

Abstract
We sequenced the genome of the strawberry poison frog, Oophaga pumilio, at a depth of 127.5× using variable insert size libraries. The total genome size is estimated to be 6.76 Gb, of which 4.76 Gb are from high copy number repetitive elements with low differentiation across copies. These repeats encompass DNA transposons, RNA transposons, and LTR retrotransposons, including at least 0.4 and 1.0 Gb of Mariner/Tc1 and Gypsy elements, respectively. Expression data indicate high levels of gypsy and Mariner/Tc1 expression in ova of O. pumilio compared with Xenopus laevis. We further observe phylogenetic evidence for horizontal transfer (HT) of Mariner elements, possibly between fish and frogs. The elements affected by HT are present in high copy number and are highly expressed, suggesting ongoing proliferation after HT. Our results suggest that the large amphibian genome sizes, at least partially, can be explained by a process of repeated invasion of new transposable elements that are not yet suppressed in the germline. We also find changes in the spliceosome that we hypothesize are related to permissiveness of O. pumilio to increases in intron length due to transposon proliferation. Finally, we identify the complement of ion channels in the first genomic sequenced poison frog and discuss its relation to the evolution of autoresistance to toxins sequestered in the skin.
Keywords: transposable elements, amphibian genomics, poison frogs, horizontal transfer
Introduction
The strawberry poison frog, Oophaga pumilio (formerly Dendrobates pumilio) is unusual because of its life history, toxicity, and variable coloration. This small, terrestrial frog is found in lowland forests along the Caribbean side of Central America from Nicaragua to Panama. Females deposit tadpoles in water pools contained in plant leaf axils or tanks, where they develop into froglets (Weygoldt 1980). These frogs display several behaviors that are unusual for an amphibian. Males and females exhibit parental care, with transporting tadpoles and feeding them with unfertilized eggs (Weygoldt 1980). Females exhibit significant color-based selectivity during courtship and mating (Summers et al. 1999; Yang et al. 2016) and brooding parents are strongly territorial with male–male combat (Yang et al. 2016). These behaviors are of interest to researchers attempting to better understand the origins of parental care (Dulac et al. 2014) and reproductive isolation (Yang et al. 2016).
Beyond these behavioral traits, O. pumilio is poisonous, sequestering alkaloid toxins from its diet (Santos et al. 2016). Its aposematic coloration advertises this toxicity to predators. These toxins disrupt ion channel function, producing noxious taste and adverse neurological effects in predators (Lander et al. 2001). The toxicity of this poison frog depends on the acquisition of toxins from the diet (Daly et al. 1994). Toxins deposited in the eggs protect offspring from predation (Stynoski, Shelton, et al. 2014; Stynoski, Torres-Mendoza, et al. 2014). The frog itself is immune to its own toxins, presumably due to interacting amino acid changes at several ion channels (Tarvin et al. 2016, 2017). The species displays remarkable variation in aposematic coloration patterns across populations, a trait known to influence mate choice (Summers et al. 1999; Yang et al. 2016). Although theory suggests that aposematic species should benefit from a consistent warning coloration (Joron and Mallet 1998), populations of O. pumilio display a wide array of aposematic coloration patterns across populations (Daly and Myers 1967). The genetic basis of these color patterns and how they might be related to toxin profiles remains an open question in evolutionary genetics (Richards-Zawacki et al. 2012; Vestergaard et al. 2015).
To facilitate research on the evolutionary genomics of this frog, we have generated a reference genome assembly for O. pumilio. Amphibian genomes have often been challenging to assemble due to large genome sizes. Salamanders have independently evolved large genome sizes up to ∼100 Gb (http://www.genomesize.com/; accessed January 2018), possibly because of low deletion rates (Sun et al. 2012; Frahry et al. 2015). Low metabolic rates may permit larger cell size in some amphibians, leading to reduced selection against large nuclei and large genomes (Licht and Lowcock 1991).
One factor that can expand genomes and complicate assembly is transposable element (TE) proliferation. TEs are selfish genetic elements that proliferate, even at the expense of host fitness. They can disrupt protein sequences, modify gene expression patterns, and result in abnormal phenotypes (Chuong et al. 2017). They further can facilitate ectopic recombination that disrupts typical chromosome structure (Montgomery et al. 1991). The average TE inserting into intergenic regions is on its own typically neutral or weakly detrimental (Lynch 2007). However, indirect effects from actively proliferating elements are thought to reduce fitness. Genomic repressors silence transposons or prevent their proliferation to keep TE content in check (McLaughlin and Malik 2017). Coevolution of TEs and repressors is thought to follow Red Queen dynamics where continuous arms races produce short bursts of TE activity followed by silencing as cells evolve to gain control of TE expression (McLaughlin and Malik 2017).
TEs that successfully evade silencing will proliferate rapidly, expanding genome content. Some classes of TEs, particularly Mariner elements are known to spread across distantly related taxa in horizontal transfer (HT) events (Schaack et al. 2010). Genomes which acquire TEs via HT are expected to be exceptionally susceptible to TE activity as these have no evolved defenses (Schaack et al. 2010). In these new genomic environments, TEs can proliferate to expand genome content. As we will show, assembly for O. pumilio is complicated by extensive TE content. Many of these TEs appear to have been acquired recently via HT. We suggest that this genome has experienced takeover by selfish genetic elements, resulting in extreme genome size.
Results
The Draft Genome
We generated an assembly from a total of 127.5× coverage of Illumina sequence data prepared with stacked insert sizes ranging from 250 bp to 20 kb (table 1). The estimated genome size is 6.76–9 Gb, and the final assembly genome size is 5.5 Gbp. The contig N50 and scaffold N50 are about 385 bb and 72 kb, respectively (table 2). Homologous proteins from human and Xenopus tropicalis were subsequently mapped to the genome assembly by using TBLASTN, with the aligned sequence being filtered and passed to GeneWise to identify accurate spliced alignments.
Table 1.
Sequence Data Used to Assemble the O. pumilio Reference Genome.
| Paired-End Libraries | Insert Size (bp) | Reads Length (bp) | Total Size (G) | High-Quality Size (G) | Sequence Coverage (X) | High-Quality Coverage (X) |
|---|---|---|---|---|---|---|
| Solexa reads | 250 | 150 | 186.75 | 149.28 | 27.87 | 22.28 |
| 500 | 150 | 133.09 | 94.45 | 19.86 | 14.10 | |
| 800 | 150 | 91.28 | 53.75 | 13.62 | 8.02 | |
| 2k | 49 | 224.25 | 140.50 | 33.47 | 20.97 | |
| 5k | 49 | 88.10 | 42.04 | 13.15 | 6.27 | |
| 10k | 49 | 66.32 | 34.22 | 9.90 | 5.11 | |
| 20k | 49 | 64.80 | 16.93 | 9.67 | 2.53 | |
| Total | 854.59 | 531.16 | 127.55 | 79.28 |
Notes: 250, 500, and 800 bp belong to the short insert size paired-end libraries and 150 bp sequencing was performed for both ends. The remaining libraries belong to the long insert size paired-end libraries and 49 bp sequencing length was conducted for both ends with which were used for the scaffold building.
Table 2.
Genome Assembly with and without Repetitive Reads.
| All Data | Repeats Masked | |
|---|---|---|
| Scaffold N50 | 72,788 bp | 61,257 bp |
| Contig N50 | 385 bp | 399 bp |
| Assembled genome | 5.0 Gb | 2.7 Gb |
We attempted de novo gene prediction with AUGUSTUS (Stank et al. 2003, 2004, 2006), producing 20,058 gene sequences. To improve annotations for genes most likely to be related to phenotypes of neurotoxicity, we de novo assembled transcriptomes for the head of one juvenile individual. Of 11,984 assembled transcripts, 11,303 transcripts can be mapped to genome assembly. Only 5112 genes in Xenopus can be found in O. pumilio. A total of 3,671 transcripts can be mapped to the homologous predictions in Xenopus and 3,594 transcripts can be mapped to the de novo predictions. Many of these were fragmented in the genome assembly.
In an attempt to identify a gene-rich genome sequence that could be useful for future work, we removed repetitive reads and reassembled the genome, but the results did not improve (table 2). The presentation of fragmented gene sequences in the assembly combined with no improvement after masking TEs, implies that TEs are scattered throughout the genome, not collected in “islands.” Furthermore, the fact that repeat masking and genome assembly with long insert libraries cannot assemble whole genes suggests that introns are very long and saturated with TEs.
We mapped all sequencing reads for 30× sequence data to the reference genome, then used samtools to call SNPs. Heterozygosity across the assembled sections of the O. pumilio genome is H = 0.0016. Assuming a mutation rate of 10−9 this would yield an estimate of Ne = 400,000. Census population sizes for O. pumilio are high, and the geographic range stretches across Central America, consistent with this high heterozygosity. These numbers could be inflated if the individual that was sequenced was the product of interbreeding between two previously isolated demes. However, this result gives a rough estimate of effective population size for O. pumilio.
Ion Channel Evolution and Autoresistance
One of the major motivations for the O. pumilio genome was to identify the genetic basis for toxin autoresistance in the strawberry poison frog. Although many genes are fragmented in this new reference assembly, a targeted effort recovered sequences for most members of two gene families known to interact with toxins. Poison frogs accumulate hundreds of diet-derived alkaloids which serve as toxic or foul-tasting predator deterrents (Daly et al. 2005; Saporito et al. 2012; Santos et al. 2016). Many of these compounds target ion-transport proteins (Daly and Spande 1986). Multiple Dendrobatid species have been shown to resist their own toxins through genetic modification of their targets (Albuquerque et al. 1973; Daly et al. 1980; Tarvin et al. 2017; Wang and Wang 2017) and resistance-conferring mutations have been identified for some toxin targets (Tarvin et al. 2016, 2017; Wang et al. 2017). However, these studies have focused on a single gene or a few paralogs. Since poison frog neurotoxins usually target multiple proteins in ion channel gene families (Daly and Spande 1986; Santos et al. 2016), resistance is expected to involve adaptation across these families. Therefore, we surveyed our genome and transcriptome assemblies, as well as those available for other frog species, for voltage-gated sodium channels (α subunit; SCNA), and nicotinic acetylcholine receptors (nACHR), which are two gene families that are common targets of poison frog alkaloids (Saporito et al. 2012; Santos et al. 2016). We then used a phylogenetic approach to gain insight on the evolution of autoresistance in O. pumilio from a multigene perspective.
We are able to retrieve autoresistance genes in spite of the highly fragmented assembly. We were able to retrieve O. pumilio sequences for 6/6 and 17/18 of the known frog SCNA and nACHR genes, respectively (fig. 1). For the CHRNE gene, we recovered two distinct orthologs for O. pumilio and Rh. marina. Phylogenetic reconstructions of the SCNA family support results from previous studies, which suggest that three of the six frog genes in this family (SCNA1-3) resulted from anuran or amphibian-specific gene expansions, independent from those that gave rise to most amniote genes (Zakon et al. 2011, 2017).
Fig. 1.

Bayesian gene trees reconstructed for annotation of O. pumilio genes in the nACHR (A) and SCNA (B) gene families, and nucleotide sequences of the CHRNE gene. Nodal support and branch labeling/coloring follow in fig. 2.
We then used ancestral sequence reconstructions to identify amino acid sites that may play a role in toxin autoresistance of O. pumilio sodium channels. We searched for two (nonexclusive) types of sites: (1) Those where three or more O. pumilio genes underwent amino acid substitutions after their split from the O. pumilio-Rh. marina ancestor (2) and sites within S6 transmembrane segments, which have been hypothesized to interact with multiple alkaloids present in O. pumilio (Tarvin et al. 2016), where substitutions took place as described above in at least one SCNA gene. Our analyses recovered 19 sites (fig. 2). Eleven of these were on S6 segments, four of them with changes in more than one gene. Notably, substitution M783L, on the S6 segment of domain DII, took place in parallel in five of the six O. pumilio sodium channels, suggesting an adaptive role for this site in terms of autoresistance. This change may have also occurred on the sixth gene (SCNA2), but our inference was ambiguous due to missing data. The remaining eight sites were all on intra or extracellular linkers (fig. 2), which could suggest a role for these regions in autoresistance. Extracellular P-loops on the S5–S6 linkers, where two of the sites are located, are of special interest, since they are known to be involved in resistance to Batrachotoxin (McGlothlin et al. 2014) and Tetrodotoxin (Geffeney et al. 2005; Jost et al. 2008; Du et al. 2009; McGlothlin et al. 2014), both neurotoxic alkaloids present in Phyllobates and some Colostethus poison frogs, respectively (Daly et al. 1994, 2005; Tokuyama et al. 1969).
Fig. 2.

Bayesian gene tree of the voltage-gated sodium channel alpha subunit (SCNA) gene family and amino acid replacements identified as possibly associated with toxin autoresistance in O. pumilio (A), with their positions on a schematic voltage-gated sodium channel (B). Branches are colored and labeled based on the closest X. tropicalis ortholog, and nomenclature follows ENSEMBL annotations. Asterisks on internodes denote Bayesian posterior probabilities and aBayes support values above 0.95. Clades resulting from amniote-specific gene family expansions were collapsed to improve visualization.
Repetitive Element Content
In view of the fragmented O. pumilio genome assembly in spite of extensive and high quality sequence data, we examined TE content using REPdenovo (Chu et al. 2016). This recently developed method quantifies genomic repeat content from raw reads without the need for a reference assembly or reference databases of known repeats. REPdenovo identifies high copy number k-mers, then assembles these into consensus contigs for highly similar copies of repetitive elements. Repeats with <1.5–2% sequence divergence among copies are collapsed into consensus sequences representing a minimum of 10 repetitive element copies. We identified over 4.76 Gb of repetitive sequences with low differentiation across copies in the genome of O. pumilio. Repeats include LTR retrotransposons, non-LTR retrotransposons, DNA transposons, as well as rRNAs and tRNAs.
We observed 1.0 Gb of Gypsy-like elements, 197 Mb of Mariner, and 181 Mb of Tc1 type elements as classified using tblastx (fig. 3). As a comparison, we used the same methods to identify repeats in the human genome, widely regarded as “repetitive.” The total repeat content of similarly identified low differentiated repeats in humans summed to 500 Mb whereas in Xenopus we identify 352 Mb of repeats, far below the level of O. pumilio. The methods implemented in REPdenovo were designed to identify young, actively proliferating repeats. Other methods would be needed to identify older, more divergent repeats that may be more common in other taxa.
Fig. 3.

Total sequence content of TEs as classified using a tblastx against the RepBase database. Among low differentiated TE families, we identified 1 Gb of Gypsy elements, 298 Mb of Copia elements, 255 Mb of hAT sequence, 197 Mb of Mariner elements, and 181 Mb of Tc1 elements in O. pumilio.
The three most abundant TE families among the low differentiated repeats include a Gypsy element at 58,354 copies (52 Mb), a Mariner element at 73,395 copies (52 Mb), and Tc1 element at 78,037 copies (86 Mb). No constraints were placed on repeats as they were matched with annotations in the RepBase database (Jurka et al. 2005; Bao et al. 2015). However, we note that 8 of the 15 most common TE families have unusually close matches with repeat sequences found in freshwater fish (supplementary table S2, Supplementary Material online). The assembled contig from REPdenovo with the highest copy number is at 9,507 copies in the cell. A total of 98 contigs are present at 100 copies or greater, and 10 are present at 1,000 copies or greater. REPdenovo collapses repeats with ∼2% differences into a single contig. High copy number with such low divergence suggests recent and rapid proliferation. Copy numbers may be slightly inflated if TEs are assembled in fragments (supplementary fig. S1, Supplementary Material online). Contigs within a single family could be aligned in a blastn with sequence similarity as low as 82%.
To determine how well repeats had been incorporated into the reference genome assembly, we used RepeatScout which identifies repetitive sequences in whole genome assemblies (Price et al. 2005). Across all classes of TEs, we can localize only 34.3 Mb representing 90,892 elements to the reference assembly fasta sequence using RepeatScout (Price et al. 2005). Hence, the majority of repeats are not localized to the reference assembly and therefore cannot be identified using RepeatScout. This result also implies that more highly diverged TEs with much greater than 2% divergence across copies are either not common in this genome or are not well integrated in this assembly. For highly repetitive genomes containing recently proliferated TEs, REPdenovo is likely to be more useful for assays of repetitive content.
Horizontal Transfer
Given the extreme prevalence of low differentiated TEs in the genome of this frog, we wondered whether HT of new TEs might explain some portion of the repetitive content. HT can be difficult to identify when phylogenies are uncertain. One approach is to identify candidate sequences that are closer matches to distant taxa than a known close relative (Tian et al. 2011). We identified such possibly horizontally transmitted elements using Xenopus as a benchmark for close alignments. We identified TEs (excluding rRNAs and tRNAs) whose closest match in the RepBase repeat database was closer than its nearest match to Xenopus repeats in RepBase or as identified using REPdenovo on Xenopus sequence data. Such cases would be likely candidates for HT from other organisms, but could also be TE families that somehow have been lost from Xenopus or not incorporated into RepBase. This latter explanation is less likely for TE families with many copy numbers in Oophaga pumilio. An advantage of this approach for identifying candidates for HT is that it is not affected by the challenges of aligning large numbers of highly repetitive sequences.
We observed 17 TE families where a TE contig from REPdenovo had at least 2% greater similarity in a BLASTn search against the RepBase database than contig matches with any known Xenopus repeat in RepBase or as identified using REPdenovo. These methods will identify any HT repeats that are currently at high copy number with low divergence across copies, more effective for recent HT (Loreto et al. 2008). They will not identify HT without copy number expansion or highly divergent TE copies. They therefore represent a minimum set of HT events. Among these 17 classes, we identified 7 Mariner elements, 7 Tc1 elements, 1 Gypsy element, and 1 hAT. Again, although no constraints were imposed when matching elements with annotations in RepBase, 15 of these 17 families matched with sequences from fish genomes. One of these matches a transposon in A. carolensis, possibly suggesting independent HT events from a species that remains unannotated rather than direct transfer from Anoles. All of these alignments matched with 80–97% nucleotide identity. Assuming a single transposition event followed by vertical inheritance of TE copies and a conservative mutation rate of 10−9 substitutions per site per million years (Crawford 2003), this would correspond to a maximum of 100 million to 15 million years divergence time. Higher mutation rates of 10−8 substitutions per site per million years would yield estimates of 10 million to 1.5 million years divergence. If TEs had minimal vertical inheritance, mutation rates during transposition could be as high as 10−4, implying divergence as short as 1000 serial transposition events. Fish diverged from tetrapods 480 million years ago (Yamanoue et al. 2006). It is exceedingly improbable that TEs would have alignments this similar if transposons were vertically inherited from a common ancestor. These candidates are therefore examples of HT since the divergence between ray-finned fish and amphibians. Multiple fish species are represented in these comparisons. HT from fish into frogs is more likely than multiple cases of HT from frogs into multiple species of fish living in different environments. Similarly, the repeats with less than 5% sequence divergence between fish and O. pumilio must have arisen by relatively recent HT. We notice that our approach for identifying HT is by no means comprehensive, and will miss all HT that does not result in increased similarity between O. pumilio and a database-represented sequence relative to Xenopus. Much intra-amphibian HT will therefore be missed. Furthermore, single HT events of silenced elements would result in only one copy of a given repeat and would not be identified using the algorithms presented here. Hence, the numbers presented are a minimum estimate of HT events.
In addition, we observed another 56 TE families that are similar to a contig in O. pumilio with no known match with a Xenopus repeat. Of these 56, 6/7 Tc1 families and 21/23 Mariner families and 5/21 other elements are annotated as repeats from freshwater or marine fish genomes. All align with 75% or greater nucleotide identity. These results suggest ongoing transfer for multiple classes of repeats to or from the O. pumilio genome and its ancestors, primarily from fish into amphibians. Although many different types of TEs are identified (DNA transposons, non-LTR retrotransposons, LTR retrotransposons), the most common candidates for HT are Mariner and Tc1 elements.
Among the elements we identify as candidates for HT, we observe many elements at high copy number. The first and second most abundant transposons in O. pumilio are identified among these candidates for elements acquired via HT. The closest match for these two highest copy-number TE families are Tc1-3_SSa (78,037 copies) and Mariner-4_DR (73,395 copies) (supplementary table S2, Supplementary Material online).
Seven of the TE families that are candidates for HT have 100 or more independently assembled repeat contigs representing thousands of independent copies, suggesting a rich diversity of TEs due to ongoing proliferation (supplementary table S2, Supplementary Material online). Such results are consistent with TEs evading silencing in a new genome that has no evolved defenses against the repetitive sequence. When we examine nucleotide similarity of Mariner-4_DR elements that are identified using REPdenovo compared with the consensus Danio rerio element in RepBase, we observe a bimodal distribution, suggestive of two rounds of HT followed by proliferation (fig. 4). The second peak has a maximum at roughly 97% identity. We infer, based on the close match of these transposons, that there has been HT of TEs from fish into frogs with at least two waves of invasion and proliferation. Resolution may be difficult with many elements if rounds of HT are not separated by long times of quiescence, so it is possible that this element and others experienced more rounds of HT than can be resolved from the data.
Fig. 4.

Bimodal distribution of nucleotide similarity for repeat contigs identified using RepDeNovo compared with Mariner-4_DR consensus sequence from D. rerio. The presence of two peaks is consistent with two waves of proliferation after HT of an element between fish and frogs. Danio and Oophaga diverged 480MYA. The distribution of nucleotide similarity is not consistent with vertical transmission from a common ancestor.
To establish the relationships of horizontally transferred elements relative to the diversity of elements in O. pumilio, we generated phylogenetic trees for a subset of horizontally transferred candidate sequences. For each candidate family with at least 100 contigs for horizontally transferred elements, we selected those sequences matching the RepBase element with at least 90% similarity and at least 200 bp in length (500 bp for the most abundant element Mariner-4_DR). We then generated alignments and UPGMA and Neighbor-Joining phylogenies using clustalw. In each case, a RepBase TE from fish lies within the diversity of TEs in O. pumilio rather than as an outgroup sequence (fig. 5; supplementary figs. S2–S11, Supplementary Material online). This observation is compatible with recent HT of TEs between fish and frogs, with subsequent proliferation in O. pumilio. All such families with 100 or more contigs are Mariner or Tc1 transposons, though biases in RepBase annotations could potentially lead to failure to identify some TEs in other classes.
Fig. 5.

Phylogenetic relationships between horizontally transmitted repetitive elements in O. pumilio (black) and annotated TE Tc1-3_SSa (red), a salmon TE in RepBase. Similar patterns are observed in other TE families. For each of the most diverse TEs that are candidates for HT, the RepBase element lies within the diversity of TEs in O. pumilio. All of these diverse and rapidly proliferating horizontally transferred elements are a close match with a TE from freshwater fish.
Repetitive Element Expression
New TE insertions from active transposons must appear in the germ line to be passed on to subsequent generations. To examine if some of the transposons HT TEs are active in the germline of O. pumilio, we gathered RNAseq data for five unfertilized ova of O. pumilio and five Xenopus laevis ova. We de novo assembled the transcriptome, and quantified expression profiles using the Tophat/Cufflinks suite (Licht and Lowcock 1991; Frahry et al. 2015) (see Materials and Methods). We estimated the sum of RNA transcription (length × FPKM) for each of the three major classes of TE in each replicate in the O. pumilio genome (fig. 6). We observe an excess of germline expression of Gypsy (t = 7.007; P = 0.006393) and Mariner sequences (t = 7.1938; P = 0.005313) in O. pumilio compared with Xenopus. Differences in Tc1 expression are not significant (t = 1.9123; P = 0.2920). These results are consistent with the abundance of transposons that have been identified in the genome of O. pumilio. Results are qualitatively similar for total expression levels (FPKM) not considering sequence length (supplementary fig. S12, Supplementary Material online).
Fig. 6.
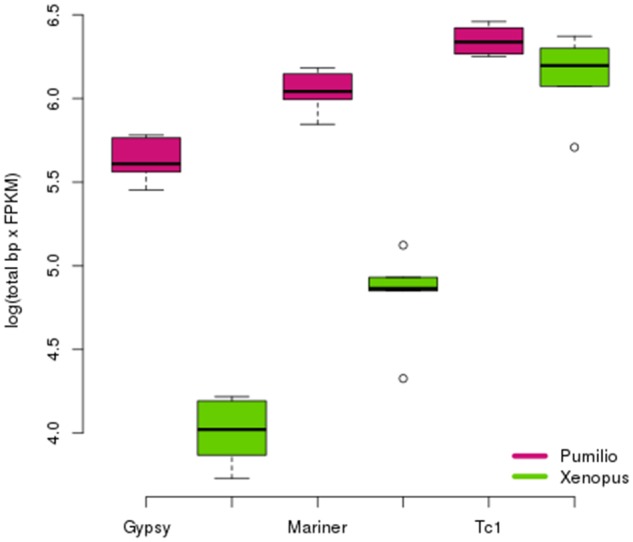
Total amount of expressed sequence for TE classes in O. pumilio and X. laevis. Gypsy and Mariner elements are expressed at significantly higher levels in O. pumilio, but Tc1 elements are not. We observe an excess of germline expression of Gypsy (t = 7.007; P = 0.006393) and Mariner sequences (t = 7.1938; P = 0.005313) in O. pumilio compared with Xenopus.
We identify highly expressed TE transcripts with FPKM > 100 for each of the three most abundant HT transposon families, Gypsy-15_Ano, Mariner-4_DR, and Tc1-3_SSa (supplementary table S3, Supplementary Material online). Although no constraints were placed on TE matches in RepBase, these recently transferred elements both have close matches with elements in the genomes of freshwater and marine fish. Mariner-4_DR elements are particularly diverse and encompass over 46,000 copies in O. pumilio (table 3, supplementary fig. S4, Supplementary Material online). To examine whether recently transferred Mariner-4_DR elements might be more highly expressed than older Mariner-4_DR elements, we tested if there was an association between transcription level and percent similarity with the Mariner-4 element in D. rerio. We observe a significant positive correlation between expression level and percent similarity (fig. 7, F = 3.956; df = 5; P = 0.002128). The most highly expressed transposons have 92% or greater similarity with the D. rerio element. These results may suggest that recently transferred Mariner-4_DR TEs are more likely to be highly expressed, however, within-genome selection acting to maintain sequence conservation on active elements only may reduce the rates of divergence for more active elements.
Table 3.
Most Common Horizontally Transmitted TE Families.
| Family | Species | Number of Contigs | Mean Length (bp) | Total Copy Number | Total Content (Mb) |
|---|---|---|---|---|---|
| Mariner-4_DR | Danio rerio (Zebrafish) | 1,441 | 1245 | 46,112 | 52.3 |
| Tc1-3_SSa | Salmo salar (Atlantic Salmon) | 1,191 | 1092 | 39,303 | 86.3 |
| TZF28C | Danio rerio (Zebrafish) | 144 | 647 | 4,032 | 25.4 |
| Mariner24_EL | Esox lucius (Northern pike) | 139 | 1110 | 6,255 | 15.3 |
| Mariner-8B_EL | Esox lucius (Northern pike) | 17 | 692 | 9,214 | 9.8 |
| Mariner-16_SSa | Salmo salar (Atlantic Salmon) | 155 | 653 | 5,425 | 9.5 |
| Tc1-1_ACar | Anolis carolenensis (Anole) | 45 | 308 | 6,390 | 5.2 |
| Mariner-9_SSa | Salmo salar (Atlantic Salmon) | 28 | 384 | 19,852 | 5.0 |
| TC1_FR5 | Takifugu rubripes (pufferfish) | 10 | 291 | 910 | 4.2 |
| Mariner-15_EL | Esox lucius (Northern pike) | 12 | 976 | 804 | 2.4 |
| Tc1-6_DR | Danio rerio (Zebrafish) | 17 | 365 | 2,686 | 2.6 |
| 5S-Sauria | Anolis carolenensis (Anole) | 16 | 1264 | 928 | 1.9 |
| TZF28B | Danio rerio (Zebrafish) | 15 | 710 | 495 | 0.9 |
| Mariner-19_EL | Esox lucius (Northern pike) | 116 | 170 | 1,856 | 0.4 |
| Mariner-7_SSa | Salmo salar (Atlantic Salmon) | 10 | 145 | 200 | 0.04 |
Fig.7.

Expression level for transcripts is positively correlated with nucleotide similarity with Mariner-4_DR (Danio rerio) consensus sequence in RepBase. Such a result is expected if recently transferred transposons invade genomes that lack repressors for the new TEs (F = 3.956; df = 5; P = 0.002128).
Piwi and piRNA Pathway Genes
One cellular defense against TEs is the piwi/pi RNA pathway, which restricts TE expression through antisense targeting and mRNA degradation. We attempted to identify transcripts for known piwi genes as well as their interacting targets in the argonaute (ago) family. In a BLASTn search at a highly permissive E-value of 1.0, we identified ago transcripts in all replicates of O. pumilio and Xenopus RNAseq data. In the piwi family, we can identify piwi2, and piwi3 in all replicates of Xenopus trinity assemblies and replicates have multiple isoforms that are identified. However, in O. pumilio, piwi2 and piwi3 are only identified in one replicate each with no evidence for isoforms or alternative splicing. These results raise the possibility that piwi genes may not be expressed in ova of O. pumilio as they are in Xenopus. Unless these proteins were acquired via maternal deposition of proteins, they might allow more permissive expression of TEs, as observed. If piwi proteins have been inactivated, it might explain some portion of the TE proliferation observed. Alternatively, it could be that piwi genes are evolving so rapidly that they evade detection based on comparisons with Xenopus.
Spliceosome Elements
Gene sequences in the O. pumilio reference assembly are often assembled in fragments. Even with 20 kb insert libraries, many exons cannot be joined to produce whole gene sequences without comparisons to well-assembled outgroups. Hence it would appear that introns in O. pumilio commonly exceed 20 kb. This pattern is unusual for a metazoan. In many species intron spicing is inefficient for long introns (Burnette et al. 2005; Swinburne and Silver 2008). These observed genomic changes made us wonder whether intron splicing machinery might have been modified to compensate for changes in genome size and organization.
In view of the high TE content of this genome, and the consequently increased intron length, we decided to explore spliceosomal elements that might influence intron evolution. We identified assembled transcripts for U1, U2, U4B, U5B1, and U6 in the assembled transcriptome of O. pumilio. We identified one transcript that contains two U1 elements, linked by a 653 bp spacer. This transcript appears to contain sequences homologous to Xenopus oocyte U1 elements, U1a and U1b. In Xenopus, these embryonic U1 elements are arrayed in a tandemly duplicated region but are transcribed and processed separately (Lund et al. 1987). If the O. pumilio fused transcript containing U1a and U1b is not processed to remove each element, this 653 bp linker sequence may be expected to interfere with U1 element function in oocytes. Assembled transcripts for Xenopus RNAseq replicates used as a comparison for expression analysis do not contain the fused transcript or the linker region.
Given this unusual change in the U1 embryonic snRNAs, we decided to identify U1-interacting proteins and their binding sites. The Sm binding site in the Xenopus U1 element is AATTTCTGG (Jarmolowski and Mattaj 1993). We observe a 1-bp substitution in the U1 element of O. pumilio to AATTTGTGG. Whether this change to the motif might alter the function of the spliceosome remains unclear. We then searched for snrnp genes in the assembled transcriptome of O. pumilio and Xenopus. Among snrnp genes, we identify U1A (snrpA) in only two of the five RNAseq replicates and U1C (snrpc) in only three of the five replicates for O. pumilio. In total, only one replicate of O. pumilio oocyte RNA contains all snrnp components necessary to assemble a full spliceosome. In contrast, transcripts for all snrnps are readily identified in the assembled transcriptome of X. laevis. How the spliceosome is assembled so that introns are processed in oocytes in O.pumilio remains unclear. Together, these results suggest that there have been unusual changes in the splieceosome of O. pumilio compared with Xenopus. Coupled with the unusually large introns observed in this genome, these changes may imply fundamental changes to intron processing in O. pumilio. It may be that unknown compensatory changes have allowed for large intron processing and consequently a tolerance for intronic TEs in this frog, a hypothesis worth exploring in future work.
Discussion
Reference Genome Assembly
We have assembled a reference genome and transcriptomes for the strawberry poison frog, O. pumilio. The reference genome appears to be highly fragmented, with a scaffold N50 of roughly 60 kb. Many genes are fragmented into single exons, in spite of high coverage data that includes 20 kb insert libraries. We show that this reference genome is riddled with repetitive element sequences, compromising genome assembly.
Ion Channel Evolution and Autoresistance
Understanding the genetic and molecular basis of toxin autoresistance in poison frogs is of major interest given the importance of toxicity in this group’s ecology and evolution. Despite the highly fragmented nature of our assembly we are able to annotate most (>90%) of the genes in the SCNA and CHRN families both known to be involved in autoresistance to several poison frog toxins. The obtained sequences for SCNA genes allowed us to identify amino acid substitutions at 19 sites that are putatively associated with toxin autoresistance in dendrobatid frogs, corroborating two previously identified substitutions in the SCN4A gene (S435A, A451D) (Tarvin et al. 2016). Among these changes, substitution M782L occurred in at least five, but possibly all six genes in parallel. Remarkably, this genotype is also present in the SCN4A gene of the Malagassy poison frog Mantella aurantiaca (Tarvin et al. 2016), which is part of a separate radiation of poison frogs that have independently evolved many of the same alkaloids sequestered by dendrobatids (Andriamaharavo et al. 2010). The highly convergent nature of its evolution strongly suggests an adaptive role for this site in toxin autoresistance of poison frogs.
Position 782 is located within the S6 segment of domain DII. Although sites across the four domains of voltage-gated sodium channels have the ability to produce resistance to poison frog toxins in vitro (Li et al. 2002; Wang et al. 2006; Wang, Tikhonov, Mitchell, et al. 2007; Wang, Tikhonov, Zhorov, et al. 2007; Wang and Wang 2017), until now all the substitutions identified in poison frogs were restricted to domains DI and DIV (Tarvin et al. 2017). Our analyses, on the other hand, pinpoint sites in all domains of all SCNA genes. This discordance is, at least in part, due to the fact that previous studies focused on identifying sites that correlated with variation in alkaloid profiles between different species of Dendrobatidae, whereas here we compare dendrobatids (represented by O. pumilio) to other tetrapods. Complementary approaches that draw from both levels of analysis are, therefore, desirable for future studies.
Furthermore, eight of the sites we identified are located on intra or extracellular loops, which had not been related to poison frog autoresistance either. Two of them (374 and 1206) are on extracellular P-loops, known to be involved in resistance to Batrachotoxin (Wang et al. 2006) and Tetrodotoxin (Geffeney et al. 2005; Jost et al. 2008; Du et al. 2009; McGlothlin et al. 2014), both neurotoxic alkaloids present in Phyllobates and some Colostethus poison frogs, respectively (Daly et al. 1994, 2005; Tokuyama et al. 1969). Curiously, neither of these alkaloids have been found in O. pumilio (Daly et al. 2005), which lends support to the suggestion that P-loops may interact with other alkaloids, for example Histrionicotoxin (Tarvin et al. 2016), which is sequestered by O. pumilio (Daly et al. 2005; Saporito et al. 2007). Alternatively these substitutions may have preceded and possibly facilitated the evolution of Tetrodotoxin or Batrachotoxin sequestration. It is worth noting that some of the convergent substitutions identified may be the product of low levels of purifying selection, and not multigene adaptation to chemical defense. This is especially true for some intra/extracellular loops, such as the DII S6-DIII S1 linker, or the C-terminal intracellular loop which exhibit low conservation and in some segments align poorly among tetrapods.
Overall, our results demonstrate that approaching the evolution of neurotoxin autoresistance from a gene family perspective provides valuable insight, even with relatively scarce taxon sampling, as in this case. Future work with increased taxon sampling that incorporates functional (e.g., electrophysiological) approaches will confirm and expand the initial patterns uncovered here.
Genome Expansion
We observed high TE content in the strawberry poison frog O. pumilio. We have identified 4.76 Gb of TEs at copy number 10× or higher with less than 2% divergence across copies in the O. pumilio genome. These repetitive elements encompass DNA transposons, LTR retrotransposons, and non-LTR retrotransposons. The most abundant and widely proliferating elements we observe include 1.0 Gb of Gypsy elements and 0.5 Gb of Mariner/Tc1. The fact that LTR retrotransposons, non-LTR retrotransposons, and DNA elements like Mariner/Tc1 are all actively proliferating and expressed in the germline speaks to widespread cellular failure to contain TE proliferation. Large genomes in salamanders are suggested to be the product of low deletion rates, with long retention times for retroelements (Sun et al. 2012). We observe large numbers of recently proliferating TEs across multiple TE families in O. pumilio. Karyotype data shows that other Dendrobatid frogs have enlarged genomes relative to neighboring taxa (Bogart 1991). Although karyotype data alone cannot distinguish the timing of TE invasion, they suggest that the proliferation that led to genome size increases occurred after the split between Epipedobates and Oophaga (Bogart 1991). Future sequencing of related species may clarify the timing and dynamics of TE invasion and proliferation.
Several hypotheses could explain this expansion in genome content. One traditional explanation for more permissibility to TE proliferation is low effective population sizes (Lynch 2007). It is possible that under nearly neutral dynamics TEs could proliferate as the average insertion has neutral or mildly detrimental effects (Bogart 1991; Lynch 2007). We observe high heterozygosity on these frogs suggesting large Ne ∼ 400,000 individuals, providing no support for the low Ne hypothesis. Moreover, census sizes for O. pumilio are high and the geographic distribution is large. However, it is possible that O. pumilio is isolated into small demes. If the individual here represents interbreeding between two previously isolated demes, then heterozygosity and consequently estimates of Ne might be inflated. Additionally, the genomic expansion is observed in karyotypes of related species, suggesting that the activation of TEs may have begun further in the past at a time where, perhaps, the ancestral species had a much lower Ne.
Some TE insertions happen to be beneficial as they can produce mutations that cause phenotypic changes (Aminetzach et al. 2005; Magwire et al. 2011; Schrader et al. 2014; Jangam et al. 2017). TEs may be a mutational force that can rewire expression on suites of genes in a short time, offering mechanisms to alter large sections of the genome when mutations are needed to adapt to a specific environmental change (Casacuberta and Gonzalez 2013; Bennetzen and Wang 2014; Lynch et al. 2015). Such domestication of TEs may lead to ongoing expression of single domesticated copies of TEs.
Combined with the unusual expansion of intron sizes and low levels of aneuploidy (Bogart 1991), it seems likely that O. pumilio has experienced some form of compensatory mutation to maintain chromosome integrity and correctly splice mRNA sequences. Irrespective of the causes of the initial TE proliferation, as genome size expands, so that the majority of the genome is repetitive, subsequent insertions may have little effect on fitness, if negative density-dependent selection leads to lower transposition rate for larger numbers of TEs (Lynch 2007). Under this scenario, repetitive sequences might accumulate further with few mechanisms to prevent even more expansion. If left unchecked, the “snowball effect” could lead to increasingly expanded genome size. Given the highly active, recently proliferating TEs in this frog, such a scenario seems unlikely for O. pumilio.
Repetitive Elements and HT
We identified 82 TE families that are candidates for HT, with an excess of data suggesting multiple cases of HT between fish and frogs. RepBase sequences from other organisms match closely to TEs in O. pumilio and lie within the diversity of assembled repeats in O. pumilio. The majority (79%) of elements that are identified as HT candidates are Mariner or Tc1 elements from freshwater fish. Both Mariner and Tc1 elements are known to transmit across species, including between fish and frogs (Stanke et al. 2004, 2006). We observe at least one candidate for LTR retrotransposons, non-LTR retrotransposons, DNA transposons, and hATs.
Among the candidates for HT, Mariner-4_DR elements appear to be common, continuously proliferating, and repeatedly transferred. Multiple copies are expressed in unfertilized oocytes, suggesting ongoing activity. Recently transferred copies with the greatest similarity to elements in the genome of D. rerio appear to be most highly expressed in oocytes. These observed dynamics are also consistent with O. pumilio eventually evolving defense mechanisms to silence copies that were acquired in the distant past. Two of the three most abundant TE contigs are Mariner-4_DR and Tc1-3_SSa, both candidates for HT. Mariner TEs are known to transfer across species and are often active on arrival in nonnative taxa (Schaack et al. 2010). O. pumilio may be unusual in that it has experienced multiple rounds of invasion and establishment by distinct and distantly related Mariner/Tc1 transposons.
It is possible that the similar environment between freshwater fish and some frogs, and the fact that the germline is exposed more directly to the environment in both frogs and fish, could facilitate repeated transfer of transposons. However, we note that O. pumilio is terrestrial in its adult form and breeds in small pools of standing water, typically in bromeliads, which would be very unlikely to contain any fish. As such, there is no directly shared aquatic environment between O. pumilio and any species of fish. This may possibly indicate that most of the HT events identified here date back to ancestors of O. pumilio that were more likely to share an aquatic environment with fish. However, given the low divergence between repeats from different species this is probably not a likely explanation. Instead, HT may be facilitated by shared pathogens or by other vectors of transmission (Sun et al. 2016). Additionally, transfer from some intermediate multicellular host between fish and frogs could produce the patterns we observe. It is also possible that the tendency to observe more TE HTs in some species than others is an observation bias caused by differences in defense mechanisms or permissibility of novel TEs. Some amphibians such as O. pumilio, may experience more TE-driven HT followed by proliferation because they lack some of the defense mechanisms employed in other organisms (Mueller 2017), because they are more permissive to increased genome size than other organisms (Licht and Lowcock 1991), or because of the more direct exposure of the germline to the environment. A lower deletion rate, lack of antisense RNAs, and a lack of methylation could all allow for greater chances of TE expansion after invasion, contributing to active elements and expanded genomes.
Changes in Gene Structure and Spliceosome Organization
We have observed evidence of very long introns longer than 20 kb with many intervening repetitive elements. This pattern is a significant departure from other highly repetitive genomes in plants or mammals in which genes with modest introns are surrounded by “seas” of transposons (Lander et al. 2001; Haberer et al. 2005). In spite of the abnormally large introns, O. pumilio is still able to produce a functional transcriptome. Our results on possible changes in the spliceosome of O. pumilio relative to Xenopus laevis may suggest that the increased genome size is accompanied by changes in the spliceosome, possibly making it more permissive to large intron sizes. U1 snRNA processing and Sm binding are essential first steps in spliceosome assembly (Matera and Wang 2014). Xenopus is atypical in that it has a tandemly arrayed duplication of the U1 element in a 1–2 kb cassette of DNA (Lund et al. 1987; Jarmolowski and Mattaj 1993). These alternate versions of U1a and U1b are expressed solely in oocytes (Lund et al. 1987). In the transcriptome of O. pumilio oocytes, we observe a fusion transcript containing two U1 elements as well as a 653-bp linker sequence. In the transcriptome of Xenopus, we identify individual transcripts for the two U1 elements but do not observe a transcript with both U1s or the linker sequence. In other organisms, U1 RNAs must be capped and processed to be transported to the nucleus for spliceosome assembly (Matera and Wang 2014). It is possible that the O. pumilio fusion transcript might interfere with such processing, unless some unknown compensatory mutation restores proper function. This modification of spliceosome machinery combined with unusually long introns of O. pumilio is novel and unexpected.
In five replicate transcriptomes of O. pumilio, we observe no evidence of independent spliceosome RNAs U1A and U1C, each considered necessary for spliceosome assembly (Matera and Wang 2014). Only one replicate contains all proteins necessary to assemble a full spliceosome, unless these proteins were acquired without transcripts via maternal deposition. Combined with the unusual modifications of the spliceosome, these results lead us to suspect that O. pumilio may have developed novel changes in intron splicing that either allowed or responded to the rapid proliferation of repetitive DNA. Assembly of the O. pumilio genome will offer one tool to design future experiments to answer these open questions about this unusual genomic system.
Materials and Methods
Sequencing
A single juvenile individual was sacrificed for genome sequencing. Samples were prepped using the NexTerra kit at Beijing Genomics Institute. To generate the reference genome of O. pumilio, we de novo sequenced an individual by using whole genome shotgun sequencing on an Illumina HiSeq 2000 platform. We constructed seven different paired-end DNA libraries with different insert sizes using sequence lengths of 150 bp length for short insert size libraries and 49 bp for long insert size libraries. In total, we generated about 855Gbp (127.5× coverage) of raw reads before filtering.
Raw genomic reads were filtered using the following steps: (1) reads containing Ns or polyA tracts over 10% or more of their total length were discarded; (2) small-insert library reads with 50 or more bases with a Q20 value of 7 or less and large-insert library reads with 15 or more bases below this threshold were removed; (3) to remove reads with adapter contamination, reads with more than 10 bp aligned to the adapter sequence (allowing up to 3 bp mismatch) were discarded; (4) small insert size reads in which read1 an read2 overlapped more than or equal to 10 bp allowing 10% mismatch were discarded; (5) PCR duplicate reads, identified when read1 and read2 of two paired-end reads are totally identical, were discarded. As a result, we obtain approx. 531Gbp (79× coverage) of high-quality reads for the subsequent genome assembly. We used SOAPdenovo (Xie et al. 2014) to assembly the genome using the sequenced data from the data that passed the quality controls mentioned above.
Homolog proteins of two species (human and Xenopus tropicalis) were mapped to the frog genome using tBLASTn with an E-value cutoff 1e-5. The aligned sequence as well as their query proteins were then filtered and passed to GeneWise to identify splice junctions. De novo prediction was performed on the repeat masked genome based on an HMM model implemented in AUGUSTUS (Stank et al. 2003, 2004, 2006). We filtered the resulting homology results using a cut-off of genewise score >75 and merged overlapping results as described below.
Repeat Assembly and Annotation
We used REPdenovo v. 0.0 (Chu et al. 2016) to identify repetitive elements at a copy number of 10× or higher in the genomes of O. pumilio, Xenopus (SRR3210976), and humans. Briefly, reads are broken into kmers. High copy number kmers are then de novo assembled, and contigs are scaffolded using paired-end reads. To obtain copy number, we mapped 6.6× coverage to the repeat contigs using bwa aln, then divided mean coverage for the contig by the genome wide average. Repetitive element sequences identified by REPdenovo were annotated using an all-by-all blastn against the RepBase database (Albuquerque et al., 1973; Daly and Spande, 1986) at an E-value of 10−10. To identify repeats that were localized to the assembled genome, we used RepeatScout v. 1.0.5 (Price et al., 2005). The genome was divided into eight segments, each of which was run independently to deal with memory constraints. Repeats identified using RepeatScout were annotated using a BLASTn against the RepBase database (Jurka et al. 2005; Bao et al. 2015). The top match for each repeat was used to identify TE families.
Horizontal Transfer
Detecting HT of TEs can be complicated in the face of uncertain phylogenies. A recent approach proposed to detect HT in absence of a well-constructed phylogeny uses genome comparisons between a close neighbor taxon with known outgroup taxa (Tian et al. 2011). Outgroup sequences that are a closer genetic matches than the known sister taxa are considered candidates for HT. We used this approach to identify repeats that might have been acquired via HT. All elements in O. pumilio were compared with RepBase and to all Xenopus contigs using a blastn search at E < 10−10. Elements with a match in a species other than Xenopus that had 2% greater sequence similarity between another organism than in Xenopus were considered a candidate for HT. All elements that had no similarity to any known Xenopus element but which did match an element in a different species were considered to be candidates for HT. Unclassified repeats with no close match in any annotated organism found in RepBase cannot be assayed for HT. Future efforts might expand such analysis as the number of taxa sequenced and annotated for TEs expands. To examine relationships among horizontally transferred elements, we took REPdenovo contigs matched to a RepBase element of a given family with 90% or greater nucleotide similarity to the matching TE sequence from RepBase that were at least 200 bp. For the most abundant family, Mariner-4_DR we required at least 500 bp. We generated alignments for these contigs and the RepBase element using clustalw v. 2.1 with default parameters. Clustalw (Thompson et al. 1994; Larkin et al. 2007) generated trees using UPGMA. Trees were visualized using drawgram.
Transcriptome Analysis
We collected RNAseq data for five replicate samples of unfertilized ova from a single clutch of O. pumilio and a single clutch of Xenopus laevis For O. pumilio, oocytes were harvested by dissection from a single individual, flash frozen in liquid nitrogen, shipped on dry ice, and then stored at −80 °C. For X. laevis, a female was induced to lay eggs, which were then stored at −80 °C. RNA was extracted from each egg using a PowerLyzer UltraClean Tissue and Cells RNA Isolation Kit (MoBio Laboratories, Inc.). Samples were homogenized in 300 μl of Solution TR1 using a PowerLyzer 24 bead beater for four cycles of 45 s at 3500 rpm and 30 s between the cycles. After the wash step with Solution TR3, the tubes were centrifuged for 2 min at 10,000 × g to remove any trace amounts of the wash buffer. A mix of 10 μl of Qiagen DNase I and 70 μl of Qiagen Buffer RDD was added directly to the spin column membrane and allowed to incubate for 15 min. Following the incubation, 350 μl of Qiagen buffer RW1 was added and the column was centrifuged at 10,000 rpm for 15 s. After these steps for the removal of DNA, the MoBio RNA extraction protocol was resumed with the addition of Solution TR4.
The intactness of the RNA was assessed using a Bioanalyzer 2100 (Agilent Technologies). The X. laevis extractions had RIN values ranging from 9.1 to 9.6. The Pumilio extractions had RIN values ranging from 7.1 to 8.4. Approximately 600 ng of RNA was taken from each sample (one O. pumilio sample had only 300 ng available), the samples were standardized to a common volume, and then the samples were concentrated with a 3× bead cleanup with KapaPure Beads. A Ribo-Zero Gold (Human/Mouse/Rat) Kit was used to remove ribosomal RNA. Libraries were then constructed using a KAPA Stranded RNA-Seq Library Preparation Kit. The samples were fragmented for 4 min at 94 °C. Half of the adapter-ligated DNA was amplified for 13 cycles, the concentration of the resulting library was assessed, and then the number of amplification cycles for other half of the adapter-ligated DNA was adjusted based on the concentration of the first amplification (range = 12–19 cycles). The two amplifications were then pooled in equal amounts by mass. Library quality was assessed with a Bioanalyzer and quantity was assessed with a Qubit. The libraries were pooled and sequenced on a lane of an Illumina HiSeq 4000 with 150 bp PE reads.
We assembled the transcriptome for each RNAseq replicate independently using Trinity 2.4.0 (https://github.com/trinityrnaseq/trinityrnaseq/wiki) under default parameters (Grabherr et al. 2011). We then mapped all RNAseq data onto the assembled transcriptome using Tophat 2.1.1 (Licht and Lowcock 1991; Frahry et al. 2015). FPKM values for each transcript and library were calculated using Cufflinks 2.2.1 (Licht and Lowcock 1991) with option –u to improve values for multiply mapped reads. Each assembled transcript was compared against Repbase using a BLASTn with an E-value cut-off of 10−10 to annotate repeat sequences. For each replicate, we calculated FPKM × sequence length to obtain an estimate of total expression for each repetitive element. Elements were classified according to the major classes represented in the genome assembly: Gypsy, Mariner, and Tc1. Total expression values of O. pumilio and X. laevis were compared using a Student’s t-test to determine significance, with a Bonferroni correction for multiple testing. Replicate was not a significant factor, suggesting no measurable batch effects on TE expression from ova taken from a single female. Mariner-4_DR elements show signatures consistent with HT between aquatic fish and frogs. We used a general linear model to determine whether there was a significant relationship between element similarity with the Danio rerio Mariner-4 element and the expression level in O. pumilio, including replicate as potential factors.
Ion Channel Evolution and Autoresistance
For each gene family (SCNA, nACHR), we queried all annotated proteins in the human and Xenopus (Silurana) tropicalis genomes against the annotations of the O. pumilio genome and transcriptome using tblastn. To include a broader representation of Anuran taxa, we also queried the annotated genome of Nanorana parkeri (V2, http://gigadb.org/dataset/100132) (Sun et al. 2015) and recently published transcriptomes of Rana pipiens (http://www.davislab.net/rana/) (Christenson et al. 2014) and Rhinella marina (http://gigadb.org/dataset/100374) (Richardson et al. 2017). We then translated all hits with an E-value below 10−10 and aligned them to publicly available sequences from other tetrapods and the coelacanth (Supplementary Material online) using MAFFT v. 7.271 (Katoh and Standley 2013) with automatic alignment parameter selection (–auto) and position-specific gap costs (–leavegappyregion). The resulting alignments were used to build gene trees for each protein family with MrBayes v. 3.2.6 (Ronquist et al. 2012) and PhyML v. 3.3 (Guindon et al. 2010). MrBayes runs were performed in duplicate for 5,000,000 steps (10% burn-in) with four chains per run, and PhyML runs searched the tree space using combined SPR and NNI moves from five random starting points. Models of protein evolution were chosen with ProtTest (Darriba et al. 2011), based on BioNJ trees optimized under each model (JTT+I + G+F SCNA; JTT+I + G for nACHR).
We conducted an initial round of phylogenetic analyzes to identify sequences from other gene families, as well as those corresponding to the same gene, such as alternate splice variants. We discarded members of other gene families, kept one randomly selected splice variant per gene, and merged sequences that overlapped with 100% identity at the protein level. When two sequences were found to nest within the same orthologous clade, but did not span overlapping segments of the alignment, they were joined and considered to represent fragments of the same gene. Finally, we looked for chimeric assemblies in transcriptome sequences by aligning each individual transcript to all X. tropicalis and N. parkeri sequences of the corresponding gene family, and visually inspecting the alignments.
After discarding these sequences, we looked for additional, unannotated genes in the O. pumilio genome assembly by querying N. parkeri, Ra. pipiens, and Rh. marina genes against it with blastn, and used the intron–exon structure for the corresponding X. tropicalis genes from the ENSEMBL database to manually annotate the identified scaffolds. The intron–exon boundaries were further refined by eye based on the closest available transcriptome sequence (usually Rh. marina). Finally, we performed a second round of phylogenetic analyzes as detailed above. We identified two distinct CHRNE orthologs in O. pumilio and Rh. marina, but their phylogenetic placement in relation to other frog sequences was ambiguous based on protein sequences. Therefore, we built a tree from the nucleotide sequences CHRNE genes using the same approach described above.
To identify sites in voltage-gated sodium channels involved in the evolution of O. pumilio’s resistance to its own toxins, we reconstructed ancestral amino acid sequences for the SCN gene family using PAML v. 4.9 g (Yang 2007), based on the JTT model, and using the MrBayes tree. We then identified sites from the PAML output at which changes had occurred along the terminal branch leading to O. pumilio genes, and selected those that were located on an S6 transmembrane segment, and/or where at least three of the six genes experienced amino acid substitutions. Amino acid numberings are based on the human SCN4A protein sequence (ENSP00000396320).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
Procedures involving animals were approved by the Institutional Animal Care and Use Committees (IACUCs) at Tulane University (protocol #0832) and the University of Pittsburgh (protocol #15106566). Thanks to Rachel Kjolby and Richard Harland for help in obtaining Xenopus eggs. We would also like to thank Hiten Madhani for advice on spliceosomes. This work was supported by NSF/IIS-1526415 to Yufeng Wu. Funding for this project was provided, in part, by grants from the National Science Foundation (nos. 0701165 and 1146370) to CLRZ and NSF grant DEB 1655336 to K.S. and R.N. Sequence data are available in the NCBI short read archive under PRJNA482685 and PRJNA482042.
References
- Albuquerque EX, Warnick JE, Sansone FM, Daly J.. 1973. The pharmacology of batrachotoxin. V. A comparative study of membrane properties and the effect of batrachotoxin on sartorius muscles of the frogs Phyllobates aurotaenia and Rana pipiens. J Pharmacol Exp Ther. 184(2):315–329. [PubMed] [Google Scholar]
- Aminetzach YT, Macpherson JM, Petrov DA.. 2005. Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila. Science 309(5735):764–767. [DOI] [PubMed] [Google Scholar]
- Andriamaharavo NR, Garraffo HM, Saporito RA, Daly JW, Razafindrabe CR, Andriantsiferana M, Spande TF.. 2010. Roughing it: a mantellid poison frog shows greater alkaloid diversity in some disturbed habitats. J Nat Prod. 73(3): 322–330. [DOI] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O.. 2015. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6:11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Wang H.. 2014. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev Plant Biol. 65:505–530. [DOI] [PubMed] [Google Scholar]
- Bogart JP. 1991. The influence of life history on karyotypic evolution in frogs In: Green DM, Sessions SK, editors. Amphibian cytogenetics and evolution. New York: Academic Press. p. 233–258. [Google Scholar]
- Burnette JM, Miyamoto-Sato E, Schaub MA, Conklin J, Lopez AJ.. 2005. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics 170(2):661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casacuberta E, Gonzalez J.. 2013. The impact of transposable elements in environmental adaptation. Mol Ecol. 22(6):1503–1517. [DOI] [PubMed] [Google Scholar]
- Christenson MK, Trease AJ, Potluri L-P, Jezewski AJ, Davis VM, Knight LA, Kolok AS, Davis PH.. 2014. De novo assembly and analysis of the northern leopard frog Rana pipiens transcriptome. J Genomics 2:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Nielsen R, Wu Y.. 2016. REPdenovo: inferring de novo repeat motifs from short sequence reads. PLoS One 11(3):e0150719.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C.. 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 18(2):71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford AJ. 2003. Relative rates of nucleotide substitution in frogs. J Mol Evol. 57(6):636–641. [DOI] [PubMed] [Google Scholar]
- Daly JW, Gusovsky F, Myers CW, Yotsu-Yamashita M, Yasumoto T.. 1994. First occurrence of tetrodotoxin in a dendrobatid frog (Colostethus inguinalis), with further reports for the bufonid genus Atelopus. Toxicon 32(3):279–285. [DOI] [PubMed] [Google Scholar]
- Daly JW, Martin Garraffo H, Spande TF, Jaramillo C, Stanley Rand A.. 1994. Dietary source for skin alkaloids of poison frogs (Dendrobatidae)? J Chem Ecol. 20(4):943–955. [DOI] [PubMed] [Google Scholar]
- Daly JW, Myers CW.. 1967. Toxicity of Panamanian poison frogs (Dendrobates): some biological and chemical aspects. Science 156(3777):970–973. [DOI] [PubMed] [Google Scholar]
- Daly JW, Myers CW, Warnick JE, Albuquerque EX.. 1980. Levels of batrachotoxin and lack of sensitivity to its action in poison-dart frogs (Phyllobates). Science 208(4450):1383–1385. [DOI] [PubMed] [Google Scholar]
- Daly JW, Spande TF.. 1986. Amphibian alkaloids: chemistry, pharmacology, and biology. Alkaloids Chem Biol Perspect. 4(1):1–274. [Google Scholar]
- Daly JW, Spande TF, Garraffo HM.. 2005. Alkaloids from amphibian skin: a tabulation of over eight-hundred compounds. J Nat Prod. 68(10):1556–1575. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D.. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27(8):1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Nomura Y, Liu Z, Huang ZY, Dong K.. 2009. Functional expression of an arachnid sodium channel reveals residues responsible for tetrodotoxin resistance in invertebrate sodium channels. J Biol Chem. 284(49):33869–33875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulac C, O'Connell LA, Wu Z.. 2014. Neural control of maternal and paternal behaviors. Science 345(6198):765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahry MB, Sun C, Chong RA, Mueller RL.. 2015. Low levels of LTR retrotransposon deletion by ectopic recombination in the gigantic genomes of salamanders. J Mol Evol. 80(2):120–129. [DOI] [PubMed] [Google Scholar]
- Geffeney SL, Fujimoto E, Brodie ED 3rd, Brodie ED Jr, Ruben PC.. 2005. Evolutionary diversification of TTX-resistant sodium channels in a predator-prey interaction. Nature 434(7034):759–763. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O.. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321. [DOI] [PubMed] [Google Scholar]
- Haberer G, Young S, Bharti AK, Gundlach H, Raymond C, Fuks G, Butler E, Wing RA, Rounsley S, Birren B, et al. 2005. Structure and architecture of the maize genome. Plant Physiol. 139(4):1612–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangam D, Feschotte C, Betran E.. 2017. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 33(11):817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmolowski A, Mattaj I.. 1993. The determinants for Sm protein binding to Xenopus U1 and U5 snRNAs are complex and non-identical. EMBO J. 12(1):223.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joron M, Mallet JL.. 1998. Diversity in mimicry: paradox or paradigm? Trends Ecol Evol. 13(11):461–466. [DOI] [PubMed] [Google Scholar]
- Jost MC, Hillis DM, Lu Y, Kyle JW, Fozzard HA, Zakon HH.. 2008. Toxin-resistant sodium channels: parallel adaptive evolution across a complete gene family. Mol Biol Evol. 25(6):1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J.. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110(1-4):462–467. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409(6822):860–921. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948. [DOI] [PubMed] [Google Scholar]
- Li HL, Hadid D, Ragsdale DS.. 2002. The batrachotoxin receptor on the voltage-gated sodium channel is guarded by the channel activation gate. Mol Pharmacol. 61(4):905–912. [DOI] [PubMed] [Google Scholar]
- Licht LE, Lowcock LA.. 1991. Genome size and metabolic rate in salamanders. Compar Biochem Physiol B Compar Biochem. 100(1):83–92. [Google Scholar]
- Loreto EL, Carareto CM, Capy P.. 2008. Revisiting horizontal transfer of transposable elements in Drosophila. Heredity 100(6):545–554. [DOI] [PubMed] [Google Scholar]
- Lund E, Bostock CJ, Dahlberg JE.. 1987. The transcription of Xenopus laevis embryonic U1 snRNA genes changes when oocytes mature into eggs. Gene Dev. 1(1):47–56. [DOI] [PubMed] [Google Scholar]
- Lynch M. 2007. The origins of genome architecture. Sunderland (MA: ): Sinauer Associates; 494. xvi, p. [Google Scholar]
- Lynch VJ, Nnamani MC, Kapusta A, Brayer K, Plaza SL, Mazur EC, Emera D, Sheikh SZ, Grützner F, Bauersachs S, et al. 2015. Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep. 10(4):551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwire MM, Bayer F, Webster CL, Cao C, Jiggins FM.. 2011. Successive increases in the resistance of Drosophila to viral infection through a transposon insertion followed by a duplication. PLoS Genet. 7(10): e1002337.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Wang ZF.. 2014. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 15(2):108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlothlin JW, Chuckalovcak JP, Janes DE, Edwards SV, Feldman CR, Brodie ED, Pfrender ME, Brodie ED Jr. 2014. Parallel evolution of tetrodotoxin resistance in three voltage-gated sodium channel genes in the garter snake Thamnophis sirtalis. Mol Biol Evol. 31(11):2836–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin RN Jr, Malik HS.. 2017. Genetic conflicts: the usual suspects and beyond. J Exp Biol. 220(Pt 1):6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery EA, Huang SM, Langley CH, Judd BH.. 1991. Chromosome rearrangement by ectopic recombination in Drosophila melanogaster: genome structure and evolution. Genetics 129(4):1085–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller RL. 2017. piRNAs and evolutionary trajectories in genome size and content. J Mol Evol. 85(5-6):169–171. [DOI] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA.. 2005. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1):i351–i358. [DOI] [PubMed] [Google Scholar]
- Richardson MF, Sequeira F, Selechnik D, Carneiro M, Vallinoto M, Reid JG, et al. 2017. Improving amphibian genomic resources: a multi-tissue reference transcriptome of an iconic invader. Gigascience 7(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards-Zawacki CL, Wang IJ, Summers K.. 2012. Mate choice and the genetic basis for colour variation in a polymorphic dart frog: inferences from a wild pedigree. Mol Ecol. 21(15):3879–3892. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP, et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JC, Tarvin RD, O’Connell LA. 2016. A review of chemical defense in poison frogs (Dendrobatidae): ecology, pharmacokinetics and autoresistance. In: Schulte BA, Goodwin TE, Ferkin MH, editors. Chemical signals in vertebrates 13. New York: Springer Science + Business Media. p. 305–337. [Google Scholar]
- Saporito RA, Donnelly MA, Jain P, Martin Garraffo H, Spande TF, Daly JW.. 2007. Spatial and temporal patterns of alkaloid variation in the poison frog Oophaga pumilio in Costa Rica and Panama over 30 years. Toxicon 50(6):757–778. [DOI] [PubMed] [Google Scholar]
- Saporito RA, Donnelly MA, Spande TF, Garraffo HM.. 2012. A review of chemical ecology in poison frogs. Chemoecology 22(3):159–168. [Google Scholar]
- Schaack S, Gilbert C, Feschotte C.. 2010. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol. 25(9):537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader L, Kim JW, Ence D, Zimin A, Klein A, Wyschetzki K, Weichselgartner T, Kemena C, Stokl J, Schultner E, et al. 2014. Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat Commun. 5:5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schoffmann O, Morgenstern B, Waack S.. 2006. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7:62.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B.. 2004. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32(web server issue): W309–W312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S.. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2):ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Stynoski JL, Shelton G, Stynoski P.. 2014. Maternally derived chemical defences are an effective deterrent against some predators of poison frog tadpoles (Oophaga pumilio). Biol Lett. 10(5):20140187.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stynoski JL, Torres-Mendoza Y, Sasa-Marin M, Saporito RA.. 2014. Evidence of maternal provisioning of alkaloid-based chemical defenses in the strawberry poison frog Oophaga pumilio. Ecology 95(3):587–593. [DOI] [PubMed] [Google Scholar]
- Summers K, Symula R, Clough M, Cronin T.. 1999. Visual mate choice in poison frogs. Proc Biol Sci. 266(1434):2141–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Li T, Xiao J, Liu L, Zhang P, Murphy RW, et al. 2016. Contribution of multiple inter-kingdom horizontal gene transfers to evolution and adaptation of amphibian-killing chytrid, Batrachochytrium dendrobatidis. Front Microbiol. 7:1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Lopez Arriaza JR, Mueller RL.. 2012. Slow DNA loss in the gigantic genomes of salamanders. Genome Biol Evol. 4(12):1340–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y-B, Xiong Z-J, Xiang X-Y, Liu S-P, Zhou W-W, Tu X-L, Zhong L, Wang L, Wu D-D, Zhang B-L, et al. 2015. Whole-genome sequence of the Tibetan frog Nanorana parkeri and the comparative evolution of tetrapod genomes. Proc Natl Acad Sci U S A. 112(11):E1257–E1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinburne IA, Silver PA.. 2008. Intron delays and transcriptional timing during development. Dev Cell 14(3):324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarvin RD, Borghese CM, Sachs W, Santos JC, Lu Y, O’Connell LA, Cannatella DC, Harris RA, Zakon HH.. 2017. Interacting amino acid replacements allow poison frogs to evolve epibatidine resistance. Science 357(6357):1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarvin RD, Santos JC, O'Connell LA, Zakon HH, Cannatella DC.. 2016. Convergent substitutions in a sodium channel suggest multiple origins of toxin resistance in poison frogs. Mol Biol Evol. 33(4):1068–1081. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ.. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22(22): 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Sun G, Ding Q, Huang J, Oruganti S, Xie B, editors. 2011. AlienG: an effective computational tool for phylogenomic identification of horizontally transferred genes. BICoB. Conference: Proceedings of the ISCA 3rd International Conference on Bioinformatics and Computational Biology, BICoB-2011. [Google Scholar]
- Tokuyama T, Daly J, Witkop B.. 1969. The structure of batrachotoxin, a steroidal alkaloid from the Colombian arrow poison frog, Phyllobates aurotaenia, and partial synthesis of batrachotoxin and its analogs and homologs. J Am Chem Soc. 91(14):3931–3938. [DOI] [PubMed] [Google Scholar]
- Vestergaard JS, Twomey E, Larsen R, Summers K, Nielsen R.. 2015. Number of genes controlling a quantitative trait in a hybrid zone of the aposematic frog Ranitomeya imitator. Proc Biol Sci. 282(1807):20141950.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Mitchell J, Tikhonov DB, Zhorov BS, Wang GK.. 2006. How batrachotoxin modifies the sodium channel permeation pathway: computer modeling and site-directed mutagenesis. Mol Pharmacol. 69(3):788–795. [DOI] [PubMed] [Google Scholar]
- Wang SY, Tikhonov DB, Mitchell J, Zhorov BS, Wang GK.. 2007. Irreversible block of cardiac mutant Na+ channels by batrachotoxin. Channels 1(3):179–188. [DOI] [PubMed] [Google Scholar]
- Wang SY, Tikhonov DB, Zhorov BS, Mitchell J, Wang GK.. 2007. Serine-401 as a batrachotoxin- and local anesthetic-sensing residue in the human cardiac Na+ channel. Pflugers Arch. 454(2):277–287. [DOI] [PubMed] [Google Scholar]
- Wang SY, Wang GK.. 2017. Single rat muscle Na(+) channel mutation confers batrachotoxin autoresistance found in poison-dart frog Phyllobates terribilis. Proc Natl Acad Sci U S A. 114(39):10491–10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weygoldt P. 1980. Complex brood care and reproductive-behavior in captive poison-arrow frogs, Dendrobates pumilio O. Schmidt. Behav Ecol Sociobiol. 7(4):329–332. [Google Scholar]
- Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, et al. 2014. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30(12): 1660–1666. [DOI] [PubMed] [Google Scholar]
- Yamanoue Y, Miya M, Inoue JG, Matsuura K, Nishida M.. 2006. The mitochondrial genome of spotted green pufferfish Tetraodon nigroviridis (Teleostei: Tetraodontiformes) and divergence time estimation among model organisms in fishes. Genes Genet Syst. 81(1):29–39. [DOI] [PubMed] [Google Scholar]
- Yang Y, Richards-Zawacki CL, Devar A, Dugas MB.. 2016. Poison frog color morphs express assortative mate preferences in allopatry but not sympatry. Evolution 70(12):2778–2788. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591. [DOI] [PubMed] [Google Scholar]
- Zakon HH, Jost MC, Lu Y.. 2011. Expansion of voltage-dependent Na+ channel gene family in early tetrapods coincided with the emergence of terrestriality and increased brain complexity. Mol Biol Evol. 28(4): 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakon HH, Li W, Pillai NE, Tohari S, Shingate P, Ren J, Venkatesh B.. 2017. Voltage-gated sodium channel gene repertoire of lampreys: gene duplications, tissue-specific expression and discovery of a long-lost gene. Proc Biol Sci. 284:1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.