a neuron-by-neuron basis to determine the inner, middle, and outer third in order to calculate the length of dendritic branching in each terminal region. In order to compute the dendritic length in str. rad. and slm for CA1 pyramidal neurons, the neurons were traced and the Sholl analysis was performed to place concentric rings around each neuron. The neuron, with the concentric rings, was then compared to the tissue to determine the start and stop locations of each layer.
Spine analysis
The spine analysis was performed at 100× magnification using the same morphometry software. Branches from the middle third of the molecular layer were used for the analysis. The middle third was estimated by eye. Z-stack images of branch segments were acquired at 0.1 μm intervals. Spines were classified as filopodia, thin, stubby, or mushroom using criteria similar to (Risher et al. 2014), with the exception that mushroom spines were classified based on a thick head and thin neck instead of using a measurement greater than 0.6 μm. The experimenter was blinded to the group.
In vivo hippocampus evoked responses
Naïve 70–100 day-old rats were anesthetized with 1.20 g/kg urethane (i.p.) and placed in a stereotaxic frame. A 16-site linear-array silicon electrode with 30 μm diameter recording contacts and 100 μm spacing (Neuronexus, Ann Arbor, MI; p/n: A 1 × 16 - 5mm - 100 - 703) was placed in the dorsal hippocampus to span all layers of CA1 and DG. A six-wire stimulating electrode bundle was placed in the contralateral ventral hippocampal commissure and a second stimulating bundle was placed in the ipsilateral perforant path. The stimulating electrodes were made by twisting six nichrome wires (75 μm) together and were cut at an angle so as to span 1 mm. The following coordinates were used to target the ventral hippocampal commissure (anteroposterior: −1.2 mm from Bregma; mediolateral: −1.0 mm; dorsoventral: −3.4 mm) and the perforant path (anteroposterior: −7.6 mm from Bregma; mediolateral: 4.1 mm, dorsoventral: 2.5–3.0 mm). The two-wire combination that evoked the largest response was selected for the stimulation experiments. A constant current stimulus isolation unit (WPI, Sarasota, FL; model: A365RC) was used to deliver individual unipolar 150 μs stimulus pulses across the electrode pair.
The response to stimulation was recorded using an Axona USBdacq recording system (St. Albans, U.K.) that was optimized for recording evoked potential responses. An attenuating resistor (either 18 kΩ or 47 kΩ) was connected to the Axona input to maximize the effective number of bits used for digitization. The signals were low-pass filtered < 5 kHz and digitized at 48 kHz. The recorded signals were multiplied by either 1 (for the 18 kΩ resistor) or 2.3 (for the 47 kΩ resistor).
Each input pathway was stimulated independently and the stimulus-response features were measured from input-output curves. We analyzed the field excitatory postsynaptic potential (fEPSP) and population spike (PS) responses from the current source density (CSD), allowing for source localization by minimizing volume conducted signals. fEPSP and PS responses were analyzed off-line using custom MATLAB software. Spike times were estimated on an animal by animal basis and used to constrict the window for estimating fEPSP. For perforant path stimulation, fEPSP responses were estimated in the time window starting 1.5 ms after the stimulation was delivered and 1 ms before any population spike activity was observed to avoid the inclusion of dendritic spikes in the synaptic activity (Herreras 1990). The input-output curves were generated from stimulus intensities ranging from 100 to 1000 μA in steps of 100 μA. At each stimulus intensity, four responses were recorded. The CSD analysis was performed by computing the second spatial derivative, without smoothing, along the depth of the probe whose sites were spaced 100 μm apart. Each fEPSP and population spike was measured from each of the four CSD responses at each stimulus and then averaged.
To analyze responses to ventral hippocampal commissure stimulation, the population spike was measured at the pyramidal layer of CA1 and the fEPSP was measured as the negative slope response in str. rad. We were unable to make reliable estimates of fEPSP for ventral hippocampal commissure stimulation when the time window was set to terminate 1 ms before the spike time because the onset of the synaptic activity occurred too close to the population spike in CA1 and the slope was too steep. We therefore estimated fEPSP by taking the steepest rising slope in the pyramidal layer and negative slope in str. rad. For stimulation intensities above the stimulation required to elicit a population spike the waveforms of these synaptic currents contain a small bump where the population spike activity occurred, but the synaptic sink persisted regardless of the population spike. This is similar to what has been reported previously as a feature of synaptic activity, whereas dendritic spikes would co-terminate with the action potential (Herreras 1990). The waveform, therefore, provides confidence that our estimate of fEPSP is not contaminated greatly by dendritic spiking activity. Finally, we performed one other estimate of the synaptic activity elicited by perforant path or ventral hippocampal commissure stimulation at a low intensity that did not elicit a spike and at the maximum stimulation intensity by taking the CSD depth at 1 ms prior to the population spike time elicited by the maximum stimulation intensity.
We also calculated the area under the curve for the pre- and post-spike sinks and sources at each layer. The pre-spike time was taken as 1.5 ms after the stimulation was delivered until the PS. The post-spike time was taken as the time from when the PS occurred until 20 ms after the stimulation was delivered. For the perforant path stimulation, the PS in the granule cell layer of the dentate gyrus was used. In the case of ventral hippocampal commissure stimulation, the timing was based on the PS in the CA1 pyramidal layer. To quantify the response at CA1 after perforant path stimulation, the negative response was quantified in slm, predominantly seen in the upper slm channel by computing the area under the curve from 1.5 to 20 ms after the stimulation was delivered.
Curve Fitting to examine multivariate relationships
For the dentate gyrus, the slope of the fEPSP-PS (E-S) coupling between the molecular layer and the granule cell layer was analyzed by fitting a Boltzman function to the data using the following equation with PS as a function of fEPSP:
Where PSmax is the estimated maximum PS observed, fEPSP50 is the fEPSP associated with the 50% PSmax, response; and S is the slope. A curve was fit for each animal and the parameters (PSmax, fEPSP50, and S) were averaged to create a group curve. All other multivariate analyses were performed using a linear fit.
Multivariate analyses were performed to determine the relationships between fEPSP at the dendritic compartments and at the soma, as well as to determine the relationship between the fEPSP at the dendritic compartment and the PS at the soma.
Paired-pulse inhibition
Stimulation to assess paired-pulse inhibition was performed at 65% of the intensity required to elicit the maximal PS response. Stimulus pairs were delivered at varying inter-stimulus intervals between the first and second pulses (5, 10, 20, 40, 80, 160, 320, and 640 ms). We allowed 30 s between each pair of stimuli. At each inter-stimulus interval, four responses were recorded. CSD analysis was performed to attenuate effects of volume conduction on each of the four responses. The responses for the first and second pulses were measured and the ratio of the second response to the first response used to determine the amount of inhibition on the second stimulation due to the initial stimulation. These ratios were then averaged across the four recordings.
Localization of the recording electrodes
Current source density analysis was used to locate sinks and sources after ventral hippocampal commissure or perforant path stimulation. The pattern of sinks and sources in each animal was used to localize the electrode recording sites for an individual rat. Stratum oriens, pyramidal layer, and str. rad. were identified using CSD from ventral hippocampal commissure evoked responses. The slm, molecular layer of the dentate gyrus, and the granule cell layer of the dentate gyrus were identified using CSD from perforant path evoked responses. We identified each layer by sinks or sources based on (Brankack et al. 1993; Wu and Leung 2003). From ventral hippocampal commissure stimulation, the pyramidal layer of CA1 was identified as a long large source that included a sink that was the population spike activity. From the pyramidal layer landmark, stratum oriens was identified as the large sink just above the pyramidal layer. The str. rad. was identified as the largest sink in the 3–4 channels below the pyramidal layer. From perforant path stimulation, the granule cell layer was identified by the largest source in the lower channels that also included a sink, which was the population spike activity. From the granule cell layer landmark, the molecular layer was identified as the largest sink in the 1–2 channels above the granule cell layer. The slm was identified by two channels: the large sources above the molecular layer and early latency sink in the 1–2 channels just above the largest source. Quantification of evoked potentials from the resulting CSD traces was performed on the localized channels. The slm response was taken as the negatively sloped response from the more upper of the two identified channels. The CSD area under the curve was measured separately for the sources and sinks within a single channel.
Verification of stimulation and recording sites
At the end of the recordings, the rats were transcardially perfused with 1X phosphate buffered saline (PBS) followed by 10% formalin. The brains were extracted and stored in formalin overnight. The brains were stored in 30% sucrose in 1X PBS until they were cut on a cryostat (40 μm) and thaw mounted onto gelatin-coated slides. The sections were dried overnight at room temperature and then Nissl stained. The slides were scanned at 10X with an Olympus VS120 microscope and the images were subsequently examined for electrode tracks to verify the stimulation and recording locations.
Statistical Analysis
Golgi Analysis
Group comparisons were made for each layer thickness using a two-tailed Student t test. Group comparisons of branching were performed using a two-tailed Student t test for the average length of branching in each of the fiber termination regions (inner 1/3rd, middle 1/3rd, and outer 1/3rd) of the molecular layer for granule cells. The same was done for the str. rad. and slm of CA1 pyramidal neurons. For the spine analysis, the GD17-MAM group was compared to the control group for each spine type and total spines using a two-tailed t test. Statistical significance was set to 0.05 for all comparisons.
Electrophysiological Analyses
Group comparisons of input/output responses were assessed by multivariate analysis of variance (MANOVA). Each response curve was evaluated separately, using stimulus intensity as a repeated measure. For the perforant path stimulation, we were unable to make reliable estimates of the fEPSP in the molecular layer for one control animal and found that the linear relationship between the fEPSP at the molecular layer and the fEPSP at the granule cell layer was a major outlier based on calculated inner and outer fences for the control group. We therefore dropped this animal from the analyses of the input-output curve at the molecular layer and the linear relationship between the molecular and granule cell layers. However, the estimates of fEPSP and PS at the granule cell layer appeared normal and this animal’s data was therefore left in for these analyses. To compare spike timing between groups, the MANOVA was performed only at the stimulation intensities at which all subjects had a population spike, which was > 100 μA for perforant path stimulation and > 300 μA for ventral hippocampal commissure stimulation.
Statistical significance was set at 0.05 for all comparisons. Two-tailed tests were performed unless stated otherwise. The statistics are included in the figure legends where possible for increased readability.
Results
Neuronal Morphology
We investigated the structural details of the GD17-MAM-altered hippocampus morphology using Golgi impregnated tissue (Fig. 1A). We assessed the thicknesses of different layers of the hippocampal circuit (Fig. 1B), as well as the morphology of individual principal cells to determine whether or not GD17-MAM effects on neuronal structure are region specific. We measured the layer thickness in DG granule cell and molecular layers, and in the CA1 stratum oriens, stratum pyramidale, str. rad., and slm layers. GD17-MAM and control rats have similar thickness of DG layers (Fig. 1A and C). In contrast, the CA1 region is markedly shrunk in GD17-MAM rats, with significantly thinned stratum oriens, pyramidal and str. rad. layers (Fig. 1C). We next evaluated the principal cell morphology, recognizing that DG and distal CA1 dendrites in slm both receive neocortical layer-specific inputs from entorhinal cortical layers II and III, respectively. CA1 also receives the intrahippocampal input from area CA3 at str. rad. dendrites (Fig. 1B). Accordingly, to determine whether GD17-MAM effects are homogeneous at the level of the principal cell or if instead GD17-MAM effects are circuit specific, we were interested to distinguish the different dendritic compartments in our evaluation.
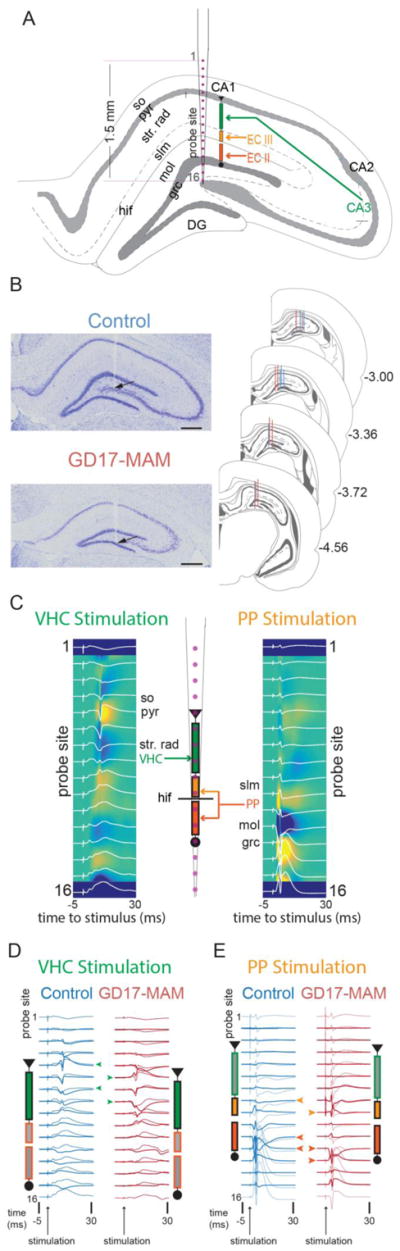
Figure 1. GD17-MAM exposure causes subfield-specific alterations of dorsal hippocampus principal cell morphology.

A) Golgi impregnated tissue was used to examine layer thicknesses, primary cell morphology, and spines. It can be seen from the tissue and the reconstructed neurons that the CA1 pyramidal layer is compressed in GD17-MAM rats. B) The hippocampus circuit receives its main input from the entorhinal cortex at both the molecular layer of DG (EC II) and in slm of CA1 (EC III). Information is transferred from DG to CA3 and then from CA3 to str. rad. of CA1. CA1 then acts as the main output of the hippocampus circuit. C) The stratum oriens, pyramidal and str. rad. of CA1 are significantly thinned in GD17-MAM rats compared to control rats (so: Group: t6 = 3.52; p = 0.01; pyr: t6 = 2.86; p = 0.03; str. rad.: t6 = 6.78; p < 0.001; slm: t6 = 1.75; p = 0.13; mol: t6 = 0.53; p = 0.61; grc: t6 = 1.40; p = 0.21. Control, n = 4; GD17-MAM, n = 4). D) Example Neurolucida reconstructions of granule cells from control and GD17-MAM rats (left panel) show similar cell morphologies. Dendritic length increases with distance from the cell body similarly in GD17-MAM and control rats (middle panel) and is not different between GD17-MAM and control rats in any sublayer of the molecular layer (right panel, inner mol: t30 = 0.70, p = 0.49; middle mol: t30 = 0.61, p = 0.55; outer mol: t30 = 0.23, p = 0.81). E) Branch order is also not different between GD17-MAM and control rats (1–3 branch order: t30 = 0.85, p = 0.40. ≥ 4 branch order: t30 = 0.28, p = 0.78. Granule cell neurons: Control, n = 16. GD17-MAM, n = 16). Spines were classified as thin, filipodia, mushroom, and stubby. In spite of similar overall morphology, GD17-MAM rats have an increased number of total spines per μm of branch length (thin: t46 = 1.25, p = 0.21; filipodia: t46 = 1.63, p = 0.11; mushroom: t46 = 0.75, p = 0.46; stubby: t46 = 1.13, p = 0.27; total: t46 = 2.39, p = 0.02. Branch segments: Control, n = 24; GD17-MAM, n = 24). F) Example Neurolucida reconstructions of CA1 pyramidal cells from control and GD17-MAM rats (left panel). Dendritic length of pyramidal neurons extends farther from the cell body in control rats (middle panel), and the total length within the str. rad. of CA1 is reduced in GD17-MAM rats while total branching in slm is not different (right panel, str. rad.: t23 = 2.30, p = 0.03. slm: t23 = 1.41, p = 0.17). Pyramidal neurons: Control, n = 14; GD17-MAM, n = 11. For D and F: Data is mean ± SEM for any concentric circle (distance from the neuron) that had more than 8 neurons. For all other graphs, data is mean ± SEM.
The structural impact of GD17-MAM on granule cell morphology (Fig. 1D, left panel) is minimal (Fig. 1D, middle panel). The molecular layer of DG receives input from the hilus, medial (MEC) and lateral (LEC) entorhinal cortex in the inner, middle and outer thirds, respectively (Amaral et al. 2007). Accordingly, we examined branching in each third separately. To account for potential variations in individual and group differences, dendritic length was normalized to the molecular layer thickness on a neuron-by-neuron basis. After normalization, there are no group differences in any third of the molecular layer (Fig. 1D, right panel). Branch order and spine density both indicate the potential receptivity of a neuron to its inputs. The branch order has been shown to correlate with place activity of granule cells; branch order ≥ 4 was more likely to be found on active cells and branch order ≤ 3 on silent cells (Diamantaki et al. 2016). While the branch order in the MEC-targeting middle third is not impacted by GD17-MAM exposure (Fig. 1E, left panel), the total number of spines (Fig. 1E, middle panel) is higher in GD17-MAM rats (Fig. 1E, right panel). In summary, GD17-MAM has relatively little impact on DG granule cell morphology, with the exception of increased spine density.
We also examined dendritic length of CA1 pyramidal cells (Fig. 1F). In control rats, CA1 pyramidal neurons extend further from the cell body than in GD17-MAM rats, in line with our observed reduction of str. rad. thickness in GD17-MAM rats (Fig. 1F, middle panel). After normalizing for layer thickness on a neuron-by-neuron basis, dendritic length within str. rad., but not slm, is reduced in GD17-MAM rats (Fig. 1F, right panel). While these data do not address potential differences in number of cells in GD17-MAM and control hippocampus (Sanderson et al. 2012), these data do indicate that individual principal cells at the hippocampus input and output of GD17-MAM rats have morphological differences that alter, and possibly constrain, their ability to receive neuronal input.
In vivo evoked synaptic responses
We evaluated synaptic responses to stimulation of the main inputs to dorsal DG (perforant path) and CA1 (perforant path and ventral hippocampal commissure) (Fig. 2A,B), where morphological abnormalities were identified. Responses to separate perforant path and ventral hippocampal commissure stimulation were recorded using a linear electrode array (Fig. 2A) and current source density (CSD) analysis (Fig. 2C, 3A, and 4A) to localize the evoked responses along the somatodendritic axis of dorsal hippocampus. The wave shapes of the responses to both perforant path and ventral hippocampal commissure stimulations are indistinguishable in GD17-MAM and control rats, suggesting similar biophysical origins (Fig. 2E,F).
Figure 2. In vivo evoked potential recording setup.

Evoked potentials were recorded from rats under urethane anesthesia. A) A 16-site silicon electrode linear array was placed into the dorsal hippocampus such that the recording sites spanned the dendritic compartments of CA1 and DG. B) Recording electrodes were identified to have been placed in the dorsal hippocampus in both Control and GD17-MAM rats. Scale bars are 500 μm. C) Stimulating wires were placed in the ventral hippocampal commissure or the angular bundle to stimulate the perforant path. Fibers from ventral hippocampal commissure terminate in str. rad. of CA1, while fibers from perforant path terminate in slm of CA1 and in the molecular layer of DG. Example evoked responses from stimulation of ventral hippocampal commissure or perforant path along the 16 sites. The voltage traces have been laid over the CSD to show how the traces correspond to sinks and sources used to identify the main dendritic compartments. For display purposes, the CSD was smoothed in the time direction. D) Example traces of evoked responses from control (blue) and GD17-MAM (red) rats after ventral hippocampal stimulation. The lighter blue and red indicate the voltage traces recorded. The darker blue and red are the CSD traces. The green arrows represent the channels that were used to quantify fEPSP and population spiking activity. E) Example traces of evoked responses control (blue) and GD17-MAM (red) rats after peforant path stimulation. The lighter blue and red indicate the voltage traces recorded. The darker blue and red are the CSD traces. The light orange arrows represent the channels that were used to quantify the area under the curve at slm and the dark orange arrows represent the channels that were used to quantify the fEPSP and PS. CA1 = cornu ammonis 1 of the hippocampus, CA2 = cornu ammonis 2 of the hippocampus, CA3 = cornu ammonis 3 of the hippocampus, so = stratum oriens, pyr = pyramidal layer, str. rad. = stratum radiatum, slm = stratum lacunosum moleculare, mol = molecular layer of DG, grc = granule cell layer of DG, hif = hippocampal fissure.
Figure 3. GD17-MAM rats have abnormal evoked synaptic network responses to perforant path stimulation.
A) Heatmaps of the average CSD across the dorsal hippocampus for control and GD17-MAM groups in response to perforant path stimulation show spiking activity in DG. For display purposes, the CSD was smoothed in the time direction. The grey dashed lines correspond to the time before and after the spiking activity that was quantified in B) and C). B) The source in the granule cell layer is stronger in GD17-MAM rats in the 1.5 ms after the stimulation was delivered until the spike occurred (t12 = 23.05, one-tailed p = 0.03). C) The source in the granule cell layer is significantly higher in GD17-MAM rats from the spike activity to 20 ms after the stimulation was delivered (t12 = 26.08, one-tailed p = 0.02). There is no difference in pre- or post-spike sink in the molecular layer (Pre-spike: t12 = 0.34, one-tailed p = 0.37; Post-spike: t12= 0.99, one-tailed p = 0.18). Average CSD depth profiles (dark lines) and SEM (shaded) were created for D) molecular layer and E) granule cell layer at low intensity stimulation (100 μA) that did not elicit a population spike and high intensity stimulation (1000 μA) that did elicit a population spike. F) The depth profiles were used to examine synaptic activity 1 ms before the population spike time that was elicited by the 1000 μA stimulation. There are no differences in synaptic population responses in the molecular layer or the granule cell layer at either stimulation intensity (mol: 100μA: t12 = 0.64, p = 0.53; 1000 μA: t12 = 0.60, p = 0.56; grc: 100μA: t12 = 0.10, p = 0.92; 1000 μA: t12 = 1.71, p = 0.11). The fEPSP slope was quantified at G) the molecular layer, where input fibers terminate, and H) the granule cell layer. There are no difference in DG fEPSPs at either the dendritic or cell layers (mol: Group: F1,12 < 0.01, p = 0.99; Stimulation Intensity: F9,4 = 2.94, p = 0.15; Interaction: F9,4 = 0.58, p = 0.77. grc: Group: F1,12 = 2.78, p = 0.12; Stimulation Intensity: F9,4 = 9.37, p = 0.02, Interaction F9,4 = 1.52, p = 0.36). J) The PS response to perforant path stimulation is indistinguishable between GD17-MAM and control rats (Group: F1,12 = 0.29, p = 0.60; Stimulation Intensity: F9,4 = 3.57, p = 0.12. Interaction: F9,4 = 0.98, p = 0.55). Calibration is the same for G, H, and J. K) The relationship between the fEPSP slope at the dendrite (molecular layer) and fEPSP slope at the soma (granule cell) is linear and L) the slope is increased in GD17-MAM rats (t12 = 2.85, p = 0.02). M) E-S coupling is right-shifted in GD17-MAM rats compared to control rats, N) as indicated by a higher fEPSP50 in GD17-MAM rats (t11 = 2.89, p = 0.02). Values are average ± SEM, except in K and M, where each individual value is plotted. Control n = 7, GD17-MAM n = 7 except for G,K, and L: Control n = 6, GD17-MAM n = 7. so = stratum oriens, pyr = pyramidal layer, str. rad. = stratum radiatum, slm = stratum lacunosum moleculare, mol = molecular layer of DG, grc = granule cell
Figure 4. Feedback inhibition appears normal in GD17-MAM rats.

A) The dentate gyrus is a complex circuit with both feedforward and feedback inhibition. Responses of control and GD17-MAM rats to pairs of pulses are shown at various inter-stimulus intervals (ISI). The response to the first stimulus is shown in black and the response to the second stimulus is shown in blue or red. The lighter lines are voltage responses and the darker responses are CSD, i.e. after correction for volume conduction. Calibration is the same for both control and GD17-MAM. There are no group differences in ratios of the second second response to the first response for B) population spiking activity (Group: F1,7 = 0.26, p = 0.63; ISI: F7,1 = 2.98×104, p < 0.01; Interaction: F7,1 = 9.20×103, p = 0.03). Post hoc analyses indicated no group differences at any inter stimulus interval. Additionally, there are no group differences in fEPSP measured at C) the granule cell layer (Group: F1,7 = 1.71, p = 0.23; Inter stimulus interval: F7,1 = 2238.25, p = 0.02; Interaction: F7,1 = 22.23, p = 0.16) and D) the molecular layer (Group: F1,7 = 1.76, p = 0.23; ISI: F7,1 = 228.14, p = 0.05; Interaction: F7,1 = 4.86, p = 0.34). There are two phases to the inhibition, an early strong inhibition that occurs until 20 ms, and a persistent inhibition that lasts up to 0.5 s, as seen by the fact that the response ratios do not recover to 1. Values are average ± SEM. Calibration is the same for B, C and D. For B and C: Control n = 5, GD17-MAM n = 5. For D: Control n = 4, GD17-MAM n = 5. mol = molecular layer of DG, grc = granule cell layer of DG.
Increased DG spine density in GD17-MAM rats predicts a greater response to perforant path stimulation. Indeed, the extracellular current source in the granule cell layer of DG of GD17-MAM rats is increased prior to the population spike (Fig. 3B) as well as after (Fig. 3C). We quantified the fEPSP from the CSD in the molecular layer, the termination zone of entorhinal cortex inputs. The synaptic response within the molecular layer is indistinguishable between GD17-MAM and control rats when measured as either the depth of the CSD (Fig. 3D,F) or as the slope of the CSD (Fig. 3G). However, when measured at the granule cell layer, there is a non-significant trend for a stronger fEPSP in GD17-MAM rats at high stimulation intensities (Fig. 3E,F, and H). The population spike at the granule cell layer is indistinguishable between GD17-MAM and control rats (Fig. 3J) and the timing of the population spike was not different between groups for any stimulation intensity (Group: F1,12 = 0.20, p = 0.66; Stimulation Intensity: F9,4 = 5.11, p = 0.07; Interaction: F9,4 = 1.95, p = 0.27).
To assess the possibility of an abnormal relationship between these measures of synaptic function, we examined how the measures covary. The linear relationship between the synaptic responses, measured as the slope of the fEPSP, at the dendritic input and the soma of granule cells is shifted in GD17-MAM rats such that the somatic response is proportionally greater for a given dendritic response (Fig. 3K,L). This shift is consistent with greater feedforward perisomatic inhibition in GD17-MAM responses to perforant path activation and, in line with this possibility, the sigmoid relationship between somatic response and the population spike is right-shifted in GD17-MAM rats (Fig. 3M,N).
To further assess inhibition, we examined E-S coupling between the dendritic response in the molecular layer and the population spike, which is not different between GD17-MAM and control rats (data not shown). We then examined paired-pulse responses (Fig. 4A), finding no group differences in any of the three responses (Fig. 4B–D). These observations indicate a complex set of changes in the entorhinal cortex-dentate physiology of GD17-MAM rats that may indicate altered excitation-inhibition balance in order to maintain DG output within normal limits.
Consistent with an apparently normal slm in GD17-MAM rats, the pre- and post-spike sinks and sources are not different between GD17-MAM and control rats after perforant path stimulation (Fig. 3B,C) and there was no difference in the area under the curve for the entire response, measured from 1.5 ms after the stimulation 20 ms after the stimulation, at any stimulation intensity (Group: F1,12 = 0.55, p = 0.47; Stimulation Intensity: F9,4 = 2.80, p = 0.17; Interaction: F9,4 = 0.72, p = 0.69).
The interaction between potentially changed cable properties and likelihood of synaptic contacts that arise from reduced dendritic length in str. rad. of CA1 complicates predictions for the functional impact of GD17-MAM treatment. CSD analysis of CA1 responses to ventral hippocampus commissure stimulation do not reveal GD17-MAM-associated abnormalities (Fig. 5A–C), although there is again a nonsignificant trend for the fEPSP responses at the pyramidal layer to be reduced in the GD17-MAM group (Fig. 5D,F,H). In str. rad, the synaptic activity was indistinguishable between GD17-MAM and control animals when measured either as the depth of the CSD profile 1 ms prior to the population spike (Fig. 5E,F) or when estimating the fEPSP as the negative slope in str. rad (Fig. 5G). The activity of stratum oriens was difficult to measure consistently in our study, perhaps because the spacing between probe sites of 100 μm was too large for consistent placement in this layer. There was no difference in the timing of the population spikes (Group: F1,8 = 1.74, p = 0.22; Stimulation Intensity: F7,2 = 0.61, p = 0.74; Interaction: F7,2 = 0.43, p = 0.83) or in the amplitude of the population spike (Fig. 5J). Nonetheless, the relationship between the synaptic responses, measured as fEPSP slopes, at the dendritic compartment and the soma is altered; although synaptic responses at str. rad. did not differ, the corresponding synaptic responses at the soma were attenuated in GD17-MAM rats compared to control (Fig. 5K). Not only is this change opposite to what is observed in DG (Fig. 3K,M), the relationship between the fEPSP slope at the dendritic input and the soma is not linear in CA1, whereas in DG there is a strong linear relationship between the efficacy of dendritic and somatic fEPSPs. The relationship between the synaptic response and the CA1 output is correspondingly reduced (Fig. 5L).
Figure 5. GD17-MAM rats have abnormal CA1 synaptic population responses to ventral hippocampal commissure stimulation.

A) Heatmaps of the average CSD in dorsal hippocampus for control and GD17-MAM groups in response to ventral hippocampal commissure stimulation show spiking activity in CA1. For display purposes, the CSD was smoothed in the time direction. The grey dashed lines correspond to the time before and after the spiking activity that was quantified in B) and C). Quantification of sinks and sources at maximum stimulation intensity within each layer of the dorsal hippocampus B) before and C) after spiking activity revealed no differences in sinks or sources in CA1 (Pre-spike: sosink: t8 = 0.16, p = 0.87; pyrsource: t 8 = 1.78, p = 0.11; str. rad.sink: t8 = 0.73, p = 0.48; Post-spike: sosink: t8 = 1.07, p = 0.31; pyrsource: t8 = 1.11, p = 0.30; str. rad.sink: t8 = 0.02, p = 0.99). Average CSD depth profiles (dark lines) and SEM (shaded) were created for D) pyramidal layer and E) str. rad at low intensity stimulation (100 μA) that did not elicit a population spike and high intensity stimulation (1000 μA) that did elicit a population spike. F) The depth profiles were used to examine synaptic activity 1 ms before the population spike time that was elicited by the 1000 μA stimulation. There are no differences in synaptic activity in the pyramidal layer or str. rad at either stimulation intensity (pyr: 100μA: t8 = 0.15, p = 0.88; 1000 μA: t8 = 1.38, p = 0.20; str. rad: 100μA: t8 = 0.75, p = 0.47; 1000 μA: t12 = 1.17, p = 0.28). The fEPSP was quantified in G) str. rad., where input fibers terminate, and H) the pyramidal cell layer. There is no difference in the slope of the fEPSP of CA1 at either the dendritic or cell layers (str. rad.: Group: F1,8 = 0.88, p = 0.38, p = 0.37. Stimulation Intensity: F1.63,13.04 = 26.81, p < 0.001; Interaction: F1.63,13.04 = 0.11, p = 0.86; pyr: Group: F1,8 = 1.32, p = 0.28; Stimulation Intensity: F1.35,10.83 = 14.36, p < 0.01; Interaction: F1.35,10.83 = 1.73, p = 0.22). J) The population spiking activity was measured at the cell layer. The CA1 population spike response to ventral hippocampal commissure stimulation is indistinguishable between GD17-MAM and control rats (Group: F1,8 = 3.21, p = 0.11; Stimulation Intenstiy: F1.80,14.35 = 9.30, p < 0.01;Interaction: F1.80,14.35 = 2.38, p = 0.15). Calibration is the same in G, H, and J. The multivariate relationships between K) fEPSP slope at the dendrite (str. rad.) and fEPSP slope at the soma (pyramidal layer) and L) the fEPSP slope at the dendrite (str. rad.) and the population spiking activity at the soma (pyramidal layer) are attenuated in GD17-MAM rats and there is a significant difference between cumulative probabilities (H: Control n = 160, GD17-MAM n = 240, Mann-Whitney U = 5.3×103, p = 10−15; J: Mann-Whitney U = 1.04×104, p = 10−15). Values are average ± SEM, except in K and L, where each individual value is plotted. Control n = 4, GD17-MAM n = 6. so = stratum oriens, pyr = pyramidal layer, str. rad. = stratum radiatum, slm = stratum lacunosum moleculare, mol = molecular layer of DG, grc = granule cell layer of DG.
Together these data indicate that in both DG and CA1, although the components of the synaptic responses appear to be normal, the relationship between the dendritic input and the somatic output is altered by GD17-MAM exposure.
Discussion
Abnormal neurodevelopment is predicted to result in abnormal adult circuits and function. Adult GD17-MAM animals have diverse abnormalities that include cognitive and motor dysfunction, with relevance to neurodevelopmental disorders, including schizophrenia and epilepsy (Lodge and Grace 2009; Le Pen et al. 2006; Gastambide et al. 2015; Gourevitch et al. 2004; O’Reilly et al. 2016; Ratajczak et al. 2015; Lucas et al. 2011; Jenks et al. 2013). The gestational timing of MAM exposure provides an animal model for testing neurodevelopmental hypotheses to explain the origins of schizophrenia. We used the GD17-MAM model to examine the impact of neurodevelopmental insult on adult brain circuitry.
We previously reported that, after accounting for motor abnormalities, adult GD17-MAM rats have relatively intact cognitive abilities, measured using the active place avoidance paradigm, but with a mild impairment to accumulate new spatial memories in this dHPC-dependent task (O’Reilly et al. 2016; Cimadevilla et al. 2001; Pastalkova et al. 2006). We therefore chose to investigate whether dHPC abnormalities are present at the levels of cellular morphology and synaptic function. The results presented here elucidate how a global neurodevelopmental insult can cause sub-circuit abnormalities, including differential effects in sub-cellular domains. As such, the findings are relevant to how we think about neurodevelopmental insult in general, and the GD17-MAM model in particular, with respect to schizophrenia and other cognitive dysfunction.
The dHPC is understudied in the GD17-MAM model, where the focus has typically been on the ventral hippocampus. The extent to which dHPC is abnormal, either grossly or specifically to sub-circuit levels, is unknown. We examined the input and output subfields and identified subfield-specific morphological abnormalities of the dorsal hippocampus in GD17-MAM rats (Fig. 1). Instead of gross, non-specific abnormality, we find shrinkage in CA1, but not DG. Within CA1, this shrinkage was restricted to the synaptically defined str. rad. sub-layer where intrahippocampal CA3 inputs terminate, indicating input-specific abnormalities result from GD17-MAM exposure. As might be expected from a neurodevelopmental insult, this circuit-specific pattern of abnormality is likely due to the sequence of developmental events in the hippocampus. Gestational day 17 exposure to MAM, which is expected to last for up to three days (Lodge 2013), coincides with peak neurogenesis in CA1 at GD19 and migration of pyramidal cells from GD18 to birth. This is also the approximate time of arrival of interneurons in str. rad. between GD15–19 that act as pioneer neurons to attract, and initially form synapses with, CA3 Shaffer collaterals (Danglot et al. 2006). In contrast, Cajal Retzius cells, which attract entorhinal inputs, are the first cells present in the hippocampus marginal zone, identifiable at GD13, the outer portion of which becomes slm (Soriano et al. 1994). This pattern of developing slm and then str. rad. is consistent with the relatively normal slm. branching versus str. rad. branching we observed in rats exposed to GD17-MAM during str. rad. development.
Because the entire developing fetus is exposed to MAM, one might expect other sub-circuit abnormalities in the brain based on the timing of the neurodevelopmental insult. The GD17-MAM model is thought to target the development of limbic and frontal cortices, the latter of which we found no obvious impact of GD17-MAM on general principal cell morphology (data not shown). This impact of timing may help to explain why neurodevelopmental mental illnesses like schizophrenia tend to be spectrum disorders, which are at once heterogeneous and difficult to differentiate from other mental illness classifications (Kirkpatrick et al. 2001; Cross-Disorder Group of the Psychiatric Genomics 2013; Vorstman et al. 2013).
The compressed str. rad. in GD17-MAM brains, accompanied by reduced dendritic length, implies a disbalance between information coming from CA3, where memories are thought to be stored, and information coming from the entorhinal cortex, which projects the neural representation of current sensory information into slm (Fries 2009; Colgin et al. 2009). Despite these morphological abnormalities, and in contrast to findings from behaviorally naïve hippocampus slice experiments (Sanderson et al. 2012) synaptic input at str. rad. appeared normal in CA1. In spite of this normal synaptic activity at str. rad, and opposite from findings in naïve hippocampus slices, the coupling between the fEPSP at str. rad. and the soma was attenuated in GD17-MAM rats (Fig. 5), a finding that could result from a number of abnormalities, such as excitation-inhibition disbalance. The attenuated dendritic-somatic functional abnormality from CA3-to-CA1 was distinctive from the amplified dendritic-somatic functional abnormality from entorhinal cortex-to-DG (Fig. 3). The amplified dendritic-somatic coupling in DG is difficult to interpret because of the feed-forward and feedback inhibition in the granule cell layer. Inhibition was not different when paired-pulse stimuli were applied using a protocol that examines feedback inhibition. Overall, these compartment specific modifications reveal a disrupted dorsal hippocampal circuit in the input, output, and input-output relationships.
The attenuated dendritic-somatic coupling in CA1 and amplified dendritic-somatic coupling in DG, as well as the increase in spines in the molecular layer, point to another level of circuit-specific abnormality in GD17-MAM rats, one that may be due to functional compensation for more primary abnormalities. The DG abnormalities are not easily explained by the timing of GD17-MAM exposure. In DG, 85% of granule cells are born after GD21 (Bayer 1980), after MAM exposure has occurred. While some of the circuit-specific abnormalities may be a primary effect of the GD17-MAM neurodevelopmental insult, other abnormalities may be a secondary result that is functional compensation, as has been observed in an acute model of hippocampus circuit disruption (Olypher et al. 2006).
The impact of GD17-MAM on structure of DG granule cells appeared minimal in spite of stronger DG activity after perforant path stimulation, whereas GD17-MAM appeared to greatly alter CA1 pyramidal neurons at the CA3 input even though the response to CA3 stimulation was not different in GD17-MAM and control rats. Consequently, there is a substantial explanatory gap between the neurobiological abnormalities associated with mechanism and the behavioral abnormalities associated with symptoms.
Although some relationships between abnormalities appear straightforward, such as increased spines in the molecular layer of DG and the increased responses to perforant path stimulation at the granule cell layer (Figs. 1 and 3), it is a substantial challenge to identify potential causal relationships to drive understanding of whether and how the majority of these particular abnormalities are related. We would like to speculate as to the causal relationships between the abnormalities we observed at the various levels of investigation and the impact these abnormalities may have on memory formation in GD17-MAM rats. However, understanding these relationships is limited because the current studies are not causal experiments. To develop causal experiments, the field would need a better understanding of the relationships between hippocampus structure, physiology and behavior. Additionally, the two-frame active place avoidance training experience alters what would have otherwise seemed to be fundamental system properties (Park et al. 2015; Pavlowsky et al. 2017; Talbot et al. 2018), making inferences from slice experiments, and whole-brain experiments sometimes difficult to compare [e.g. compare Fig. 5J and Fig. 3 from (Sanderson et al. 2012)]. There may be new opportunities to better understand mental illness by conducting investigations that integrate across levels of biological function (Mitchell et al. 2013). Given the naturally occurring wide variation that is prevalent in biological systems, how tightly constrained a parameter has to be to yield normal function varies from parameter to parameter (Prinz et al. 2004). The key insight in differentiating two groups of subjects might be that one group occupies the margins close to the function/dysfunction border of the parameter space that is typically defined by the healthy subpopulation (Marder and Bucher 2007; Schulz et al. 2007).
Acknowledgments
This work was supported by NIH-R01MH084038 and NIH-R25NS080686-06. The authors would like to thank Drs. Hsin-Yi Kao and Edith Lesburgueres for help generating control and GD17-MAM rats. We would also like to thank Yi-Wen Chen and Dr. Chiye Aoki for assistance with the Golgi studies. Preliminary reports of these data have been present at the Annual meetings for Society for Neuroscience (2016) and the Canadian Association for Neuroscience (2016).
Footnotes
Author Contributions: KCO, AVP, and MIP collected data, KCO and ERJL performed analyses, KCO, and AAF designed research and wrote the manuscript.
Compliance with Ethical Standards
Conflicts of Interest: The authors have no conflicts to report.
Animal Welfare: All methods complied with Public Health and Service Policy on Humane Care and Use of Laboratory Animals and were approved by the New York University Animal Welfare Committee.
References
- Amaral DG, Scharfman HE, Lavenex P. The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies) Prog Brain Res. 2007;163:3–22. doi: 10.1016/S0079-6123(07)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA. Development of the hippocampal region in the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. J Comp Neurol. 1980;190(1):87–114. doi: 10.1002/cne.901900107. [DOI] [PubMed] [Google Scholar]
- Brankack J, Stewart M, Fox SE. Current source density analysis of the hippocampal theta rhythm: associated sustained potentials and candidate synaptic generators. Brain Res. 1993;615(2):310–327. doi: 10.1016/0006-8993(93)90043-m. [DOI] [PubMed] [Google Scholar]
- Cimadevilla JM, Wesierska M, Fenton AA, Bures J. Inactivating one hippocampus impairs avoidance of a stable room-defined place during dissociation of arena cues from room cues by rotation of the arena. Proc Natl Acad Sci U S A. 2001;98(6):3531–3536. doi: 10.1073/pnas.05162839898/6/3531. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL, Denninger T, Fyhn M, Hafting T, Bonnevie T, Jensen O, Moser MB, Moser EI. Frequency of gamma oscillations routes flow of information in the hippocampus. Nature. 2009;462(7271):353–357. doi: 10.1038/nature08573. [DOI] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danglot L, Triller A, Marty S. The development of hippocampal interneurons in rodents. Hippocampus. 2006;16(12):1032–1060. doi: 10.1002/hipo.20225. [DOI] [PubMed] [Google Scholar]
- Diamantaki M, Frey M, Berens P, Preston-Ferrer P, Burgalossi A. Sparse activity of identified dentate granule cells during spatial exploration. Elife. 2016:5. doi: 10.7554/eLife.20252. [DOI] [PMC free article] [PubMed]
- Fries P. The model- and the data-gamma. Neuron. 2009;64(5):601–602. doi: 10.1016/j.neuron.2009.11.024. [DOI] [PubMed] [Google Scholar]
- Gastambide F, Taylor AM, Palmer C, Svard H, Karjalainen M, Janhunen SK, Tricklebank M, Bannerman DM. Alterations in spatial memory and anxiety in the MAM E17 rat model of hippocampal pathology in schizophrenia. Psychopharmacology (Berl) 2015;232(21–22):4099–4112. doi: 10.1007/s00213-014-3862-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourevitch R, Rocher C, Le Pen G, Krebs MO, Jay TM. Working memory deficits in adult rats after prenatal disruption of neurogenesis. Behav Pharmacol. 2004;15(4):287–292. doi: 10.1097/01.fbp.0000135703.48799.71. [DOI] [PubMed] [Google Scholar]
- Hernan AE, Mahoney JM, Curry W, Richard G, Lucas MM, Massey A, Holmes GL, Scott RC. Environmental enrichment normalizes hippocampal timing coding in a malformed hippocampus. PLoS One. 2018;13(2):e0191488. doi: 10.1371/journal.pone.0191488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreras O. Propagating dendritic action potential mediates synaptic transmission in CA1 pyramidal cells in situ. J Neurophysiol. 1990;64(5):1429–1441. doi: 10.1152/jn.1990.64.5.1429. [DOI] [PubMed] [Google Scholar]
- Insel TR. Rethinking schizophrenia. Nature. 2010;468(7321):187–193. doi: 10.1038/nature09552. nature09552 [pii] [DOI] [PubMed] [Google Scholar]
- Jaaro-Peled H, Hayashi-Takagi A, Seshadri S, Kamiya A, Brandon NJ, Sawa A. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 2009;32(9):485–495. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenks KR, Lucas MM, Duffy BA, Robbins AA, Gimi B, Barry JM, Scott RC. Enrichment and training improve cognition in rats with cortical malformations. PLoS One. 2013;8(12):e84492. doi: 10.1371/journal.pone.0084492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick B, Buchanan RW, Ross DE, Carpenter WT., Jr A separate disease within the syndrome of schizophrenia. Arch Gen Psychiatry. 2001;58(2):165–171. doi: 10.1001/archpsyc.58.2.165. [DOI] [PubMed] [Google Scholar]
- Le Pen G, Gourevitch R, Hazane F, Hoareau C, Jay TM, Krebs MO. Peri-pubertal maturation after developmental disturbance: a model for psychosis onset in the rat. Neuroscience. 2006;143(2):395–405. doi: 10.1016/j.neuroscience.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK Clinical Antipsychotic Trials of Intervention Effectiveness I. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Lodge DJ. The MAM rodent model of schizophrenia. Curr Protoc Neurosci. 2013;Chapter 9(Unit9):43. doi: 10.1002/0471142301.ns0943s63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27(42):11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Gestational methylazoxymethanol acetate administration: a developmental disruption model of schizophrenia. Behav Brain Res. 2009;204(2):306–312. doi: 10.1016/j.bbr.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas MM, Lenck-Santini PP, Holmes GL, Scott RC. Impaired cognition in rats with cortical dysplasia: additional impact of early-life seizures. Brain. 2011;134(Pt 6):1684–1693. doi: 10.1093/brain/awr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Bucher D. Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu Rev Physiol. 2007;69:291–316. doi: 10.1146/annurev.physiol.69.031905.161516. [DOI] [PubMed] [Google Scholar]
- Matricon J, Bellon A, Frieling H, Kebir O, Le Pen G, Beuvon F, Daumas-Duport C, Jay TM, Krebs MO. Neuropathological and Reelin deficiencies in the hippocampal formation of rats exposed to MAM; differences and similarities with schizophrenia. PLoS One. 2010;5(4):e10291. doi: 10.1371/journal.pone.0010291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KJ, O’Donnell P, Durstewitz D, Fenton AA, Gingrich JA, Gordan JA, Kelsch W, Moghaddam B, Phillips WA, Sawa A. A framework for the use of models in schizophrenia. In: Silverstein SM, Moghaddam B, Wykes T, editors. Schizophrenia: Evoluation and Synthesis. MIT Press; Cambridge, MA: 2013. pp. 212–226. [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60(3):253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KC, Perica MI, Fenton AA. Memory deficits with intact cognitive control in the methylazoxymethanol acetate (MAM) exposure model of neurodevelopmental insult. Neurobiol Learn Mem. 2016 doi: 10.1016/j.nlm.2016.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olypher AV, Klement D, Fenton AA. Cognitive disorganization in hippocampus: a physiological model of the disorganization in psychosis. J Neurosci. 2006;26(1):158–168. doi: 10.1523/JNEUROSCI.2064-05.2006. 26/1/158 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EH, Burghardt NS, Dvorak D, Hen R, Fenton AA. Experience-dependent regulation of dentate gyrus excitability by adult-born granule cells. Journal of Neuroscience. 2015 doi: 10.1523/JNEUROSCI.0885-15.2015. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313(5790):1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Pavlowsky A, Wallace E, Fenton AA, Alarcon JM. Persistent modifications of hippocampal synaptic function during remote spatial memory. Neurobiol Learn Mem. 2017;138:182–197. doi: 10.1016/j.nlm.2016.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz AA, Bucher D, Marder E. Similar network activity from disparate circuit parameters. Nat Neurosci. 2004;7(12):1345–1352. doi: 10.1038/nn1352. [DOI] [PubMed] [Google Scholar]
- Ratajczak P, Kus K, Murawiecka P, Slodzinska I, Giermaziak W, Nowakowska E. Biochemical and cognitive impairments observed in animal models of schizophrenia induced by prenatal stress paradigm or methylazoxymethanol acetate administration. Acta Neurobiol Exp (Wars) 2015;75(3):314–325. [PubMed] [Google Scholar]
- Risher WC, Ustunkaya T, Singh Alvarado J, Eroglu C. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS One. 2014;9(9):e107591. doi: 10.1371/journal.pone.0107591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TM, Cotel MC, O’Neill MJ, Tricklebank MD, Collingridge GL, Sher E. Alterations in hippocampal excitability, synaptic transmission and synaptic plasticity in a neurodevelopmental model of schizophrenia. Neuropharmacology. 2012;62(3):1349–1358. doi: 10.1016/j.neuropharm.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Goaillard JM, Marder EE. Quantitative expression profiling of identified neurons reveals cell-specific constraints on highly variable levels of gene expression. Proc Natl Acad Sci U S A. 2007;104(32):13187–13191. doi: 10.1073/pnas.0705827104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano E, Del Rio JA, Martinez A, Super H. Organization of the embryonic and early postnatal murine hippocampus. I. Immunocytochemical characterization of neuronal populations in the subplate and marginal zone. J Comp Neurol. 1994;342(4):571–595. doi: 10.1002/cne.903420406. [DOI] [PubMed] [Google Scholar]
- Talbot ZN, Sparks FT, Dvorak D, Curran BM, Alarcon JM, Fenton AA. Normal CA1 Place Fields but Discoordinated Network Discharge in a Fmr1-Null Mouse Model of Fragile X Syndrome. Neuron. 2018;97(3):684–697e684. doi: 10.1016/j.neuron.2017.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Breetvelt EJ, Thode KI, Chow EW, Bassett AS. Expression of autism spectrum and schizophrenia in patients with a 22q11.2 deletion. Schizophr Res. 2013;143(1):55–59. doi: 10.1016/j.schres.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44(7):660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. On the plausibility of “the neurodevelopmental hypothesis” of schizophrenia. Neuropsychopharmacology. 1996;14(3 Suppl):1S–11S. doi: 10.1016/0893-133X(95)00199-N0893133X9500199N. [pii] [DOI] [PubMed] [Google Scholar]
- Wu K, Leung LS. Increased dendritic excitability in hippocampal ca1 in vivo in the kainic acid model of temporal lobe epilepsy: a study using current source density analysis. Neuroscience. 2003;116(2):599–616. doi: 10.1016/s0306-4522(02)00567-5. [DOI] [PubMed] [Google Scholar]