

Abstract
Introduction:
Clozapine (CLZ) is a second-generation antipsychotic FDA-approved for refractory schizophrenia, and glucuronidation is an important pathway in its metabolism. The goal of this study was to fully characterize the CLZ glucuronidation pathway and examine whether polymorphisms in active glucuronidating enzymes could contribute to variability in CLZ metabolism.
Methods:
Cell lines over-expressing wild-type or variant UGT enzymes were used to determine which UGTs exhibit activity against CLZ and its major active metabolite, N-desmethyl-CLZ (dmCLZ). Human liver microsomes (HLM) were used to comparing hepatic glucuronidation activity against UGT genotype.
Results:
Several UGTs including 1A1 and 1A4 were active against CLZ; only UGT1A4 exhibited activity against dmCLZ. UGT1A1 exhibited a 2.1-fold (p<0.0001) higher Vmax/KM for formation of the CLZ-N+-glucuronide than UGT1A4; UGT1A4 was the only UGT for which CLZ-5-N-glucuronide kinetics could be determined. The UGT1A424Pro/48Val variant exhibited a 5.2-, 2.0-, and 3.4-fold (p<0.0001 for all) higher Vmax/KM for formation of the CLZ-5-N-glucuronide, CLZ-N+-glucuronide, and dmCLZ-5-N-glucuronide, respectively, as compared to wild-type UGT1A424Pro/48Leu. There was a 37% (p<0.05) decrease in the rate of CLZ-N+-glucuronide formation in HLM with the UGT1A1 (*28/*28)/UGT1A4 (*1/*1) genotype, and a 2.2- and 1.8-fold (p<0.05 for both) increase in formation of CLZ-5-N-glucuronide and CLZ-N+-glucuronide formation in UGT1A1 (*1/*1)/UGT1A4 (*3/*3) HLM, compared to UGT1A1 (*1/*1)/UGT1A4 (*1/*1) HLM. The UGT1A1*28 allele was a significant (p=0.045) predictor of CLZ-N+-glucuronide formation; the UGT1A4*3 allele was a significant (p<0.0001) predictor of CLZ-5-N- and dmCLZ-glucuronide formation.
Conclusion:
These data suggest that the UGT1A1*28 and UGT1A4*3 alleles contribute significantly to inter-individual variability in CLZ and dmCLZ metabolism.
Keywords: UDP-glucuronosyltransferases, UGT, clozapine, N-desmethylclozapine, variant alleles, UGT1A1, UGT1A4, drug metabolism
Introduction
Clozapine (CLZ) is a second-generation antipsychotic that is FDA (U.S.)-approved for the treatment of refractory schizophrenia [1]. It demonstrates unique efficacy and tolerability compared to other antipsychotics due in part to its binding affinities at dopaminergic, serotonergic, and glutaminergic receptors and to the actions of its active metabolite, N-desmethylclozapine (dmCLZ) [2, 3]. Despite its superior efficacy and motoric tolerability, CLZ is a second-line drug due to its risk of hematologic side effects [4]. Furthermore, CLZ elicits severe weight gain [5–10], contributing to metabolic dysfunction, dyslipidemia, overweight/obesity, type II diabetes mellitus, heart disease, and mortality [11, 12].
Previous studies have demonstrated that CLZ and dmCLZ plasma concentrations are associated with therapeutic efficacy and toxicity. Approximately 30% of patients fail to respond to CLZ treatment despite adequate dosing and the relative risk of attrition due to side effects is higher for patients taking CLZ than other second-generation antipsychotics [13]. Studies suggest there is a therapeutic window for CLZ plasma concentrations; however, for a given dose of CLZ, there is wide inter-individual variability in CLZ and dmCLZ plasma concentrations (3–1507 ng/ml). There is evidence to suggest that therapeutic response is related to plasma concentrations rather than drug dose [14–16]. Thus, CLZ and dmCLZ plasma concentrations and therapeutic response cannot be predicted based on dose alone, suggesting that genetic variability in drug metabolizing enzyme (DME) genes could affect their efficacy and tolerability [17].
DMEs are classified as Phase I or Phase II depending on the type of reaction they catalyze. The uridine diphosphate (UDP)-glucuronosyltransferases (UGTs) are one group of Phase II enzymes that mediate greater than one-third of Phase II drug metabolism [18] and influence the plasma levels and clearance of a given compound [19]. UGTs are important to the biotransformation and clearance of several second-generation antipsychotics as evidenced by plasma and urine metabolites in humans [20–24]. CLZ undergoes extensive biotransformation, with 97% of the administered dose being metabolized [15]. Seven CLZ-glucuronides have been identified in humans ([20, 21, 25]; see Figure 1). One of these glucuronides is a dmCLZ conjugate [25] suggesting that glucuronidation has a role in the metabolism and clearance of this active metabolite as well. UGT activity against CLZ has been partially characterized. In vitro, UGT1A4 catalyzes the formation of tertiary and quaternary CLZ-glucuronide metabolites, and the UGT1A4L48V polymorphism was found to be twice as efficient as wild-type UGT1A4 [26]. This genetic difference, along with the extensive role that UGTs play in the metabolism and clearance of CLZ [15, 17, 20], suggest that UGT pharmacogenetic factors influence CLZ plasma levels. However, the UGTs with enzymatic activity against CLZ and dmCLZ have either not been characterized or only partially characterized. Additionally, the effects of UGT polymorphisms on CLZ and dmCLZ metabolism are largely unknown in vitro or in vivo.
Figure 1. Schematic of CLZ metabolism.
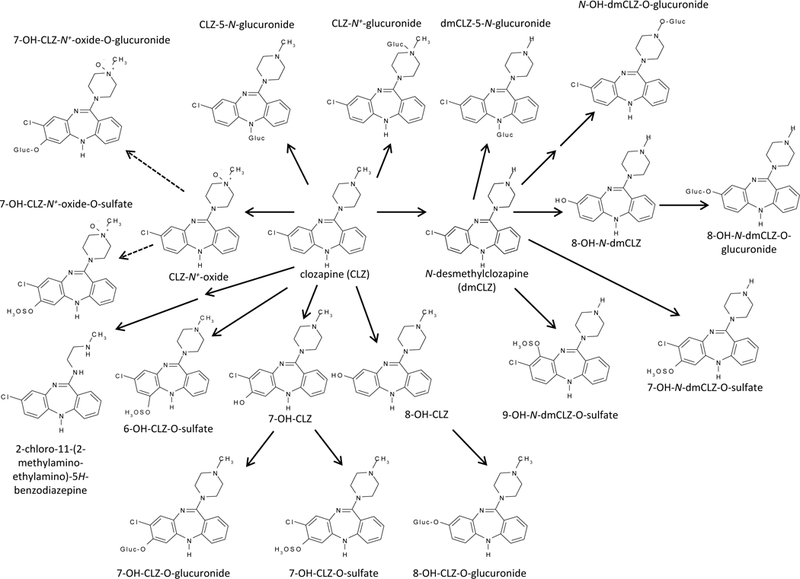
Previous studies identified two primary conjugates of CLZ, the 5-N-glucuronide and the N+-glucuronide, and one primary conjugate of dmCLZ, dmCLZ-5-N-glucuronide, in urine and bile [20, 21, 25]. gluc=glucuronide. Dashed line represents multiple reaction pathways needed to generate final metabolite.
The goal of the present study was to characterize the glucuronidation activity of known UGT1A and 2B family enzymes against CLZ and dmCLZ in vitro and examine the effects of prevalent SNPs in active UGTs on formation of individual CLZ and dmCLZ glucuronide metabolites. The results of this study demonstrate that prevalent missense SNPs in UGTs 1A1 and 1A4 may be important in the overall metabolism of CLZ and dmCLZ.
Materials and Methods
Chemicals and Materials.
Alamethicin, β-glucuronidase, bovine serum albumin, anti-ß-actin monoclonal antibody, CLZ, dmCLZ, hecogenin, and niflumic acid were purchased from Sigma-Aldrich (St. Louis, MO). DMEM, Dulbecco’s PBS (minus calcium chloride and magnesium chloride), fetal bovine serum, penicillin-streptomycin, geneticin (G418), Platinum Pfx DNA polymerase, and the pcDNA3.1/V5-His-TOPO mammalian expression vector were all obtained from Invitrogen (Carlsbad, CA). The BCA protein assay kit was purchased from Pierce (Rockford, IL). PCR primers were purchased from Integrated DNA Technologies (Coralville, IA). The human UGT1A western blotting kit that includes the anti-UGT1A polyclonal antibody was purchased from Gentest (Woburn, MA). All other chemicals were purchased from Fisher Scientific (Waltham, MA) unless specified otherwise.
Tissues.
As described previously [27, 28] normal (non-cancerous) adjacent liver specimens were obtained from individuals undergoing surgery for resection of hepatocellular carcinoma at the H. Lee Moffitt Cancer Center (Tampa, FL) and were quick-frozen at −80°C within 2 h post surgery. Matching genomic DNA was obtained from each subject. HLM were prepared through differential centrifugation as previously described [29] and stored (10–20 mg microsomal protein/mL) at −80°C. Microsomal protein concentrations were measured using the BCA assay. All protocols involving the analysis of tissue specimens were approved by the institutional review board at the Penn State College of Medicine and in accordance with assurances filed with and approved by the United States Department of Health and Human Services.
Cell lines.
The cell lines over-expressing the UGT1A and UGT2B isoforms used in this study were described previously [30–35]. All UGT over-expressing cell lines were grown in DMEM to 80% confluence before preparing cell homogenates by re-suspending the cell pellet in Tris-buffered saline (25 mM Tris base, 138 mM NaCl, and 2.7 mM KCl, pH 7.4) and subjecting them to three rounds of freeze-thaw before gentle homogenization. Total homogenate protein concentrations were measured using the BCA protein assay. Homogenates were stored at −80°C in 50-μL aliquots to minimize freeze-thaws.
Western blot analysis.
UGT1A1 and 1A4 protein levels were determined by western blot analysis for UGT1A1 and variant UGT1A4-over-expressing cell lines as described previously [33, 34]. Briefly, UGT1A1 and 1A4 variant -over-expressing cell homogenate protein (25–100 μg), HEK 293 homogenate protein (100 μg), and UGT1A standard (50 ng; BD Gentest, Woburn, MA) were adjusted to contain equal volumes of loading buffer and heated at 100°C for 10 min. Samples were run at 93 V on a 10% acrylamide gel then transferred to a polyvinylidene difluoride (PVDF) membrane for 2 h at 30 V. PVDF membranes were then probed with the rabbit polyclonal UGT1A antibody (1:5000 dilution) for 1 h at 23°C, washed three times, followed by horseradish peroxidase-conjugated goat anti-rabbit IgG (1:5000 dilution). Housekeeping protein levels were assayed using a 1:5000 dilution of β-actin. UGT1A protein was visualized using the SuperSignal West Dura Extended Duration Substrate (ThermoScientific, Waltham, MA) and Hyblot CL autoradiography film (Denville Scientific, Metuchen, NJ).UGT1A protein levels were determined by western blot by densiometric analysis of X-ray film exposure (2–4 min exposures) of western blots using ImageJ software. After exposure, blots were stripped and re-probed using the β-actin antibody. Quantification of UGT1A-over-expressing cell lines was performed first by normalizing to the B-actin loading control, then by quantifying protein levels relative to known amounts of UGT1A loaded on the same gel.
UGT genotyping.
HLMs were genotyped to for the UGT1A1*28 allele that encodes the (A(TA)7TAA) sequence in its TATAA box promoter element or the functional variants encoded by the UGT1A4 *2 (Pro24Thr) and *3 (Leu48Val) alleles. Genotyping for UGT1A1 and UGT1A4 has been previously described for all of the HLM specimens described in this study [36, 37]. Briefly, UGT1A1 and 1A4 genotypes were determined by direct sequencing of PCR-amplified PCR products from liver genomic DNA spanning the UGT1A1 TATAA box promoter element of UGT1A1 and spanning both codons 24 and 48 for UGT1A4. The same primers were used for both PCR amplification and sequencing of UGT1A1: sense (1A1S, 5′-GTCACGTGACACAGTCAAAC-3′) and antisense (1A1AS, 5′-TTTGCTCCTGCCAGAGGTT-3′) primers corresponding to nucleotides −104 through −85 and −25 through −7, respectively, relative to the translation start site in UGT1A1 exon 1 (GenBank accession no. AF297093), and UGT1A4: sense, 5’-GGCTTCTGCTGAGATGGCCAG-3’, and antisense, 5’-CCTTGAGTGTAGCCCAGCGT-3’, corresponding to nucleotides located –13 to +8 and +277 to +306, respectively, relative to the UGT1A4 translation start site (Genbank accession no. NM_007120). Sequencing was performed using an ABI 3130 Capillary Sequencer at the Genomics Core Facility at the University Park campus of Penn State University.
Glucuronidation assays.
UGT-over-expressing cell line homogenates or HLM were incubated with alamethicin (50 μg/mg protein) for 15 min on ice similar to that described previously [34]. Glucuronidation assays were performed in 50 mmol/L Tris buffer (pH 7.5), 10 mmol/L MgCl2, 4 mmol/L UDP-glucuronic acid (UDPGA), and 7.8 μmol/L to 4 mmol/L of substrate at 37°C in a water bath for 30 min for UGT1A4-over-expressing cell line kinetic reactions and 2 h for all other reactions. HLM (20 μg of protein) or human UGT-over-expressing cell homogenate (1 mg of protein) were screened for glucuronidation activity against CLZ using 160 μM of CLZ or 320 μM dmCLZ in a 20 or 50 μL reaction, respectively. Kinetic assays were performed in 20 μL reactions using a range of 10–612 μM of CLZ and 10–640 μM of dmCLZ, and 250 μg and 50 μg of UGT1A1- and UGT1A4-over-expressing cell homogenate protein, respectively, or 12.5 μg of HLM protein. Reactions were terminated by the addition of the same volume of cold acetonitrile as the initial reaction volume. Reactions were centrifuged at 13,000 g for 10 min at 4°C and supernatants were collected. Glucuronidation assays (5 μL) were analyzed for CLZ glucuronide formation using a Waters ACQUITY ultra pressure liquid chromatography (UPLC) system (Milford, MA) as previously described [22, 28, 38] using a 100 × 2.1 mm inner diameter Acquity UPLC ethylene bridged hybrid (BEH) C18 column with 1.7 μm particles (Waters) and a 0.2 μm prefilter installed before the column. Elution consisted of a gradient elution starting with 10% buffer B (100% acetonitrile) and 90% buffer A [20mmol/L ammonium acetate (pH 7.0)] for 5 min, a linear gradient to 90% buffer B over 1 min and held for 2 min, then a linear gradient back to initial conditions and held for 2 min for a total run time of 10 min. The flow rate was maintained at 0.5 mL/min. The amount of glucuronide formed was determined based on the ratio of CLZ- or dmCLZ-glucuronide versus unconjugated CLZ or dmCLZ after calculating the area under the curve for the CLZ or dmCLZ and CLZ- or dmCLZ-glucuronide peaks. CLZ and dmCLZ peaks were quantified by comparison to a standard curve using known amounts of CLZ or dmCLZ (10–612 μM and 10–640 μM respectively). CLZ- or dmCLZ-glucuronide was confirmed by sensitivity to treatment with 1,000 U β-glucuronidase at 37°C for 12–16 h as previously described [27], by treatment with 3 M HCl at 50°C for 24 h [25], and by mass spectrometry (described below). As controls, glucuronidation assays were performed using HLM as a positive control for glucuronidation activity and untransfected HEK 293 cell homogenate protein as a negative control for glucuronidation activity. Four independent experiments were performed for kinetic analysis and CLZ-5-N-glucuronide to CLZ-N+-glucuronide peak ratios of UGT-over-expressing cell homogenates, with all assays within each experiment performed in duplicate; two independent experiments were performed for rate determination assays and three for kinetic analysis for HLM specimens.
Inhibition studies were performed as described above using 160 μM of CLZ or 320 μM dmCLZ in a 20 μL reaction containing 10 μM hecogenin [39] or 100 μM niflumic acid [40] dissolved in DMSO (with DMSO less than 1% of the final reaction volume). For inhibition studies in HLM, analysis were performed for five HLM exhibiting the UGT1A1 (*1/*1)/UGT1A4 (*1/*1) genotype, with the same HLM used for studies with hecogenin or niflumic acid.
Mass spectrometry.
Triple-quadrupole tandem mass spectrometric detection was performed using an ACQUITY SQD (Waters Corp.) with electrospray ionization interface and an UPLC system consisting of a binary gradient pump, an auto sampler (4°C), and a column oven (40°C). UPLC was operated under the same conditions as described above for glucuronidation assays. Peaks were detected at 254 nm wavelength. The mass spectrometer operated in positive mode was set up to scan the daughter ion of m/z 327. The optimized mass spectrometry parameters used were as follows: capillary voltage, 0.57 kV; cone voltage, 30 V; collision energy, 15 V; source temperature, 450°C; and desolvation temperature, 140°C. Nitrogen was used as the desolvation and cone gas with a flow rate of 760 L/h. Argon was used as the collision gas at a flow rate of 0.1 mL/min. Data acquisition and analysis were performed using the MassLynx NT 4.1 software with QuanLynx program (Waters Corp.).
Statistical analysis.
Michaelis-Menten kinetic constants were determined using Prism Version 5 software (La Jolla, CA). The Students t-test (two-sided) was used to compare kinetic values of glucuronide formation for the UGT1A1 and 1A4 isoforms against CLZ and dmCLZ in cell lines, and between HLM for genotypes of different UGT enzymes. One-way ANOVA using Tukey’s post-hoc test was used for comparative analysis of CLZ and dmCLZ glucuronidation rates and kinetic values for HLM of different genotypes for the same UGT gene. To perform the most conservative comparison, unequal variances were assumed when comparing levels of CLZ-glucuronide formation rates in HLM with wild-type alleles versus HLM with one or two polymorphic alleles. Analysis of CLZ glucuronidation in HLM stratified by UGT1A1 genotypes was performed only for those specimens also exhibiting the wild-type UGT1A4 (*1/*1) genotype (n=94); analysis of CLZ glucuronidation in HLM stratified by UGT1A4 genotypes was performed using all 113 HLM for the CLZ-5-N-glucuronide and the dmCLZ-5-N-glucuronide and only for those specimens also exhibiting the wild-type UGT1A1 (*1/*1) genotype for the CLZ-N+-glucuronide (n=49). Regression analysis of CLZ-glucuronide and dmCLZ-glucuronide formation against UGT1A1 or 1A4 genotype was performed using the data from these samples (Minitab Corporation, State College, PA). All data were normally distributed as determined by the Kolmogorov-Smirnov test.
Results
Characterization of CLZ and dmCLZ glucuronidation.
Figure 1 outlines the metabolic pathways and enzymes reported previously to be involved in CLZ and dmCLZ metabolism [20, 21, 41]. In human subjects taking CLZ unchanged CLZ comprises ~6% of the total recovered CLZ and dmCLZ accounts for ~3% of recovered metabolites, while CLZ and dmCLZ glucuronides comprise between 33–38% of all recovered metabolites in urine and bile [20, 21, 25] . While UGT1A3 and UGT1A4 were shown to be active against CLZ and UGT1A4 against dmCLZ [42, 43], several UGTs were not previously screened. To better characterize the UGT enzymes responsible for CLZ and dmCLZ glucuronidation, glucuronidation assays were performed and CLZ- and dmCLZ-glucuronides were separated by UPLC. CLZ eluted at 5.75 min and, when incubated with HLM, formed two additional peaks eluting at 3.3 and 3.75 min (Figure 2, panel A). The 3.75 min peak was highly sensitive to treatment with β-glucuronidase, while the earlier eluting peak at 3.3 min was extremely sensitive to treatment with 3 M HCl solution, a pattern that was previously reported for CLZ glucuronides (Figure 2, panel B) [25]. The peaks at 3.3 and 3.75 min both demonstrated a [M+] peak at m/z 503 (the glucuronide conjugate of CLZ) by mass spectrometry (MS/MS) analysis (Figure 2, panel E, F). The peak at 3.3 min showed a [M+H]+ peak at m/z 327 for CLZ after loss of the glucuronic acid moiety (molecular weight = 176 g/mol) and an m/z at 447 and 270 representing a loss of the CH2=CH-NH-CH3 group from the methyl piperazine moiety with and without a glucuronic acid moiety (Figure 2, panel E). This is identical to the pattern identified for the CLZ-5-N-glucuronide reported previously [25]. The peak at 3.75 min (Figure 2, panel F) demonstrated a [M+H]+ peak at m/z 327 (CLZ) and at m/z 296 due to the loss of CH3NH2 and glucuronic acid, but no peak at m/z 447, which is characteristic of the CLZ-N+-glucuronide [21, 25].
Figure 2. UPLC and MS/MS analysis of CLZ and dmCLZ glucuronides formed by HLM and UGT-over-expressing cell lines.
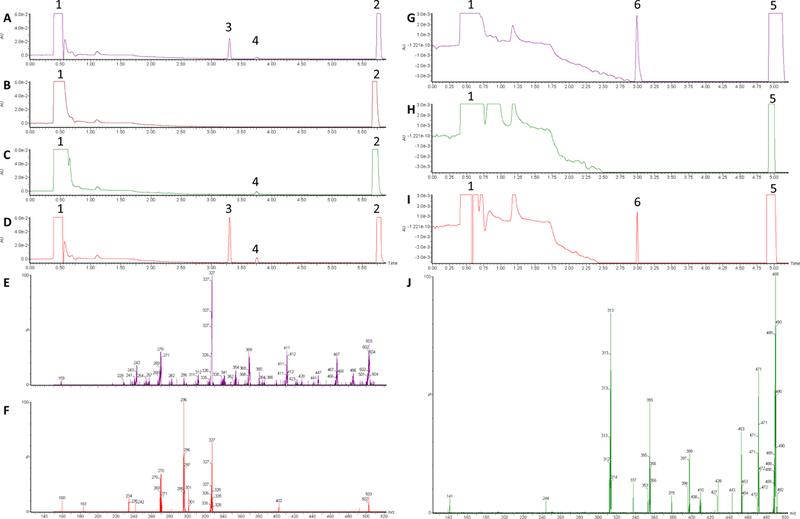
Glucuronidation assays were performed using 12.5 μg HLM protein, 250 μg UGT1A1- or 50 μg UGT1A4-over-expressing cell homogenates, and 160 μM CLZ or 320 μM dmCLZ, and incubated at 37°C for 2 h with 4 mM UDP-glucuronic acid prior to analysis by UPLC as described in the Materials and Methods. Panel A, HLM + CLZ; panel B, HLM + CLZ after treatment with 1,000 U of β-glucuronidase for 24h followed by treatment with 3 M HCl for 24 h; panel C, UGT1A1 + CLZ; panel D, UGT1A4 + CLZ; panel E, mass spectra of UPLC peak 3 from panel A; panel F, mass spectra of peak 4 from panel A; Panel G, HLM + dmCLZ; panel H, HLM + dmCLZ after treatment with 3 M HCl for 24 h; panel I, UGT1A4 + dmCLZ; Panel J, mass spectra of UPLC peak 6 from panel G. Peak 1, UDP-glucuronic acid; peak 2, CLZ; peak 3, CLZ-5-N-glucuronide; peak 4, CLZ-N+-glucuronide; peak 5, dmCLZ; peak 6, dmCLZ-5-N-glucuronide. AU, absorbance (arbitrary units); x-axis = retention time (min).
dmCLZ eluted at 4.95 min and formed one glucuronide eluting at 3.0 min as detected by UPLC (Figure 2, panel G). The peak at 3.0 min was not sensitive to treatment with β-glucuronidase (results not shown) but was extremely sensitive to treatment with 3 M HCl solution (Figure 2, panel H), suggesting it is the dmCLZ-5-N-glucuronide previously detected in human urine [25, 44]. The 3.0 min peak demonstrated a [M+] peak at m/z 489 (the glucuronide conjugate of dmCLZ) by mass spectrometry (MS/MS) analysis. The peak at 3.0 min showed a [M+H]+ peak at m/z 313 for dmCLZ after loss of the glucuronic acid moiety (molecular weight = 176 g/mol) and an m/z at 243 fragment after loss of CH2=CH-NH-CH=CH2 from the piperazine moiety of the parent compound (Figure 2, panel J). This is identical to the pattern identified for the dmCLZ-5-N-glucuronide reported previously [25].
Previous studies of selected UGT enzymes suggested that UGT1A3 and 1A4 were active against CLZ and UGT1A4 was active against dmCLZ. To fully characterize all of the UGTs responsible for glucuronidation of CLZ and its active metabolite, a comprehensive screening of CLZ and dmCLZ glucuronidation activity by homogenates from HEK 293 cells over-expressing wild-type UGTs 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B11, 2B15 and 2B17 was performed. Four UGTs exhibited detectable levels of activity against CLZ by UPLC-MS/MS using multiple reaction monitoring (MRM) - the hepatic UGTs 1A1, 1A3, 1A4 and 2B10 (Figure 3 panels A, B, C, and D, respectively). None of the other UGTs screened in our assays (UGTs 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B11, 2B15 or 2B17) exhibited any detectable glucuronidation activity against CLZ using up to 1 mg of UGT-over-expressing cell homogenate. The UPLC retention times for potential CLZ-glucuronide peaks for UGTs 1A1, 1A3, 1A4 and 2B10 were identical to that observed for HLM and showed similar sensitivity to treatment with β-glucuronidase and 3 M HCl solution (data not shown). Similar to that observed for CLZ in incubations with HLM, the UGT1A1-, 1A3-, 1A4- and UGT2B10-generated peaks were identified as CLZ-5-N-glucuronide at 3.3 min and CLZ-N+-glucuronide at 3.75 min as determined by UPLC/MS/MS analysis (data not shown). Only the 3.3 and 3.75 min peaks generated by UGT1A4 and the 3.75 min peak generated by UGT1A1 were detectable by UPLC-UV (Figure 2, panels C, D); the activity exhibited by UGTs 1A3 and 2B10 as well as UGT1A1-induced CLZ-5-N-glucuronide formation were only detected by the more sensitive MRM by MS analysis. The ratio of CLZ-5-N-glucuronide to CLZ-N+-glucuronide in UGT1A4-over-expressing cell homogenates as determined by UV absorbance was 18.7 (using 160 μM CLZ, which was within the linear range of UGT1A4 glucuronidation of CLZ; results not shown); using MRM analysis, this ratio was 0.3 for homogenates from UGT1A1-over-expressing cells. Addition of the UGT1A4-specific inhibitor hecogenin [39] inhibited CLZ-5-N-glucuronide and CLZ-N+-glucuronide formation by 77% and 82%, respectively, in assays with UGT1A4-over-expressing cell homogenates, while the UGT1A1-specific inhibitor, niflumic acid [40] , decreased CLZ-N+-glucuronide formation by 84% in assays with homogenates from UGT1A1-over-expressing cells.
Figure 3. MRM analysis of UGT-over-expressing cell lines.
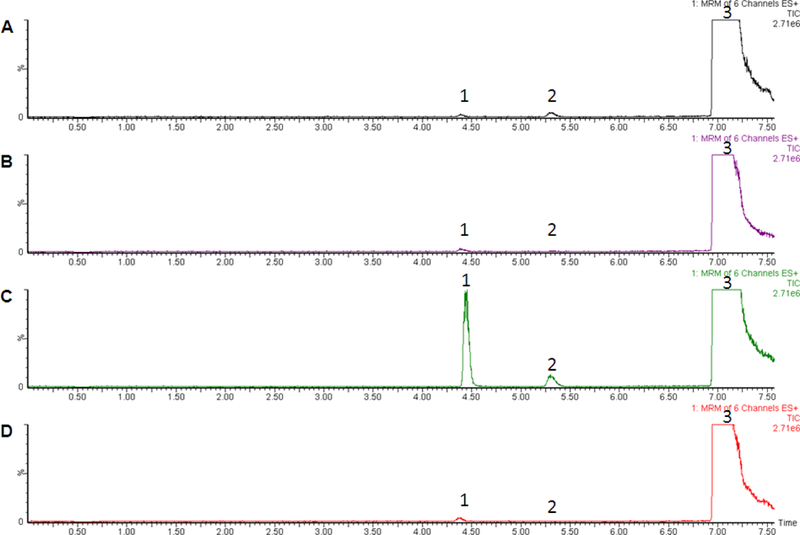
Glucuronidation assays were performed using 20 μg HLM protein or 1 mg UGT-over-expressing cell lines and incubated at 37°C for 2 h with 4 mM UDP-glucuronic acid prior to analysis by MS for MRM analysis as described in the Materials and Methods. Panel A, UGT1A1 + CLZ; panel B, UGT1A3 + CLZ; panel C, UGT1A4 + CLZ; panel D, UGT2B10 + CLZ. Peak 1, CLZ-5-N-glucuronide; peak 2, CLZ-N+-glucuronide; peak 3, CLZ. Y-axis = signal intensity; x-axis = retention time (min).
A single UGT exhibited detectable levels of activity against dmCLZ - the hepatic UGT1A4 (Figure 2, panel I). None of the other UGTs screened in our assays (UGTs 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B11, 2B15 or 2B17) exhibited any glucuronidation activity against dmCLZ using up to 1 mg of UGT-over-expressing cell homogenate either by UV-detection or by MRM analysis. The UPLC retention times for peaks resulting from dmCLZ incubation with UGT1A4 over-expressing cell line homogenate was identical to that observed for HLM and showed similar sensitivity to treatment with 3 M HCl; sensitivity to β-glucuronidase was not observed (data not shown). Similar to that observed for incubations of dmCLZ with HLM, the UGT1A4-generated peak at 3.0 min retention time was identified as dmCLZ-5-N-glucuronide as determined by UPLC/MS analysis (data not shown). Addition of the UGT1A4-specific inhibitor hecogenin inhibited UGT1A4-mediated dmCLZ-5-N-glucuronide formation by 81%.
Representative kinetic plots of CLZ and dmCLZ glucuronidation rate versus substrate concentration are shown in Figure 4 for wild-type UGTs 1A1 (panel A) and 1A4 (UGT1A424Pro/48Leu; panels B-D), as well as for HLM exhibiting the wild-type UGT1A1 (*1/*1)/UGT1A4 (*1/*1) genotype (panels E-G). While UGT1A4 was the only UGT to catalyze formation of the CLZ-5-N-glucuronide and the dmCLZ-5-N-glucuronide as detected by UV spectra, UGT1A4 exhibited a 2.2-fold larger KM for the formation of the dmCLZ-5-N-glucuronide compared to the formation of CLZ-5-N-glucuronide (Table 1). Both UGTs 1A1 and 1A4 catalyzed the formation of CLZ-N+-glucuronide, with UGT1A1 exhibiting a significant (p<0.0001) 2.1-fold higher Vmax/KM, manifested primarily by a significantly (p<0.0001) lower KM, than UGT1A4 (Table 1). UGT1A3- and UGT2B10-generated CLZ glucuronides as well as the UGT1A1-induced CLZ-5-N-glucuronide were not within the limits of the UV detection system, therefore kinetic analysis was not performed for these metabolites.
Figure 4. Kinetic curves for HLM and UGT1A1 and UGT1A424Pro/48Leu-over-expressing cell homogenates against CLZ and dmCLZ.
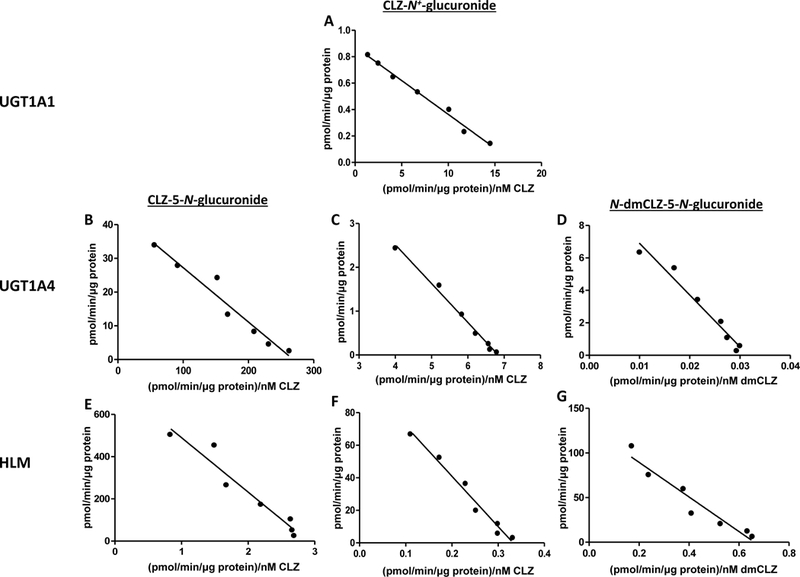
Representative Eadie-Hofstee plots are shown for kinetic analysis of HLM and UGT1A1- and UGT1A424Pro/48Leu-over-expressing cell homogenates, performed as described in the Materials and Methods using CLZ concentrations of 10, 20, 40, 80, 160, 306, and 612 μM and dmCLZ concentrations of 10, 20, 40, 80, 160, 320, and 640 μM. HLM were from subjects exhibiting the UGT1A4 (*1/*1)/UGT1A1 (*1/*1) genotype. CLZ, clozapine; dmCLZ, desmethylclozapine.
Table 1.
Kinetic analysis of UGTs active against CLZ in vitro.
| CLZ-5-N-glucuronide | CLZ-N+-glucuronide | dmCLZ-5-N-glucuronide | |||||||
|---|---|---|---|---|---|---|---|---|---|
| UGT variant | Vmax (pmol·min−1·μg−1)* |
KM (μM) |
Vmax/KM (nl·min−1·μg−1)* |
Vmax (pmol·min−1·μg−1)* |
KM (μM) |
Vmax/KM (nl·min−1·μg−1)* |
Vmax (pmol·min−1·μg−1)* |
KM (μM) |
Vmax/KM (nl·min−1·μg−1)* |
| UGT1A1 | nd | nd | nd | 0.86 ± 0.02a,b | 53 ± 2a,b | 17 ± 0.6a | no activity detected | ||
| UGT1A424Pro/48Leu | 42 ± 3 | 173 ± 19 | 246 ± 31 | 6.3 ± 0.6 | 768 ± 62 | 8.2 ± 0.8 | 10 ± 0.7 | 379 ± 46 | 28 ± 2.8 |
| UGT1A424Thr/48Leu | 33 ± 2b,c | 167 ± 19d | 196 ± 14b,c | 3.6 ± 0.1a,b | 459 ± 23e | 7.8 ± 0.3b | 6.4 ± 0.9b,e | 440 ± 45b | 15 ± 1.5b,e |
| UGT1A424Pro/48Val | 93 ± 9a | 73 ± 8e | 1284 ± 77a | 7.6 ± 0.1c | 481 ± 9e | 16 ± 0.2a | 17 ± 1.3d | 173 ± 22e | 96 ± 8.0a |
Michaelis-Menten kinetic values represent per μg UGT protein. nd = not detected, below the limits of UPLC detection.
p<0.0001,
p<0.0001,
p<0.05,
p<0.002 versus corresponding value for UGT1A424Pro/48Val-over-expressing cell homogenate, calculated using the Students t-test (two-sided).
p<0.001 versus corresponding value for UGT1A424Pro/48Leu-over-expressing cell homogenate, calculated using the Students t-test (two-sided).
Analysis of CLZ and dmCLZ glucuronidation by hepatic UGT variants.
UGT1A4 is a hepatic enzyme that exhibits prevalent functional missense polymorphisms [34, 45, 46]. The UGT1A424Thr/48Leu and UGT1A424Pro/48Val variant HEK 293 over-expressing cell lines described previously [27, 33, 36, 46] were utilized to examine the effect of these functional variants on CLZ and dmCLZ glucuronidation in vitro. After normalization of UGT1A4 wild-type and UGT1A4 variant protein levels in UGT1A4-over-expressing cell homogenates by western blot analysis, the Vmax/KM for CLZ-5-N-glucuronide formation was 20% (p=0.04) lower for the UGT1A424Thr/48Leu variant mediated primarily by a 21% (p=0.002) decrease in Vmax compared to wild-type UGT1A424Pro/48Leu. The UGT1A424Pro/48Val variant exhibited a 5.2- and 6.6-fold (p<0.0001 for both) higher Vmax/KM for CLZ-5-N-glucuronide formation compared to the wild-type UGT1A424Pro/48Leu and UGT1A424Thr/48Leu variant, respectively (Table 1). These increases were mediated by both a 2.2- and 2.8-fold (p<0.0001 for both) increase in Vmax and a 58% (p<0.0001) and 56% (p=0.0002) decrease in KM for the UGT1A424Pro/48Val variant as compared to the wild-type UGT1A424Pro/48Leu and UGT1A424Thr/48Leu variant, respectively. The Vmax/KM of the UGT1A424Pro/48Val variant was 2.0- and 2.1-fold higher than that observed for wild-type UGT1A424Pro/48Leu and the UGT1A424Thr/48Leu variant (p<0.0001 for both), respectively, for formation of the CLZ-N+-glucuronide.
Compared to cell homogenates over-expressing wild-type UGT1A424Pro/48Leu, homogenates over-expressing the UGT1A424Thr/48Leu variant exhibited a 46% (p<0.0001) lower Vmax/KM for the formation of dmCLZ-5-N-glucuronide mediated primarily by a 36% (p=0.0002) decrease in Vmax, while homogenates over-expressing the UGT1A424Pro/48Val variant exhibited a 3.4-fold (p<0.0001) higher Vmax/KM, mediated by a 1.7-fold (p<0.0001) increase in Vmax and a 54% (p<0.0001) decrease in KM. Furthermore, the UGT1A424Pro/48Val variant exhibited a 6.4-fold (p<0.0001) increase in Vmax/KM compared to the UGT1A424Thr/48Leu variant, which was due to a 2.7-fold (p<0.0001) increase in Vmax and a 61% (p<0.0001) decrease in KM exhibited by the UGT1A424Pro/48Val variant. At a concentration of 160 μM CLZ (within the linear range of UGT1A4 glucuronidation of CLZ), the ratio of CLZ-5-N-glucuronide to CLZ-N+-glucuronide in UGT1A424Thr/48Leu- and UGT1A424Pro/48Val-over-expressing cell homogenates was 17.4 and 41.5 (results not shown).
Analysis of CLZ and dmCLZ glucuronidation by HLM stratified by UGT1A1 or UGT1A4 genotypes.
The prevalence of the UGT1A1*28 (A(TA)7TAA), UGT1A4*2 (P24T), and UGT1A4*3 (L48V) allelic variants is approximately 30%, 8%, and 9% in Caucasians, respectively [34, 37, 45–48]. To explore a possible in vivo relationship between CLZ and dmCLZ glucuronidation and the UGT1A1 and UGT1A4 polymorphisms, a series of 113 HLM were examined for their glucuronidation activity against CLZ and dmCLZ. The rate of CLZ-glucuronide and dmCLZ-glucuronide formation was determined by UPLC using 160 μM CLZ or 320 μM dmCLZ, which were concentrations found to be within 10–20% of the KM for the formation of the major glucuronide product for CLZ and dmCLZ, respectively, in HLM.
For HLM with the wild-type UGT1A1 (*1/*1) genotype (n=44), no significant difference in formation of any CLZ or dmCLZ glucuronide was observed when comparing those homozygous for the UGT1A4*2 allele (n=2) to HLM homozygous for the UGT1A4*1 allele (n=33), even after excluding HLM with one or more UGT1A4*3 alleles (results not shown). Therefore, for further analysis of the UGT1A1*28 and UGT1A4*3 alleles, HLM were not sub-stratified based on the UGT1A4*2 allele.
For HLM with the homozygous wild-type UGT1A4 (*1/*1) genotype, CLZ-N+-glucuronide product formation decreased significantly (p=0.028, ANOVA) with increasing numbers of the UGT1A1*28 allele (Figure 5, panel A). HLM with the UGT1A1 (*28/*28) genotype (n=11) exhibited a significant 37% (p<0.05) decrease in formation of the CLZ-N+-glucuronide as compared with HLM with the UGT1A1 (*1/*1) genotype (n=37). While a 25% decrease in CLZ-5-N-glucuronide formation was also observed for HLM with the UGT1A1 (*28/*28) genotype versus HLM with the UGT1A1 (*1/*1) genotype; this difference was not significant (p=0.21, ANOVA; Figure 5, panel B). Regression analysis showed that the UGT1A1*28 allele was a significant predictor for formation of the CLZ-N+-glucuronide (p=0.045). For each UGT1A1*28 allele, product formation decreased by 3.1 pmol.min−1.mg protein−1 for the CLZ-N+-glucuronide, representing a 16% decrease in glucuronide formation with each UGT1A1*28 allele compared to the mean CLZ-N+-glucuronide-product formed by wild-type HLM. Although not significant (p=0.14), the UGT1A1*28 allele was predicted to decrease CLZ-5-N-glucuronide formation by 21 pmol.min−1.mg protein−1, a decrease of 10% per UGT1A1*28 allele compared to the mean CLZ-5-N-glucuronide-product formed by wild-type alleles. In five UGT1A1- and UGT1A4-wild-type HLM, the addition of niflumic acid inhibited CLZ-5-N-glucuronide and CLZ-N+-glucuronide formation by 9 ± 2% and 55 ± 5%, respectively.
Figure 5. HLM activity stratified by UGT genotypes.
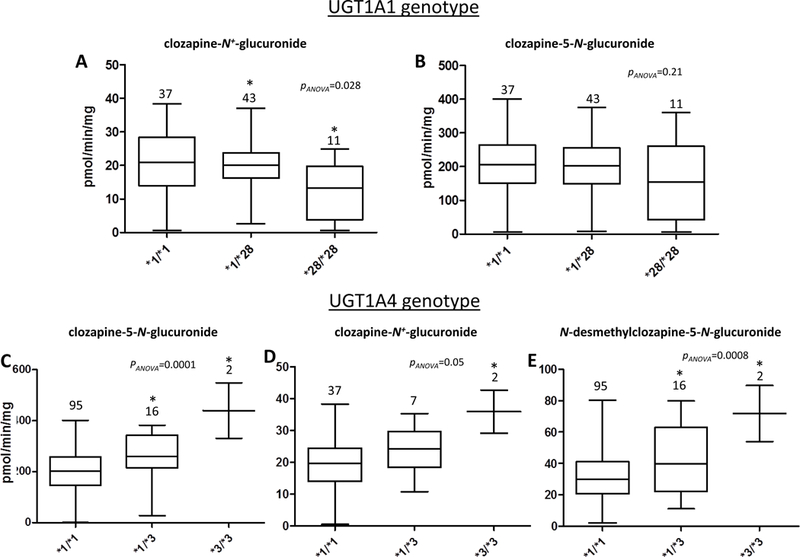
Shown are box and whisker plots of the levels of CLZ-5-N-glucuronide, CLZ-N+-glucuronide, and dmCLZ-5-N-glucuronide formation versus UGT1A1 or UGT1A4 genotypes in HLM. Glucuronidation activity assays were performed using 160 μM CLZ or 320 μM dmCLZ and 12.5 μg of HLM protein, and CLZ or dmCLZ glucuronides were detected and separated by UPLC as described in the Materials and Methods. Using genomic DNA from the same liver specimens for which HLM were prepared, UGT1A1 and UGT1A4 genotypes were determined by DNA sequencing. Panels A-B, CLZ glucuronidation in HLM stratified by UGT1A1 genotypes; panels C-E, CLZ and dmCLZ glucuronidation in HLM stratified by UGT1A4 genotypes. Panel A and D, CLZ-N+-glucuronide formation; panels B and C, CLZ-5-N-glucuronide formation; panel E, dmCLZ-5-N-glucuronide formation. Analysis of CLZ glucuronidation in HLM stratified by UGT1A1 genotypes was performed only for those specimens also exhibiting the wild-type UGT1A4 (*1/*1) genotype (n=94). Analysis of CLZ and dmCLZ glucuronidation in HLM stratified by UGT1A4 genotypes was performed only for those specimens also exhibiting the wild-type UGT1A1 (*1/*1) genotype (n=49) for CLZ-N+-glucuronide formation. All HLM (n=113) were used for analysis of CLZ-5-N-glucuronide and dmCLZ-5-N-glucuronide formation. Whiskers = min and max; horizontal line = median. Statistical analysis was performed using ANOVA, with *=p<0.05 using Tukey’s post-hoc correction for comparison of HLM with variant genotypes with the corresponding wild-type (*1/*1) HLM group.
The CLZ-5-N-glucuronide was detectable only by MRM analysis in UGT1A1 over-expressing cell lines, and the UGT1A1*28 allele was not a significant predictor of CLZ-5-N-glucuronide formation by HLM. Therefore, the effect of the UGT1A4*3 allele on HLM CLZ-5-N-glucuronide formation was not sub-stratified by UGT1A1 genotype. CLZ-5-N-glucuronide product formation increased significantly with each UGT1A4*3 allele (p=0.0001, ANOVA) with a 2.2-fold (p<0.05 Figure 5, panel C) increase in CLZ-5-N-glucuronide formation in HLM with the UGT1A4 (*3/*3) genotype compared to HLM with the UGT1A4 (*1/*1) genotype. While the UGT1A4*3 allele was only marginally significant (p=0.05, ANOVA) with CLZ-N+-glucuronide formation, there was a significant 1.8-fold (p<0.05; panel D) increase in CLZ-N+-glucuronide formation in HLM with the UGT1A4 (*3/*3) genotype as compared to HLM with the UGT1A4 (*1/*1) genotype.
As UGT1A1 does not catalyze the formation of dmCLZ-5-N-glucuronide, the effect of the UGT1A4*3 allele on HLM dmCLZ-5-N-glucuronide formation was not sub-stratified by UGT1A1 genotype. dmCLZ-5-N-glucuronide formation increased significantly (p=0.0008, ANOVA) in HLM with each additional UGT1A4*3 allele. HLM with the UGT1A4 (*1/*3) and (*3/*3) genotypes exhibited a 1.3-fold (p<0.05; panel E) and 2.3-fold (p<0.05; panel E) increase in dmCLZ-5-N-glucuronide formation, respectively, as compared to HLM with the UGT1A4 (*1/*1) genotype.
Regression analysis showed that the UGT1A4*3 allele is a significant predictor for formation of the CLZ-5-N-glucuronide (p<0.0001), the CLZ-N+-glucuronide (p=0.02), and the dmCLZ-5-N-glucuronide (p=0.001). For each UGT1A4*3 allele, product formation increased by 82, 5.9, and 13.4 pmol.min−1.mg protein−1 for the CLZ-5-N-glucuronide, the CLZ-N+-glucuronide, and the dmCLZ-5-N-glucuronide, respectively, representing a 41, 29, and 43% increase in glucuronidation, respectively, for each UGT1A4*3 allele compared to the mean glucuronide-product formation by wild-type HLM. There was insufficient power to examine the effect of combined UGT1A1 and UGT1A4 genotypes on CLZ or dmCLZ glucuronidation in this series of HLM specimens. Hecogenin inhibited CLZ-5-N-glucuronide formation by 89 ± 3% and inhibited CLZ-N+-glucuronide formation by 46 ± 5%, respectively, in HLM (results not shown).
The average ratio of CLZ-5-N-glucuronide to CLZ-N+-glucuronide in all screened HLM (n=113) was 9.9 ± 2.9, with a range of 2.2 – 23.1. The CLZ-5-N-glucuronide to CLZ-N+-glucuronide ratio of 10.1 ± 3.6 for HLM wild-type for UGT1A1 and UGT1A4 (n=38) was not significantly higher (p=0.6) than that observed for HLM wild-type for UGT1A4 and homozygous for the UGT1A1*28 allele (9.3 ± 4.2; n=11), nor was it significantly lower (p=0.14) than that observed for HLM wild-type for UGT1A1 and homozygous for the UGT1A4*3 allele (12.1 ± 1.1; n=2). There was a non-significant (p=0.64) 2.4% decrease in the ratio of CLZ-5-N-glucuronide to CLZ-N+-glucuronide with each addition of the UGT1A1*28 allele, and there was a nearly significant (p=0.07) increase of 16% in the ratio of CLZ-5-N-glucuronide to CLZ-N+-glucuronide with each addition of the UGT1A4*3 allele.
Analysis of CLZ and dmCLZ glucuronidation kinetics by HLM stratified by UGT1A1 or UGT1A4 genotypes.
Results of kinetic analysis of HLM with varying UGT1A1 and UGT1A4 genotypes are shown in Table 2. For UGT1A4 (*1/*1) HLM stratified by UGT1A1 genotype, a non-significant trend (p=0.054) of decreasing Vmax was observed for CLZ-5-N-glucuronide formation with each additional UGT1A1*28 allele. There was also a significant (p<0.05) 39% decrease in Vmax/KM for formation of the CLZ-N+-glucuronide in UGT1A4 (*1/*1) HLM with the UGT1A1 (*28/*28) genotype as compared to UGT1A4 (*1/*1) HLM with the UGT1A1 (*1/*1) genotype. This was mediated primarily by a decrease in Vmax, with UGT1A4 (*1/*1) HLM with the UGT1A1 (*1/*28) and UGT1A1 (*28/*28) genotypes exhibiting a significant 20% (p<0.05) and 37% (p<0.05) lower Vmax, respectively, for CLZ-N+-glucuronide formation.
Table 2.
Kinetic analysis of CLZ and dmCLZ glucuronidation in HLMs of varying UGT1A1 and UGT1A4 genotypes.
| CLZ-5-N-glucuronide | CLZ-N+-glucuronide | dmCLZ-glucuronide formation | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HLM genotype | Vmax (pmol·min−1·mg−1) |
KM (μM) |
Vmax/KM (μl·min−1·mg−1) |
Vmax (pmol·min−1·mg−1) |
KM (μM) |
Vmax/KM (μl·min−1·mg−1) |
Vmax (pmol·min−1·mg−1) |
KM (μM) |
Vmax/KM (μl·min−1·mg−1) |
| UGT1A1(*1/*1)/ UGT1A4(*1/*1)a | 758 ± 22 | 238 ± 5 | 3.2 ± 0.2 | 112 ± 15 | 311 ± 40 | 0.36 ± 0.0 | 108 ± 6 | 357 ± 21 | 0.30 ± 0.01 |
| UGT1A1(*1/*28)/ UGT1A4(*1/*1)a | 698 ± 43c | 217 ± 44c | 3.3 ± 0.7g | 88 ± 2e,h | 311 ± 19c | 0.29 ± 0.02g,e | not analyzed | ||
| UGT1A1(*28/*28)/UGT1A4(*1/*1)a | 684 ± 61d | 234 ± 26c | 2.9 ± 0.1g | 69 ± 1h,e | 325 ± 27i | 0.22 ± 0.02i | not analyzed | ||
| UGT1A4(*1/*3)/ UGT1A1(*1/*1)b |
1088 ± 93e | 187 ± 22e | 5.9 ± 0.3e | 156 ± 22e | 234 ± 14e | 0.67 ± 0.12e | 120 ± 11e | 214 ± 23e | 0.57 ± 0.11e |
| UGT1A4(*3/*3)/ UGT1A1(*1/*1)b |
1573 ± 202f | 121 ± 22f | 13 ± 0.7f | 161 ± 36e | 174 ± 20e | 1.1 ± 0.083f | 175 ± 13e | 128 ± 4f | 1.4 ± 0.11f |
Three HLM with the UGT1A1 (*1/*1), the UGT1A1 (*1/*28), and the UGT1A4 (*3/*3) genotype were examined in this analysis. Only HLM with the UGT1A4 (*1/*1) genotype were used to compare varying UGT1A1 genotypes. HLM of each genotype were chosen at random. UGT1A1 (*1/*28)/UGT1A4 (*1/*1) and UGT1A1 (*28/*28)/UGT1A4 (*1/*1) HLM were not analyzed for dmCLZ-glucuronide formation since UGT1A1 exhibits no activity towards dmCLZ.
Three HLM with the UGT1A4 (*1/*1) genotype, three HLM with the UGT1A4 (*1/*3) genotype, and two HLM with the UGT1A4 (*3/*3) genotype were examined in this analysis [only two HLM exhibited the UGT1A1 (*3/*3) genotype in this series of specimens]. Only HLM with the UGT1A1 (*1/*1) genotype were used to compare varying UGT1A4 genotypes. HLM of each genotype were chosen at random.
p<0.005,
p<0.05,
Significantly (p<0.05) different than UGT1A1 (*1/*1)/UGT1A4 (*1/*1) after Tukey’s post-hoc correction.
Significantly (p<0.05) different than UGT1A1 (*1/*1)/UGT1A4 (*1/*1) and UGT1A1 (*1/*1)/UGT1A4 (*1/*3) after Tukey’s post-hoc correction.
p<0.001, and
p<0.01 versus the corresponding value for HLM with the UGT1A1 (*1/*1)/UGT1A4 (*3/*3) genotype as determined using the Students t-test (2-sided).
Significantly (p<0.05) different than UGT1A1 (*1/*1)/UGT1A4 (*1/*1) and UGT1A1 (*1/*1)/UGT1A4 (*1/*28) after Tukey’s post-hoc correction.
UGT1A4 genotype affected the kinetics of HLM-mediated glucuronidation of CLZ-and dmCLZ. An increased Vmax (p=0.001) and Vmax/KM (p<0.0001) and a decreased KM (p=0.02) was observed for the formation of the CLZ-5-N-glucuronide in UGT1A1 (*1/*1) HLM with increasing numbers of the UGT1A4*3 allele. There was a 1.8- and 4.1-fold (p<0.05 for both) higher Vmax/KM for formation of the CLZ-5-N-glucuronide in UGT1A1 (*1/*1) HLM with the UGT1A4 (*1/*3) and UGT1A4 (*3/*3) genotypes, respectively, as compared to UGT1A1 (*1/*1) HLM with the UGT1A4 (*1/*1) genotype, an effect that was manifested primarily by a 1.4- and 2.1-fold (p<0.05 for both) increase in Vmax, but also by a 21% and 49% (p<0.05 for both) decrease in KM. There was a significant increase in Vmax (p=0.04), decrease in KM (p=0.008), and increase in Vmax/KM (p=0.001) for formation of the CLZ-N+-glucuronide in UGT1A1 (*1/*1) HLM with increasing numbers of the UGT1A4*3 allele. UGT1A1 (*1/*1) HLM with the UGT1A4 (*1/*3) and UGT1A4 (*3/*3) genotypes exhibited a 1.9- and 3.1-fold (p<0.05 for both) increase in Vmax/KM for formation of the CLZ-N+-glucuronide compared to UGT1A1 (*1/*1) HLM with the UGT1A4 (*1/*1) genotype. This was due to a 1.4-fold (p<0.05 for both) increase in Vmax for both UGT1A4 (*1/*3) and UGT1A4 (*3/*3) HLM and a 25% and 44% (p<0.05 for both) decrease in KM for UGT1A4 (*1/*3) and UGT1A4 (*3/*3) HLM, respectively.
For dmCLZ-5-N-glucuronide formation, UGT1A1 (*1/*1) HLM with the UGT1A4 (*1/*3) and UGT1A4 (*3/*3) genotypes exhibited a 1.9- and 4.7-fold (p<0.05 for both) increase in Vmax/KM, respectively, as compared to UGT1A1 (*1/*1) HLM with the UGT1A4 (*1/*1) genotype. This was mediated primarily by a 40% (p<0.05) decrease in KM for HLM with the UGT1A4 (*1/*3) genotype and a 1.6-fold (p<0.05) increase in Vmax and a 64% (p<0.05) decrease in KM for HLM with the UGT1A4 (*3/*3) genotype. There was a significant increase in Vmax (p=0.02), decrease in KM (p<0.0001), and increase in Vmax/KM (p<0.0001) for formation of the dmCLZ-5-N-glucuronide in UGT1A1 (*1/*1) HLM with increasing numbers of the UGT1A4*3 allele as compared to UGT1A1 (*1/*1) HLM with the UGT1A4 (*1/*1) genotype.
Compared to HLM with the UGT1A1 (*1/*1)/UGT1A4 (*3/*3) genotype, HLM with the UGT1A1 (*28/*28)/UGT1A4 (*1/*1) genotype exhibited a 78% and 80% (p<0.0001 and p=0.0003, respectively) decrease in Vmax/KM for the formation of the CLZ-5-N-glucuronide and CLZ-N+-glucuronide, respectively. These differences were mediated primarily by significant differences in Vmax, which were 57% (p=0.005) and 63% (p=0.009) lower for HLM with the UGT1A4 (*28/*28)/UGT1A4 (*1/*1) genotype as compared to HLM with the UGT1A4 (*1/*1)/UGT1A4 (*3/*3) genotype.
Discussion
In agreement with in vivo studies, two primary CLZ-glucuronides and one primary dmCLZ-glucuronide were observed in these in vitro studies using HLM - the CLZ-5-N-glucuronide, CLZ-N+-glucuronide, and dmCLZ-5-N-glucuronide [20, 21, 25] . Previous studies reported that both UGTs 1A3 and 1A4 exhibited glucuronidation activity against CLZ, with the activity of UGT1A3 being 25-fold lower than that of UGT1A4 [42, 43] . However, no other UGTs were reportedly examined in these studies [42, 43] . In the present study, all of the known human UGT1A and UGT2B enzymes except UGTs 1A5 and 2B28 were screened for activity against CLZ and dmCLZ using UGT-over-expressing HEK 293 cell homogenates. In addition to UGTs 1A3 and 1A4, UGTs 1A1 and 2B10 also exhibited glucuronidation activity against CLZ in this study, with both UGTs 1A3 and 2B10 exhibiting activity that was barely above the limits of detection (>0.01 μmol) in our study. Of the UGTs screened in the present study, UGT1A4 was the sole UGT that exhibited glucuronidation activity against dmCLZ. UGT1A4 was less efficient at glucuronidating dmCLZ than CLZ as reflected by a 10-fold lower Vmax/KM (comparing dmCLZ-5-N-glucuronide formation with CLZ-5-N-glucuronide formation), which may have implications for the metabolism and clearance of this active metabolite compared to CLZ metabolism and clearance in vivo. The Vmax and KM of the reaction mediated by UGT1A4-over-expressing cell lines against CLZ were slightly higher than that reported previously [43]. This is likely due to the use of an optimum enzyme pH (8.5) in previous studies rather than the physiological pH (7.4) used in the present study.
In vitro kinetic studies using individual UGT-over-expressing cell lines suggest that UGT1A4 is more efficient at catalyzing the formation of CLZ-5-N-glucuronide than the other active UGTs as reflected by a larger Vmax and lower KM. In addition, the UGT1A4 inhibitor, hecogenin, resulted in large decreases in CLZ-5-N-glucuronide formation in HLM, suggesting an important role for UGT1A4 in the formation of this CLZ metabolite. However, the ratio of CLZ-5-N-glucuronide to CLZ-N+-glucuronide in all HLM was below that measured for cell homogenates over-expressing UGT1A4, suggesting that UGTs other than UGT1A4 may be important in making the two products in HLM. For example, UGT1A1 is likely accounting for ~10% of total hepatic CLZ-5-N-glucuronide formation since the UGT1A1-specific inhibitor, niflumic acid, decreased CLZ-5-N-glucuronide formation by 9% in HLM. While UGT1A1 exhibits low CLZ-5-N-glucuronide formation activity in vitro, these data are consistent with the fact that UGT1A1 is expressed at 1.5–2.0-fold higher levels in human liver than UGT1A4 [49, 50].
For CLZ-N+-glucuronide formation, the Vmax/KM was 2.1-fold higher for UGT1A1 versus UGT1A4, with a KM for UGT1A4 that was 14.5-fold higher than UGT1A1 in vitro, indicating that UGT1A1 has a greater affinity for making this metabolite. The KM for CLZ-N+-glucuronide formation by UGT1A1 (*1/*1)/UGT1A4 (*1/*1) HLM (311 μM) was between the KM observed for UGT1A1- (53 μM) and UGT1A4- (768 μM) over-expressing cell homogenates. Furthermore, CLZ-N+-glucuronide formation in HLM was decreased by ~46–55% when using either the UGT1A1-specific inhibitor, niflumic acid, or the UGT1A4-specific inhibitor, hecogenin, suggesting that both UGTs 1A1 and 1A4 contribute fairly equally to CLZ-N+-glucuronide formation. While not a major metabolite of CLZ, due to the instability of the CLZ-5-N-glucuronide in acidic conditions such as urine [25], the CLZ-N+-glucuronide is an important marker for CLZ metabolism and clearance.
The present study is the first to examine the functional importance of missense SNPs in relevant UGTs on CLZ and dmCLZ in vitro at physiological conditions. The UGT1A1 A(TA)7TAA promoter polymorphism has been described to decrease the rate of transcription initiation of the UGT1A1 gene and is associated with decreased substrate glucuronidation and increased drug-induced toxicity [37, 51]. Furthermore, individuals homozygous for the UGT1A1*28 allele reportedly have a 30% reduction in UGT1A1 expression [51]. In the present study, a significant association was observed between the rate of CLZ-N+-glucuronide formation and HLM genotype, and HLM with the UGT1A1 (*28/*28)/UGT1A4 (*1/*1) genotype exhibited significant decreases in Vmax and Vmax/KM for CLZ-N+-glucuronide formation as compared to HLM with the UGT1A1 (*1/*1)/UGT1A4 (*1/*1) genotype. In addition, while only a small non-significant association was observed between rate of CLZ-5-N-glucuronide formation and UGT1A1 genotype in HLM, the KM for CLZ-5-N-glucuronide formation by HLM with the UGT1A1 (*1/*1)/UGT1A4 (*1/*1) genotype was 1.4- (p=0.007) fold higher than that observed in cell lines over-expressing UGT1A4 in vitro (238 versus 173 μM). These data further support a role for UGT1A1 as an important enzyme in CLZ metabolism and suggest that the UGT1A1*28 allele is a modifier of CLZ metabolism in vivo.
While cell homogenates over-expressing the UGT1A424Thr/48Leu variant had significantly lower efficiency in forming the CLZ-5-N-glucuronide compared with the wild-type UGT1A424Pro/48Leu in over-expressing cell line homogenate studies, no effect on the formation of other CLZ glucuronides were observed in vitro and no association was observed between the formation of the two CLZ glucuronides and the UGT1A4*2 allele in HLM. These data suggest that the UGT1A4 Pro24Thr polymorphism may not exhibit a significant effect on overall CLZ metabolism in vivo.
The data presented in this study suggest that the UGT1A4 Leu48Val polymorphism is a prevalent UGT1A4 missense SNP that may be linked to altered hepatic CLZ glucuronidation activity. The UGT1A424Pro/48Val variant increased the formation of CLZ-5-N-glucuronide, CLZ-N+-glucuronide, and dmCLZ-5-N-glucuronide products as measured by rate of formation as well as by kinetic parameters in UGT-over-expressing cell lines and in HLM with increasing numbers of the UGT1A4*3 allele as compared to UGT1A424Pro/48Leu-over-expressing cell lines and wild-type HLM. The fact that the CLZ-5-N-glucuronide to CLZ-N+-glucuronide ratio was significantly higher for cell homogenates over-expressing UGT1A424Pro/48Val versus UGT1A424Pro/48Leu suggests that this polymorphism preferentially affects CLZ-5-N-glucuronide formation. These data are consistent with other studies which reported increased detoxifying activity with the UGT1A424Pro/48Val-encoding *3 allele [22, 34, 46, 52]. The kinetics of the UGT1A424Pro/48Val variant were previously studied in vitro for CLZ-5-N-glucuronide and CLZ-N+-glucuronide combined [53]. In the present study, the reported enzyme efficiency for overall CLZ-glucuronidation (CLZ-5-N-glucuronide and CLZ-N+-glucuronide combined) is in the same direction of change but greater than that reported previously (data not shown). This may be due to the different experimental parameters used previously compared to the present study.
The UGT1A424Pro/48Val variant also exhibited significantly higher levels of dmCLZ-5-N-glucuronide formation and was associated with a significantly increase both in the rate of dmCLZ-5-N-glucuronide formation as well as in kinetic measures of HLM-mediated glucuronidation with increasing numbers of the UGT1A4*3 allele. dmCLZ is the active metabolite of CLZ, with circulating levels between 30–60% of the CLZ dose in humans, and is thought to be responsible for some of the superior therapeutic efficacy compared to other SGA compounds. While dmCLZ is currently not FDA-approved for patient treatment, a number of publications have highlighted its potential as a therapy [16, 54–57]. However, dmCLZ is associated with the development of agranulocytosis, a life-threatening adverse effect occurring in ~1% of individuals that take CLZ. It is thought that a reactive intermediate of dmCLZ is toxic to leukocytes in a dose-dependent manner [58–60]. It is possible that decreased dmCLZ clearance could contribute to the development of agranulocytosis and that inactivation and clearance of dmCLZ by UGTs may lower the risk of an individual developing agranulocytosis.
The effect of the UGT1A3*3 and UGT1A1*28 alleles was most profound when comparing UGT1A1 (*28/*28)/UGT1A4 (*1/*1) HLM versus UGT1A1 (*1/*1)/UGT1A4 (*3/*3) HLM, with a 78–82% decrease in Vmax/KM observed for formation of the two CLZ glucuronides in UGT1A1 (*28/*28)/UGT1A4 (*1/*1) HLM. Therefore, UGT1A4 and UGT1A1 polymorphisms may contribute to the wide inter-individual variability that occurs in CLZ plasma levels [14, 16, 25] and may contribute to an individual’s likelihood of treatment efficacy or risk of developing adverse effects, most notably weight gain, metabolic dysfunction, and agranulocytosis. A limitation of the present study, however, was the fact that only two HLM homozygous for the UGT1A4*3 allele were examined. Further studies with additional HLMs exhibiting this genotype will be necessary to better establish the trends observed for UGT1A4 variants.
In conclusion, the data presented in this study suggests that the UGT1A1A(TA)7TAA and UGT1A4 Leu48Val polymorphisms significantly alter CLZ and/or dmCLZ glucuronidation in vitro and could potentially be important in determining inter-individual differences in CLZ and dmCLZ metabolism in vivo. Other factors such as drug-induced UGT-gene expression, cigarette smoking, age, ethnicity, and sex may also contribute to inter-individual differences in drug metabolism. Therapeutic drug monitoring (TDM) that can be used to ensure patient serum CLZ levels are within the therapeutic window and accounts for all causes of differences in CLZ and dmCLZ drug metabolism. However, there are significant costs associated with TDM and it cannot provide information regarding which therapies would be more efficacious and result in less adverse effects for a given patient before starting a therapy. The results presented in this study may provide new targets that could lead to new screening strategies to help determine dosing, patient response, or risk of adverse effects to CLZ before starting treatment of refractory schizophrenia.
Acknowledgements
We thank Diane McCloskey for careful review and editing of the manuscript and Samuel J. Ridout for help with statistical analysis. We also thank the Genomics Core Facility at the University Park campus of Penn State University for their excellent DNA sequencing services.
Funding for this study was provided in part by a grant from the American Diabetes Association grant # 7–08-CST-01
Footnotes
This material is original research, has not been previously published and has not been submitted for publication elsewhere while under consideration
Conflict of Interest Statement: None of the authors have conflict of interests to report
References (updated)
- 1.FDA. Electronic Orange Book. U.S. Department of Health & Human Services; 2009. [updated 6/10/2009; cited 2009 3/10/2009]; Available from: http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm. [Google Scholar]
- 2.Swartz MS, Perkins KO, Stroup TS, Davis SM, Capuano G, Rosenheck RA, et al. Effects of antipsychotic medications on psychosocial functioning in patients with chronic schizophrenia: Findings from the NIMH CATIE study. Am J Psychiatry. 2007;164:428–36. [DOI] [PubMed] [Google Scholar]
- 3.Swartz MS, Stroup TS, McEvoy JP, Davis SM, Rosenheck RA, Keefe RSE, et al. What CATIE Found: Results From the Schizophrenia Trial. Psychiatr Serv. 2008 May 1, 2008;59:500–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Psychiatric A. Practice guidelines for the treatment of patients with schizophrenia. 2004. [Google Scholar]
- 5.Gothelf DFB, Singer P, Kairi M, Phillip M, Zigel L et al. Weight gain associated with increased food intake and low habitual activity levels in male adolescent schizophrenic inpatients treated with olanzapine. Am J Psychiatry. 2002; 159:1055–7. [DOI] [PubMed] [Google Scholar]
- 6.Kinon BJ BB, Gilmore JA, Tollefson GD. Longterm olanzapine treatment: weight change and weight-related health factors in schizophrenia. J Clin Psychiatry. 2001;62:92–100. [PubMed] [Google Scholar]
- 7.Wirshing DA WW, Kysar L, Berisford MA, Goldstein D, Pashdag J et al. Novel antipsychotics: comparison of weight gain liabilities. J Clin Psychiatry. 1999;60:358–63. [PubMed] [Google Scholar]
- 8.Parsons B, Allison DB, Loebel A, Williams K, Giller E, Romano S, et al. Weight effects associated with antipsychotics: a comprehensive database analysis. Schizophr Res. 2009. May;110:103–10. [DOI] [PubMed] [Google Scholar]
- 9.Rosenheck RA, Davis S, Covell N, Essock S, Swartz M, Stroup S, et al. Does switching to a new antipsychotic improve outcomes? Data from the CATIE Trial. Schizophr Res. 2009. January;107:22–9. [DOI] [PubMed] [Google Scholar]
- 10.Komossa K, Rummel-Kluge C, Hunger H, Schmid F, Schwarz S, Duggan L, et al. Olanzapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. 2010;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteleone P, Martiadis V, Maj M. Management of schizophrenia with obesity, metabolic, and endocrinological disorders. Psychiatr Clin North Am. 2009. December;32:775–94. [DOI] [PubMed] [Google Scholar]
- 12.Laurent S, Boutouyrie P. Arterial stiffness and stroke in hypertension: therapeutic implications for stroke prevention. CNS Drugs. 2005;19:1–11. [DOI] [PubMed] [Google Scholar]
- 13.Asenjo Lobos C, Komossa K, Rummel-Kluge C, Hunger H, Schmid F, Schwarz S, et al. Clozapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. 2010;10:CD006633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rostami-Hodjegan A, Amin AM, Spencer EP, Lennard MS, Tucker GT, Flanagan RJ. Influence of Dose, Cigarette Smoking, Age, Sex, and Metabolic Activity on Plasma Clozapine Concentrations: A Predictive Model and Nomograms to Aid Clozapine Dose Adjustment and to Assess Compliance in Individual Patients. Journal of Clinical Psychopharmacology. 2004;24:70–8. [DOI] [PubMed] [Google Scholar]
- 15.Spina E, Avenoso A, Facciolà G, Scordo MG, Ancione M, Madia AG, et al. Relationship between plasma concentrations of clozapine and norclozapine and therapeutic response in patients with schizophrenia resistant to conventional neuroleptics. Psychopharmacology. 2000;148:83–9. [DOI] [PubMed] [Google Scholar]
- 16.Raedler TJ, Hinkelmann K, Wiedemann K. Variability of the In Vivo Metabolism of Clozapine. Clinical Neuropharmacology. 2008;31:347–52 10.1097/WNF.0b013e31815cba61. [DOI] [PubMed] [Google Scholar]
- 17.Hiemke C, Dragicevic A, Gründer G, Hätter S, Sachse J, Vernaleken I, et al. Therapeutic Monitoring of New Antipsychotic Drugs. Therapeutic Drug Monitoring. 2004;26:156–60. [DOI] [PubMed] [Google Scholar]
- 18.Evans WE, Relling MV. Pharmacogenomics: Translating Functional Genomics into Rational Therapeutics. Science. 1999 October 15, 1999;286:487–91. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Giambrone NE, Dluzen DF, Muscat JE, Berg A, Gallagher CJ, et al. Glucuronidation Genotypes and Nicotine Metabolic Phenotypes: Importance of Functional UGT2B10 and UGT2B17 Polymorphisms. Cancer Research. October 1, 2010;70:7543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dain JG, Nicoletti J, Ballard F. Biotransformation of Clozapine in Humans. Drug Metabolism and Disposition. 1997 May 1, 1997;25:603–9. [PubMed] [Google Scholar]
- 21.Schaber G, Wiatr G, Wachsmuth H, Dachtler M, Albert K, Gaertner I, et al. Isolation and Identification of Clozapine Metabolites in Patient Urine. Drug Metabolism and Disposition. 2001 June 1, 2001;29:923–31. [PubMed] [Google Scholar]
- 22.Erickson-Ridout KK, Zhu J, Lazarus P. Olanzapine metabolism and the significance of UGT1A448V and UGT2B1067Y variants. Pharmacogenetics and genomics. September;21:539–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersson ML, Bjorkhem-Bergman L, Lindh JD. Possible drug-drug interaction between quetiapine and lamotrigine--evidence from a Swedish TDM database. British journal of clinical pharmacology. July;72:153–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van de Wetering-Krebbers SF, Jacobs PL, Kemperman GJ, Spaans E, Peeters PA, Delbressine LP, et al. Metabolism and excretion of asenapine in healthy male subjects. Drug metabolism and disposition: the biological fate of chemicals. April;39:580–90. [DOI] [PubMed] [Google Scholar]
- 25.Breyer-Pfaff U, Wachsmuth H. Tertiary N-Glucuronides of Clozapine and Its Metabolite Desmethylclozapine in Patient Urine. Drug Metabolism and Disposition. 2001 October 1, 2001;29:1343–8. [PubMed] [Google Scholar]
- 26.Mori A, Maruo Y, Iwai M, Sato H, Takeuchi Y. UDP-GLUCURONOSYLTRANSFERASE 1A4 POLYMORPHISMS IN A JAPANESE POPULATION AND KINETICS OF CLOZAPINE GLUCURONIDATION. Drug Metabolism and Disposition. 2005 May 2005;33:672–5. [DOI] [PubMed] [Google Scholar]
- 27.Wiener D, Doerge DR, Fang JL, Upadhyaya P, Lazarus P. Characterization of N-glucuronidation of the lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human liver: importance of UDP-glucuronosyltransferase 1A4. Drug metabolism and disposition: the biological fate of chemicals. 2004. January;32:72–9. [DOI] [PubMed] [Google Scholar]
- 28.Sun D, Chen G, Dellinger RW, Sharma AK, Lazarus P. Characterization of 17-dihydroexemestane glucuronidation: potential role of the UGT2B17 deletion in exemestane pharmacogenetics. Pharmacogenetics and genomics. 2010. October;20:575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coughtrie MW, Burchell B, Bend JR. A general assay for UDPglucuronosyltransferase activity using polar amino-cyano stationary phase HPLC and UDP[U-14C]glucuronic acid. Anal Biochem. 1986. November;159:198–205. [DOI] [PubMed] [Google Scholar]
- 30.Chen G, Dellinger RW, Gallagher CJ, Sun D, Lazarus P. Identification of a prevalent functional missense polymorphism in the UGT2B10 gene and its association with UGT2B10 inactivation against tobacco-specific nitrosamines. Pharmacogenetics and genomics. 2008;18:181–91. [DOI] [PubMed] [Google Scholar]
- 31.Chen G, Dellinger RW, Sun D, Spratt TE, Lazarus P. Glucuronidation of tobacco-specific nitrosamines by UGT2B10. Drug metabolism and disposition: the biological fate of chemicals. 2008 May 1, 2008;36:824–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren Q, Murphy SE, Zheng Z, Lazarus P. O-Glucuronidation of the lung carcinogen 4-(methylnitrosamino)-1- (3-pyridyl)-1-butanol (NNAL) by human UDP-glucuronosyltransferases 2B7 and 1A9. Drug metabolism and disposition: the biological fate of chemicals. 2000. November;28:1352–60. [PubMed] [Google Scholar]
- 33.Dellinger RW, Fang JL, Chen G, Weinberg R, Lazarus P. Importance of UDP-glucuronosyltransferase 1A10 (UGT1A10) in the detoxification of polycyclic aromatic hydrocarbons: decreased glucuronidative activity of the UGT1A10 139Lys isoform. Drug Metabolism and Disposition. 2006 June 2006;34:943–9. [DOI] [PubMed] [Google Scholar]
- 34.Sun D, Chen G, Dellinger RW, Duncan K, Fang JL, Lazarus P. Characterization of tamoxifen and 4-hydroxytamoxifen glucuronidation by human UGT1A4 variants. Breast Cancer Res. 2006;8:R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazarus P, Blevins Primeau AS, Zheng Y, Sun D. Potential role of UGT pharmacogenetics in cancer treatment and prevention: Focus on tamoxifen. Ann N Y Acad Sci. 2009. February;1155:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blevins-Primeau AS, Sun D, Chen G, Sharma AK, Gallagher CJ, Amin S, et al. Functional significance of UDP-glucuronosyltransferase variants in the metabolism of active tamoxifen metabolites. Cancer Res. 2009 March 1, 2009;69:1892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang JL, Lazarus P. Correlation between the UDP-glucuronosyltransferase (UGT1A1) TATAA box polymorphism and carcinogen detoxification phenotype: significantly decreased glucuronidating activity against benzo(a)pyrene-7,8-dihydrodiol(−) in liver microsomes from subjects with the UGT1A1*28 variant. Cancer Epidemiol Biomarkers Prev. 2004. January;13:102–9. [DOI] [PubMed] [Google Scholar]
- 38.Gallagher CJ, Muscat JE, Hicks AN, Zheng Y, Dyer A-M, Chase GA, et al. The UDP-Glucuronosyltransferase 2B17 Gene Deletion Polymorphism: Sex-Specific Association with Urinary 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanol Glucuronidation Phenotype and Risk for Lung Cancer. Cancer Epidemiol Biomarkers Prev. 2007 April 1, 2007;16:823–8. [DOI] [PubMed] [Google Scholar]
- 39.Uchaipichat V, Mackenzie PI, Elliot DJ, Miners JO. Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) “probes” for human udp-glucuronosyltransferases. Drug metabolism and disposition: the biological fate of chemicals. 2006. March;34:449–56. [DOI] [PubMed] [Google Scholar]
- 40.Miners JO, Bowalgaha K, Elliot DJ, Baranczewski P, Knights KM. Characterization of niflumic acid as a selective inhibitor of human liver microsomal UDP-glucuronosyltransferase 1A9: application to the reaction phenotyping of acetaminophen glucuronidation. Drug metabolism and disposition: the biological fate of chemicals. April;39:644–52. [DOI] [PubMed] [Google Scholar]
- 41.Devane CL, Markowitz JS. Antipsychotics. 1st ed Levy RH, editor Ambler: Lippencott Williams & Wilkins; 2000. [Google Scholar]
- 42.Green MD, King CD, Mojarrabi B, Mackenzie PI, Tephly TR. Glucuronidation of amines and other xenobiotics catalyzed by expressed human UDP-glucuronosyltransferase 1A3. Drug metabolism and disposition: the biological fate of chemicals. 1998. June;26:507–12. [PubMed] [Google Scholar]
- 43.Green MD, Tephly TR. Glucuronidation of amines and hydroxylated xenobiotics and endobiotics catalyzed by expressed human UGT1.4 protein. Drug Metabolism and Disposition. 1996 March 1996;24:356–63. [PubMed] [Google Scholar]
- 44.Luo H, McKay G, Midha KK. Identification of clozapine N(+)-glucuronide in the urine of patients treated with clozapine using electrospray mass spectrometry. Biol Mass Spectrom. 1994. March;23:147–8. [DOI] [PubMed] [Google Scholar]
- 45.Ehmer U, Vogel A, Schütte JK, Krone B, Manns MP, Strassburg CP. Variation of hepatic glucuronidation: Novel functional polymorphisms of the UDP-glucuronosyltransferase UGT1A4. Hepatology. 2004;39:970–7. [DOI] [PubMed] [Google Scholar]
- 46.Wiener D, Fang JL, Dossett N, Lazarus P. Correlation between UDP-glucuronosyltransferase genotypes and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone glucuronidation phenotype in human liver microsomes. Cancer Res. 2004. February 1;64:1190–6. [DOI] [PubMed] [Google Scholar]
- 47.Saeki M, Saito Y, Jinno H, Sai K, Hachisuka A, Kaniwa N, et al. Genetic variations and haplotypes of UGT1A4 in a Japanese population. Drug metabolism and pharmacokinetics. 2005. April;20:144–51. [DOI] [PubMed] [Google Scholar]
- 48.Gulcebi MI, Ozkaynakci A, Goren MZ, Aker RG, Ozkara C, Onat FY. The relationship between UGT1A4 polymorphism and serum concentration of lamotrigine in patients with epilepsy. Epilepsy research. June;95:1–8. [DOI] [PubMed] [Google Scholar]
- 49.Izukawa T, Nakajima M, Fujiwara R, Yamanaka H, Fukami T, Takamiya M, et al. Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers. Drug Metabolism and Disposition. 2009 August 2009;37:1759–68. [DOI] [PubMed] [Google Scholar]
- 50.Ohno S, Nakajin S. Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug metabolism and disposition: the biological fate of chemicals. 2009. January;37:32–40. [DOI] [PubMed] [Google Scholar]
- 51.Guillemette C Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 2003;3:136–58. [DOI] [PubMed] [Google Scholar]
- 52.Ghotbi R, Mannheimer B, Aklillu E, Suda A, Bertilsson L, Eliasson E, et al. Carriers of the UGT1A4 142T>G gene variant are predisposed to reduced olanzapine exposure—an impact similar to male gender or smoking in schizophrenic patients. European Journal of Clinical Pharmacology. 2010;66:465–74. [DOI] [PubMed] [Google Scholar]
- 53.Mori A, Maruo Y, Iwai M, Sato H, Takeuchi Y. UDP-glucuronosyltransferase 1A4 polymorphisms in a Japanese population and kinetics of clozapine glucuronidation. Drug Metab Dispos. 2005. May;33:672–5. [DOI] [PubMed] [Google Scholar]
- 54.Li Z, Huang M, Ichikawa J, Dai J, Meltzer HY. N-desmethylclozapine, a major metabolite of clozapine, increases cortical acetylcholine and dopamine release in vivo via stimulation of M1 muscarinic receptors. Neuropsychopharmacology. 2005. November;30:1986–95. [DOI] [PubMed] [Google Scholar]
- 55.Lameh J, Burstein ES, Taylor E, Weiner DM, Vanover KE, Bonhaus DW. Pharmacology of N-desmethylclozapine. Pharmacology & Therapeutics. 2007;115:223–31. [DOI] [PubMed] [Google Scholar]
- 56.Sur C, Mallorga PJ, Wittmann M, Jacobson MA, Pascarella D, Williams JB, Brandish PE, Pettibone DJ, Scolnick EM, Conn PJ N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc Natl Acad Sci U S A. 2003;100:13674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Couchman L, Morgan PE, Spencer EP, Flanagan RJ. Plasma clozapine, norclozapine, and the clozapine:norclozapine ratio in relation to prescribed dose and other factors: data from a therapeutic drug monitoring service, 1993–2007. Ther Drug Monit. 2010. August;32:438–47. [DOI] [PubMed] [Google Scholar]
- 58.Williams DP, Pirmohamed M, Naisbitt DJ, Uetrecht JP, Park BK. Induction of metabolism-dependent and -independent neutrophil apoptosis by clozapine. Mol Pharmacol. 2000. July;58:207–16. [DOI] [PubMed] [Google Scholar]
- 59.Williams DP, Pirmohamed M, Naisbitt DJ, Maggs JL, Park BK. Neutrophil cytotoxicity of the chemically reactive metabolite(s) of clozapine: possible role in agranulocytosis. J Pharmacol Exp Ther. 1997. December;283:1375–82. [PubMed] [Google Scholar]
- 60.Maggs JL, Williams D, Pirmohamed M, Park BK. The metabolic formation of reactive intermediates from clozapine, a drug associated with agranulocytosis in man. J Pharmacol Exp Ther. 1995. December;275:1463–75. [PubMed] [Google Scholar]