

Abstract
Background
In sub-Saharan Africa, individuals infected with HIV who are severely immunocompromised have high mortality (about 10%) shortly after starting antiretroviral therapy (ART). This group also has the greatest risk of morbidity and mortality associated with immune reconstitution inflammatory syndrome (IRIS), a paradoxical response to successful ART. Integrase inhibitors lead to significantly more rapid declines in HIV viral load (VL) than all other ART classes. We hypothesised that intensifying standard triple-drug ART with the integrase inhibitor, raltegravir, would reduce HIV VL faster and hence reduce early mortality, although this strategy could also risk more IRIS events.
Methods and findings
In a 2×2×2 factorial open-label parallel-group trial, treatment-naive adults, adolescents, and children >5 years old infected with HIV, with cluster of differentiation 4 (CD4) <100 cells/mm3, from eight urban/peri-urban HIV clinics at regional hospitals in Kenya, Malawi, Uganda, and Zimbabwe were randomised 1:1 to initiate standard triple-drug ART, with or without 12-week raltegravir intensification, and followed for 48 weeks. The primary outcome was 24-week mortality, analysed by intention to treat. Of 2,356 individuals screened for eligibility, 1,805 were randomised between 18 June 2013 and 10 April 2015. Of the 1,805 participants, 961 (53.2%) were male, 72 (4.0%) were children/adolescents, median age was 36 years, CD4 count was 37 cells/mm3, and plasma viraemia was 249,770 copies/mL. Fifty-six participants (3.1%) were lost to follow-up at 48 weeks. By 24 weeks, 97/902 (10.9%) raltegravir-intensified ART versus 91/903 (10.2%) standard ART participants had died (adjusted hazard ratio [aHR] = 1.10 [95% CI 0.82–1.46], p = 0.53), with no evidence of interaction with other randomisations (pheterogeneity > 0.7) and despite significantly greater VL suppression with raltegravir-intensified ART at 4 weeks (343/836 [41.0%] versus 113/841 [13.4%] with standard ART, p < 0.001) and 12 weeks (567/789 [71.9%] versus 415/803 [51.7%] with standard ART, p < 0.001). Through 48 weeks, there was no evidence of differences in mortality (aHR = 0.98 [95% CI 0.76–1.28], p = 0.91); in serious (aHR = 0.99 [0.81–1.21], p = 0.88), grade-4 (aHR = 0.88 [0.71–1.09], p = 0.29), or ART-modifying (aHR = 0.90 [0.63–1.27], p = 0.54) adverse events (the latter occurring in 59 [6.5%] participants with raltegravir-intensified ART versus 66 [7.3%] with standard ART); in events judged compatible with IRIS (occurring in 89 [9.9%] participants with raltegravir-intensified ART versus 86 [9.5%] with standard ART, p = 0.79) or in hospitalisations (aHR = 0.94 [95% CI 0.76–1.17], p = 0.59). At 12 weeks, one and two raltegravir-intensified participants had predicted intermediate-level and high-level raltegravir resistance, respectively. At 48 weeks, the nucleoside reverse transcriptase inhibitor (NRTI) mutation K219E/Q (p = 0.004) and the non-nucleoside reverse transcriptase inhibitor (NNRTI) mutations K101E/P (p = 0.03) and P225H (p = 0.007) were less common in virus from participants with raltegravir-intensified ART, with weak evidence of less intermediate- or high-level resistance to tenofovir (p = 0.06), abacavir (p = 0.08), and rilpivirine (p = 0.07). Limitations of the study include limited clinical, radiological, and/or microbiological information for some participants, reflecting available services at the centres, and lack of baseline genotypes.
Conclusions
Although 12 weeks of raltegravir intensification was well tolerated and reduced HIV viraemia significantly faster than standard triple-drug ART during the time of greatest risk for early death, this strategy did not reduce mortality or clinical events in this group and is not warranted. There was no excess of IRIS-compatible events, suggesting that integrase inhibitors can be used safely as part of standard triple-drug first-line therapy in severely immunocompromised individuals.
Trial registration
ClinicalTrials.gov NCT01825031.
Trial registration
International Standard Randomised Controlled Trials Number ISRCTN 43622374.
In the factorial randomised REALITY trial, Sarah Walker and colleagues document outcomes of intensified initial antiretroviral treatment for people with advanced HIV disease.
Author summary
Why was this study done?
Individuals in Africa who are HIV positive and initiating treatment with severe immunosuppression (CD4 < 100 cells/mm3) have high risks of dying shortly after starting treatment.
It would be expected that adding an integrase inhibitor (raltegravir) to standard triple-drug antiretroviral therapy for 12 weeks would reduce HIV viral load faster: we wanted to find out whether this would reduce high early mortality.
What did the researchers do and find?
We randomly allocated 1,805 adults, adolescents, and older children to receive standard antiretroviral therapy with or without 12 weeks of adjunctive raltegravir.
We found that the group receiving 12 weeks of adjunctive raltegravir had significantly faster declines in HIV viral load.
However, we found no significant difference in deaths within 24 weeks of starting treatment between those receiving adjunctive raltegravir (97/902 [10.9%]) or not (91/903 [10.2%]); nor were there differences in clinical disease progression, immune reconstitution, inflammatory syndrome, or adverse events.
What do these findings mean?
In this study, we found that intensifying the potency of initial antiretroviral therapy did not reduce early mortality on treatment, providing no support for its widespread use in individuals initiating treatment with severe immunosuppression.
However, it also did not increase the rates of clinically important immune reconstitution inflammatory syndrome, suggesting that integrase inhibitors could replace other components of standard first-line antiretroviral therapy safely.
Introduction
Despite World Health Organisation (WHO) guidelines recommending universal antiretroviral therapy (ART) regardless of cluster of differentiation 4 (CD4) cell count [1], 20%–25% of individuals infected with HIV in sub-Saharan Africa continue to present for care with severe immunosuppression (CD4 count < 100 cells/mm3) [2]. Of these, about 10% die in the first 3 months after ART initiation [3–6]. Causes of early death are multifactorial and are similar for adults and older children [5]. Possible strategies to reduce this excess mortality could aim to accelerate immune restoration by controlling HIV viraemia more rapidly; to control or prevent overt clinical infections; or to provide nutritional support to enhance immune response to infection and reverse metabolic deficiencies.
Standard WHO-recommended ART [1] consists of two nucleos(t)ide reverse transcriptase inhibitors (N(t)RTI), with a non-nucleoside reverse transcriptase inhibitor (NNRTI). Another key drug class is integrase strand-transfer inhibitors (INSTIs), which result in faster decline in HIV viral load (VL) over the first 3 months of combination therapy [7,8]. Several studies have investigated how this might impact chronic inflammation and immune restoration; in some, INSTIs appeared to improve markers of inflammation (including microbial translocation) and/or T-cell activation [9–14], but in others there were no clear differences in inflammatory and other markers (i.e., D-dimer, interleukin 6 [IL-6], and T-cell activation) [15]. The clinical importance of intensified or quadruple ART in patients with VL >100,000 copies/mL or advanced disease remains unclear.
To our knowledge, no randomised trial has been powered to test the hypothesis that the INSTI-associated rapid reduction in HIV viraemia translates into clinical benefit, mortality reductions in particular, or, conversely, is associated with harm through increased risk of immune reconstitution inflammatory syndrome (IRIS). We therefore conducted the Reduction of EArly mortaLITY (REALITY) randomised clinical-endpoint trial (NCT01825031; ISRCTN43622374) to compare three interventions to reduce early mortality in adults and older children initiating ART with CD4 <100 cells/mm3 in four sub-Saharan African countries: adjunctive intensification with raltegravir [16] (the first licenced INSTI), enhanced anti-infective prophylaxis, and food supplementation. Here, we report on the impact of adjunctive raltegravir intensification.
Materials and methods
Adults, adolescents, and children aged ≥5 years infected with HIV, diagnosed through national screening programmes, not on ART and reporting no previous ART, and with CD4 <100 cells/mm3 were approached consecutively for screening from inpatient and outpatient facilities at clinics at eight urban/peri-urban regional hospitals in Zimbabwe, Uganda, Malawi, and Kenya. When CD4 counts were not routinely performed at diagnosis, those with new HIV diagnoses were approached consecutively for CD4 testing and study screening. Participants were enrolled if they had screening CD4 count <100 cells/mm3, were ART-naive, were not pregnant/breastfeeding, had not used single-dose nevirapine to prevent mother-to-child transmission, had no contraindications to trial drugs, and provided written informed consent. The trial was approved by Ethics Committees in Zimbabwe, Uganda, Malawi, Kenya, and the United Kingdom. The protocol and CONSORT checklist are provided as S1 Protocol and S1 CONSORT checklist.
Participants were randomised 1:1 to initiate open-label ART with 2NRTI+NNRTI either alone (standard ART) or with 12 weeks’ raltegravir (raltegravir intensification) using standard doses (see S1 Text). Standard ART was tenofovir-disoproxil-fumarate+emtricitabine or zidovudine+lamivudine in adults and adolescents and abacavir+lamivudine or zidovudine+lamivudine in children aged 5–<13 years, with nevirapine or efavirenz, according to physician choice and local standard of care. Participants were also factorially randomised 1:1 to 12 weeks enhanced anti-infection prophylaxis versus standard-of-care co-trimoxazole prophylaxis [17], and 1:1 to 12 weeks ready-to-use supplementary food (RUSF) versus no routine supplementation [18] (see S1 Text for details).
Randomisation was stratified by centre, age (</≥13 years), and the other factorial randomisations. A computer-generated sequential randomisation list using variably sized permuted blocks was prepared by the trial statistician and incorporated securely into the online trial database. The list was concealed until allocation after eligibility was confirmed by local centre staff, who then performed the randomisation.
Nurse visits at weeks 2, 4, 8, 12, 18, 24, 36, and 48 included symptom checklist, self-reported adherence assessment (with standard adherence support), medication dispensing, and body composition assessment using bioimpedance (TANITA BC-420MA). At weeks 4, 12, 24, 36, and 48, history and examination were undertaken by a physician; haematology, biochemistry, and CD4/CD8 assays were performed at local laboratories; and plasma was stored for retrospective VL and genotyping (results not available in real time). Nurses/physicians were unblinded. Laboratory tests were assayed blind to randomisation. Toxicity substitution and/or second-line switch were at physician discretion, following WHO guidelines [19]. Participants exited the trial after 48 weeks. Consent was obtained to ascertain vital status in all participants at trial closure (May 2016, i.e., beyond 48 weeks from randomisation) from the ART programmes where they had transferred to care at trial exit.
The primary outcome was all-cause mortality to 24 weeks. Secondary outcomes to 48 weeks were all-cause mortality; serious adverse events (SAEs) following International Committee on Harmonization definitions, grade-4 adverse events (AEs), and AEs leading to modification of ART/trial drugs; specific mechanisms of each intervention (CD4 count change; incidence of tuberculosis, cryptococcosis, candidiasis, severe bacterial infections); changes in weight/BMI; hospitalisations; and self-reported ART adherence/acceptability. Other prespecified outcomes included VL suppression (originally planned to be assayed only in a random subset; ultimately assayed in all participants), genotypic resistance (assayed in samples from Kenya, Uganda, and Zimbabwe, see S1 Text), and body composition. All AEs were graded following established guidelines [20,21]. HIV integrase genotypes were assayed for raltegravir-intensified participants with VL >1,000 c/mL at 12 weeks, and reverse transcriptase genotypes were assayed for all participants with VL >1,000 c/mL at 48 weeks. An Endpoint Review Committee (ERC) (majority independent members), blind to trial drugs received, adjudicated secondary clinical endpoints and trial-drug relatedness against protocol-defined criteria and assessed compatibility of clinical events with IRIS. Clinical endpoints were predominantly clinically defined, as microbiological and other diagnostic facilities were limited in most centres. Because of this known limitation, IRIS was not a secondary outcome and was considered an exploratory analysis. However, IRIS was part of the standardised ERC assessment and ERC case record form from the start of the trial and was prospectively ascertained on all reviewed events. Events were considered IRIS compatible if there was atypical or exaggerated presentation of an opportunistic infection or tumour soon after ART initiation. VLs were only measured retrospectively on stored plasma and so were not available contemporaneously for assessment of IRIS compatibility; the earliest post-randomisation CD4 count was done at 4 weeks and so was not available for events before this. As access to diagnostic testing was limited, previously published definitions [22,23] were used but in modified form; the ERC adjudication relied heavily on the description of the clinical presentation and any radiology/microbiology/histology provided by site, and the timing of the presentation and its evolution in relation to ART initiation.
A total of 1,800 adults/children provided >80% power to detect 50% relative reductions in 24-week all-cause mortality from 7% to 3.5% (two-sided alpha = 0.05), allowing 5% lost to follow-up. Interim data were reviewed by an independent Data Monitoring Committee (three annual meetings) using the Haybittle-Peto criterion (p < 0.001). Randomised groups were compared following the intention to treat principle using log-rank tests for time-to-event outcomes (censoring those lost to follow-up), Fisher exact tests for binary outcomes, and generalized estimating equations (GEEs) with independent working correlation for global tests of repeated measures (logit/normal distributions for binary/continuous outcomes, respectively). Primary analyses were stratified by randomisation stratification factors (details in S1 Text). Analyses used Stata v15.1.
Results
Between 18 June 2013 and 10 April 2015, 1,805 participants were randomised to standard ART (n = 903) or raltegravir-intensified ART (n = 902) (Fig 1).
Fig 1. Trial profile.

ART, antiretroviral therapy; CD4, cluster of differentiation 4; RUSF, ready-to-use supplementary food.
Baseline characteristics were well balanced between randomised groups (Table 1). Median age was 36 years; 72 (4.0%) participants were aged 5–17 years. Median CD4 count was 37 cells/mm3 and VL was 249,770 copies/mL; 1,334/1,804 (73.9%) had VL ≥ 100,000 copies/mL. Despite this, 854 (47.3%) participants were WHO stage 1/2. A total of 56 (3.1%) participants were lost to follow-up (no clinic attendance for >91 days). Before death/loss to follow-up, 12,664/12,944 (97.8%) scheduled visits were completed.
Table 1. Characteristics at enrolment.
| Factor | Standard ART (N = 903) |
Raltegravir-intensified (N = 902) |
All (N = 1,805) |
|---|---|---|---|
| Male | 482 (53.4%) | 479 (53.1%) | 961 (53.2%) |
| Age at last birthday (years) | 36 (29–42) [5–77] | 35 (29–42) [6–72] | 36 (29–42) [5–77] |
| 5–17 years | 33 (3.7%) | 39 (4.3%) | 72 (4.0%) |
| HIV VL (c/mL) (N = 1,804) | 250,000 (95,450–631,560) |
246,700 (94,000–578,080) |
249,770 (95,280–606,360) |
| ≥100,000 c/mL | 667/902 (73.9%) | 667/902 (73.9%) | 1,334/1,804 (73.9%) |
| <1,000 c/mL** | 7/902 (0.8%) | 7/902 (0.8%) | 14/1,804 (0.8%) |
| CD4 count* (cells/mm3) | 36 (16–61) | 38 (16–64) | 37 (16–63) |
| 0–24 cells/mm3 | 335 (37.1%) | 321 (35.6%) | 656 (36.3%) |
| 25–49 cells/mm3 | 256 (28.3%) | 253 (28.0%) | 509 (28.2%) |
| Weight* (kg) (N = 1,800) | 52.9 (46.7–59.8) | 52.3 (45.8–59.0) | 52.5 (46.3–59.3) |
| BMI (kg/m2) (N = 1,797) | 19.3 (17.5–21.6) | 19.0 (17.1–21.2) | 19.2 (17.2–21.4) |
| <18 kg/m2 | 302/900 (33.6%) | 323/897 (36.0%) | 625/1,797 (34.8%) |
| WHO stage | |||
| 1 | 145 (16.1%) | 155 (17.2%) | 300 (16.6%) |
| 2 | 290 (32.1%) | 264 (29.3%) | 554 (30.7%) |
| 3 | 341 (37.8%) | 350 (38.8%) | 691 (38.3%) |
| 4 | 127 (14.1%) | 133 (14.7%) | 260 (14.4%) |
| Current tuberculosis disease | 137 (15.2%) | 134 (14.9%) | 271 (15.0%) |
| Haemoglobin (g/L) (N = 1,800) | 112 (96–127) | 111 (95–127) | 112 (96–127) |
| ≤80 g/L | 86 (9.6%) | 90 (10.0%) | 176 (9.8%) |
| NRTIs | |||
| Tenofovir/emtricitabine | 719 (79.6%) | 703 (77.9%) | 1,422 (78.8%) |
| Zidovudine/lamivudine | 154 (17.1%) | 169 (18.7%) | 323 (17.9%) |
| Abacavir/lamivudine | 30 (3.3%) | 30† (3.3%) | 60 (3.3%) |
| NNRTI | |||
| Efavirenz | 816‡ (90.4%) | 803 (89.0%) | 1,619 (89.7%) |
| Nevirapine | 87 (9.6%) | 99 (11.0%) | 186 (10.3%) |
| Randomised to receive enhanced anti-infection prophylaxis | 451 (49.9%) | 455 (50.4%) | 906 (50.2%) |
| Randomised to receive RUSF | 449 (49.7%) | 448 (49.7%) | 897 (49.7%) |
*Mean of screening and enrolment values. Eligibility required screening CD4 to be <100 cells/mm3, so baseline can be above 100, depending on the CD4 at enrolment.
**Potentially indicating undisclosed prior ART: median CD4 was 76 cells/mm3 in these participants.
†One child was mistakenly initiated on Aluvia (lopinavir/ritonavir) rather than abacavir/lamivudine (plus efavirenz and raltegravir); substituted with abacavir/lamivudine after 4 weeks.
‡One adult took tenofovir/emtricitabine alone for 4 days in error before adding efavirenz on day 4.
Note: Showing n (%) or median (IQR) [range].
Abbreviations: CD4, cluster of differentiation 4; NNRTI, non-nucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; RUSF, ready-to-use supplementary food; VL, viral load.
Randomised interventions
A total of 901 (99.9%) participants randomised to raltegravir-intensified ART were prescribed raltegravir. Overall, in the first 12 weeks on ART the raltegravir-intensified ART group spent 98.2% of person-time prescribed raltegravir versus 0.0% in the standard ART group, compared with 0.6% versus 0.1%, respectively, from 12 to 48 weeks (S1 Fig). Self-reported ART adherence was high but was significantly poorer in the raltegravir-intensified ART group during the first 12 weeks only (p < 0.001) (overall p = 0.08, S2A Fig), particularly in those taking other ART once daily (S2B Fig). At last follow-up, 885 (98.0%) standard ART versus 880 (97.6%) raltegravir-intensified ART participants remained on first-line ART (exact p = 0.53), with 67 (7.4%) standard ART versus 52 (5.8%) raltegravir-intensified ART participants having made within-class substitutions (exact p = 0.18).
VL suppression
Despite slightly poorer self-reported adherence, early VL suppression was significantly and substantially faster with raltegravir intensification (Fig 2A), with 41.0% (343/836), 71.9% (567/789), 76.7% (594/774), and 80.7% (661/757) participants, respectively, recording <50 copies/mL 4, 12, 24, and 48 weeks after randomisation versus 13.4% (113/841), 51.7% (415/803), 74.7% (586/784), and 79.2% (591/746) in the standard ART group, respectively (p < 0.001, p < 0.001, p = 0.36, and p = 0.47, respectively). Overall, therefore, 59.4% versus 43.6% of time at risk through 24 weeks was spent with VL <50 copies/mL in raltegravir-intensified versus standard ART, respectively. At higher thresholds, differences at week 4 were even greater and attenuated more rapidly but persisted through 24 weeks (S3 Fig). At 48 weeks, there was no evidence of differential suppression at 50–5,000 copies/mL (p > 0.4). Mean change in log10 VL to week 4 was −3.4 (standard error [SE] 0.03) versus −2.7 (SE 0.03) in raltegravir-intensified ART versus standard ART (p < 0.001), with geometric mean VL 80 c/mL and 480 c/mL, respectively.
Fig 2. HIV viral load, CD4 cell count, and weight.
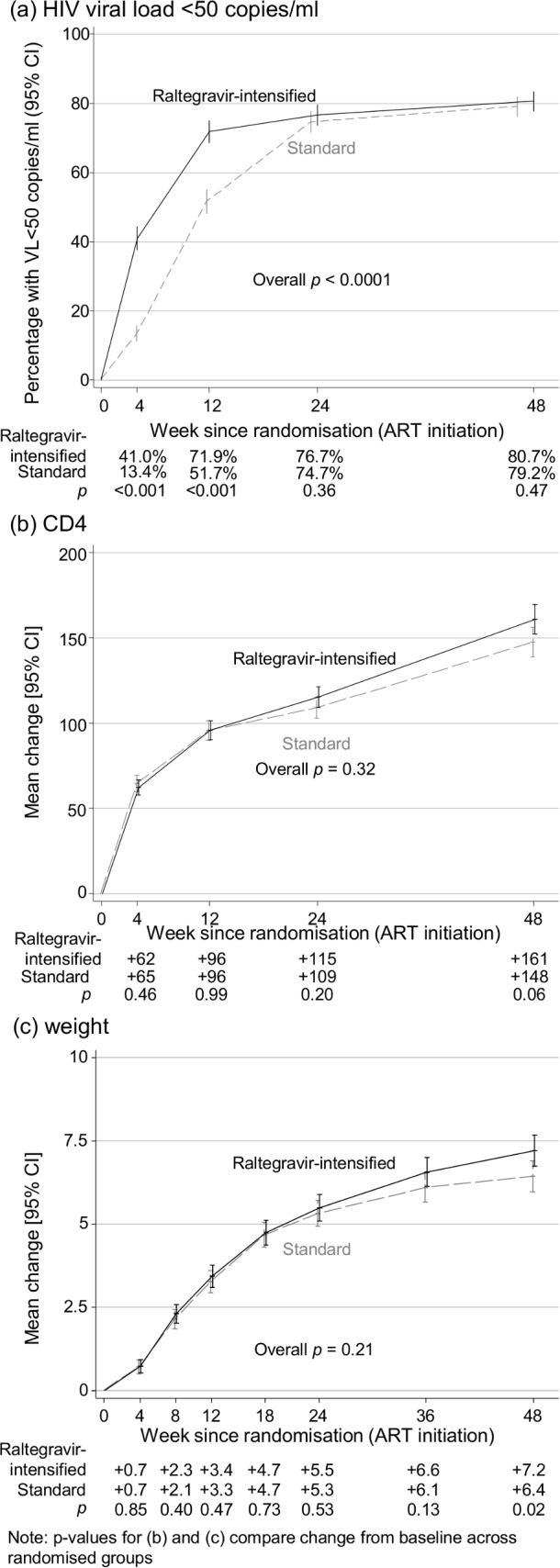
ART, antiretroviral therapy; CD4, cluster of differentiation 4; VL, viral load.
Mortality
By 24 weeks (primary endpoint), 97 (10.9%) raltegravir-intensified ART versus 91 (10.2%) standard ART participants died (adjusted hazard ratio [aHR] = 1.10 [95% CI 0.82–1.46], log-rank p = 0.53, Fig 3A), with no evidence of interaction with other randomisations (pheterogeneity > 0.7). Absolute 24-week mortality difference was +0.6% (−2.2%, +3.5%). By 48 weeks, 110 (12.4%) versus 115 (13.0%) participants, respectively, had died (aHR = 0.98 [0.76–1.28], log-rank p = 0.91; absolute difference −0.6% [−3.7%, +2.5%]) with no evidence of differences by cause (Table 2). There was no difference in longer-term (after week 48) mortality (aHR = 0.95 [0.74–1.21], log-rank p = 0.69, S4 Fig). Estimated mortality rates decreased sharply from day 19 through week 12 (Fig 3B), with 71 (31.6%) of the 225 deaths occurring by 4 weeks, and 147 (65.3%) by 12 weeks. There was no evidence of early differences in mortality rates (S5 Fig) and no evidence of heterogeneity in the impact of raltegravir intensification over time on ART (pheterogeneity = 0.14, comparing 0–24 versus 24–48 versus 48+ weeks on ART). Of nine preplanned and five exploratory subgroup analyses (S6 Fig; details in S1 Text), only one had weak evidence for variation across dual NRTIs (pheterogeneity = 0.04, suggesting harm from raltegravir intensification in combination with tenofovir-emtricitabine; pheterogeneity = 0.06 comparing tenofovir-emtricitabine versus zidovudine-lamivudine [excluding abacavir-lamivudine]). Specifically, there was no evidence that mortality differences depended on pre-ART VL or pre-ART CD4 (pheterogeneity = 0.84/0.49 and 0.22/0.27, respectively, using categorical/continuous interactions). No subgroup analyses suggested heterogeneity in the impact of raltegravir intensification on suppression <50 copies/mL at week 4 (pheterogeneity > 0.05) (S7 Fig).
Fig 3. Mortality.

ART, antiretroviral therapy; PY, person-years.
Table 2. Secondary and other clinical outcomes through 48 weeks.
| Outcome |
Standard ART N participants (% of 903) [events] |
Raltegravir-intensified ART N participants (% of 902) [events] |
Total N participants (% of 1,805) [events] |
Hazard ratio† (95% CI) | p |
|---|---|---|---|---|---|
| Death* | 115 (13.0%) [115] | 110 (12.4%) [110] | 225 (12.5%) [225] | 0.98 (0.76, 1.28) | 0.91 |
| - from tuberculosis | 24 (2.7%) [24] | 18 (2.0%) [18] | 42 (2.3%) [42] | 0.75 (0.40, 1.37)† | 0.35 |
| - from cryptococcal disease | 6 (0.7%) [6] | 11 (1.2%) [11] | 17 (0.9%) [17] | 1.84 (0.68, 4.97)† | 0.23 |
| - from severe bacterial infections | 16 (1.8%) [16] | 17 (1.9%) [17] | 33 (1.8%) [33] | 1.07 (0.54, 2.11)† | 0.86 |
| - from other causes | 23 (2.5%) [23] | 22 (2.4%) [22] | 45 (2.5%) [45] | 0.96 (0.53, 1.71)† | 0.88 |
| - cause unknown | 46 (5.1%) [46] | 42 (4.7%) [42] | 88 (4.9%) [88] | 0.91 (0.60, 1.38)† | 0.66 |
| New WHO 4 event or death | 164 (18.2%) [216] | 155 (17.2%) [203] | 319 (17.7%) [419] | 0.97 (0.77, 1.20) | 0.75 |
| New WHO 3 or 4 event or death | 197 (21.8%) [310] | 206 (22.8%) [301] | 403 (22.3%) [611] | 1.08 (0.89, 1.31) | 0.44 |
| New tuberculosis disease* | 80 (8.9%) 103 | 76 (8.4%) 93 | 156 (8.6%) [196] | 0.95 (0.70, 1.31) | 0.77 |
| New cryptococcal disease* | 18 (2.0%) 25 | 14 (1.6%) 26 | 32 (1.8%) [51] | 0.78 (0.38, 1.58) | 0.49 |
| New candida disease* | 16 (1.8%) 18 | 17 (1.9%) 18 | 33 (1.8%) [36] | 1.06 (0.54, 2.11) | 0.86 |
| New presumptive severe bacterial infection* | 33 (3.7%) 49 | 42 (4.7%) 62 | 75 (4.2%) [111] | 1.28 (0.81, 2.02) | 0.28 |
| IRIS | 86 (9.5%) [89] | 89 (9.9%) [91] | 175 (9.7%) [180] | 1.04 (0.78, 1.40) | 0.79 |
| Any SAE* | 207 (22.9%) 287 | 203 (22.5%) 251 | 410 (22.7%) [538] | 0.99 (0.81, 1.21) | 0.88 |
| New hospitalisation* | 175 (19.4%) [233] | 165 (18.3%) [198] | 340 (18.8%) [431] | 0.94 (0.76, 1.17) | 0.59 |
| Grade-4 AE* | 188 (20.8%) [267] | 165 (18.3%) [237] | 353 (19.6%) [504] | 0.88 (0.71, 1.09) | 0.29 |
| Grade-3 or -4 AE | 327 (36.2%) [550] | 331 (36.7%) [503] | 658 (36.5%) [1053] | 1.03 (0.88, 1.20) | 0.72 |
| Grade-4 AE definitely, probably, or possibly related to ART | 67 (7.4%) [73] | 59 (6.5%) [65] | 126 (7.0%) [138] | 0.89 (0.63, 1.27) | 0.52 |
| Grade-4 AE definitely or probably related to ART | 28 (3.1%) [28] | 16 (1.8%) [18] | 44 (2.4%) [46] | 0.57 (0.31, 1.06) | 0.07 |
| AE leading to ART modification* | 66 (7.3%) [73] | 59 (6.5%) [66] | 125 (6.9%) [139] | 0.90 (0.63, 1.27) | 0.54 |
| Grade-4 AE definitely, probably, or possibly related to raltegravir | - | 31 (3.4%) [34] | - | - | |
| Grade-4 AE definitely or probably related to raltegravir | - | 1‡ (0.1%) [1] | - | - | |
| AE leading to raltegravir modification | - | 19 (2.1%) [19] | - | - |
*Secondary outcome prespecified in the protocol.
†For causes of death, competing risks subhazard ratio accounting for other causes of death.
‡Stevens-Johnson syndrome that was adjudicated as definitely/probably related to efavirenz, raltegravir, and co-trimoxazole and possibly related to fluconazole, tenofovir, and emtricitabine.
Note: Table shows number of patients with one or more episode (percentage of patients) [number of episodes] (e.g., ‘2 (20.0%) [3]’ would indicate a total of three episodes in two patients). No evidence of interaction with other factorial randomisations (pheterogeneity > 0.1; 34 tests; testing not conducted for grade-4 AE definitely or probably related to raltegravir as only one event).
Abbreviations: AE, adverse event; ART, antiretroviral therapy; IRIS, immune reconstitution inflammatory syndrome; SAE, serious adverse event.
Other clinical efficacy outcomes
There was no evidence of an impact of raltegravir intensification on any disease progression clinical outcome (Table 2).
Genotypic resistance
Integrase genotypes were obtained in 33 raltegravir-intensified ART participants with VL >1,000 (median 47,724) copies/mL at 12 weeks (see S2 Text). One participant had predicted intermediate-level (mutations T97A+R263K) and two predicted high-level (N155H, T97A+Y143R/C) raltegravir resistance mutations (0.6% of those randomised, accounting for missing genotypes using probability weights). No patient had predicted intermediate- or high-level dolutegravir resistance.
Reverse-transcriptase genotypes were available for 75 raltegravir-intensified ART versus 87 standard ART participants with VL >1,000 (median 89,815) copies/mL at 48 weeks. The NRTI mutation K219E/Q (p = 0.004) and the NNRTI mutations K101E/P (p = 0.03) and P225H (p = 0.007) were less common in raltegravir-intensified ART, with no evidence of differences for other mutations (p > 0.1, S8 Fig). There was marginal evidence suggesting less intermediate- or high-level resistance with raltegravir-intensified ART to tenofovir (24.0% [18/75] raltegravir-intensified ART versus 37.9% [33/87] standard ART, p = 0.06), abacavir (40.0% [30/75] raltegravir-intensified ART versus 54.0% [47/87] standard ART, p = 0.08), and rilpivirine (38.7% [29/75] raltegravir-intensified ART versus 52.9% [46/87] standard ART, p = 0.07) (S9 Fig) (see S2 Text for details).
CD4 and CD8 count and body composition
Absolute CD4 count increases were similar in both groups through 24 weeks (p = 0.76; Fig 2B); however, there was marginal evidence of a small difference at 48 weeks (+161 [standard deviation ±4.4] cells/mm3 raltegravir-intensified ART versus +148 [±4.4] cells/mm3 standard ART, adjusted difference +11.4 [95% CI −0.4 to +23.1], p = 0.06). There was similar weak evidence of slightly (2%–5%) greater percentages with CD4 ≥ 200 cells/mm3 at later time points (p = 0.049 at week 24, p = 0.07 at week 48, S10 Fig). Changes in weight were also similar through week 24 (p = 0.52; Fig 2C), with small but significant differences appearing from 36–48 weeks. Similar late differences were observed for BMI in adolescents/adults (data not shown), fat mass (S11A and S11B Fig), and muscle mass (S11C and S11D Fig). There was no evidence of heterogeneity in these differences by age (S12 Fig). Absolute CD8 count increases were similar in both groups through 48 weeks (p = 0.82, S13 Fig).
Safety outcomes
There was no evidence for differences in time to first SAEs (log-rank p = 0.88; S1 Table), grade-4 AEs (log-rank p = 0.29), grade-3/4 AEs (log-rank p = 0.72), grade-4 AEs definitely/probably/possibly related to ART (log-rank p = 0.52), grade-4 AEs definitely/probably related to ART (log-rank p = 0.07), AEs leading to ART modification (log-rank p = 0.51) or new hospitalisations (log-rank p = 0.59) (Table 2). Raltegravir was modified for AEs in 19 (2.1%) participants (S2 Table), including renal (n = 6) events, hepatic (n = 6) events, hypersensitivity reactions (n = 3; two discontinued with efavirenz, which was subsequently restarted; one discontinued with tenofovir+emtricitabine+efavirenz, subsequently started lopinavir+ritonavir+tenofovir+emtricitabine+raltegravir), and Stevens-Johnson syndrome in one participant (discontinued lamivudine+zidovudine+efavirenz+raltegravir at week 7). Only one participant experienced a grade-4 AE adjudicated as definitely/probably related to raltegravir (Table 2). There was no evidence of any difference in total hospitalisation-days (2,367 raltegravir-intensified ART versus 2,685 standard ART, rank-sum p = 0.56) or total hospitalisations (198 versus 233, respectively, Poisson p = 0.09).
IRIS (exploratory analysis)
Fatal or nonfatal events judged compatible with IRIS occurred in 89 (9.9%) raltegravir-intensified ART versus 86 (9.5%) standard ART participants (log-rank p = 0.79) (Table 2); 36 (4.0%) versus 31 (3.4%), respectively, experienced fatal IRIS events (p = 0.54). Tuberculosis IRIS events occurred in 53 (5.9%) participants with raltegravir-intensified ART versus 54 (6.0%) with standard ART (exact p = 1.00) and cryptococcal IRIS events in 15 (1.7%) versus 16 (1.8%) participants, respectively (exact p = 1.00) (S3 Table). IRIS events occurred a median 3.4 (IQR 2.0–6.3) weeks from randomisation, with rates declining from the third week on ART (S14 Fig). IRIS was more common in participants initiating ART at older ages (p = 0.005), with lower CD4 counts (p < 0.001) or with pre-existing TB (p = 0.007); IRIS was less common in those initiating ART with enhanced prophylaxis against opportunistic infections (p = 0.001) (S4 Table). There was no evidence that raltegravir intensification was associated with IRIS after adjusting for these factors (p = 0.63).
Discussion
In this large trial in adults and older children with CD4 <100 cells/mm3 in sub-Saharan Africa, we found that 12-week raltegravir intensification of standard ART was well tolerated and reduced plasma HIV VL substantially faster than standard ART alone, but that this had no discernible clinical benefit and no effect on mortality; however, nor was there any evidence of increased rates of IRIS. This is important given the current move to first-line INSTI-based ART; all INSTIs have similarly rapid VL reductions [24], so this result can likely be extrapolated to other INSTIs.
The underpinning hypothesis was that mortality might be reduced by accelerating plasma HIV RNA decline with INSTIs, because initiating ART reduces mortality compared with rates observed pre-ART [25]. One reason this did not occur could be that the differential VL suppression, although significant and substantial, was still too small to reduce mortality. As VL reductions are similar across INSTIs [24], other INSTIs would likely produce similar results. Other explanations for lack of mortality benefit despite significantly faster VL declines may be because there was a lag in, or no, improvement in early functional CD4 recovery or because the disease may simply have been too severe to be reversed despite rapid VL decline. CD4 and CD8 T cells were the only markers of immune restoration measured in real time in all participants, so we cannot investigate further; but there was no differential effect on CD4 (or CD8) counts, at least in the first 24 weeks. This might be expected, as immune restoration does not only depend on HIV viraemia driving inflammation and T-cell turnover [26]. What is intriguing in this context is the small but significant difference in CD4 count at week 48 following only 12 weeks’ raltegravir intensification, which is consistent with previous findings of modestly greater longer-term CD4 increases in HIV seroconverters initiating integrase-inhibitor-containing ART [27] and the STARTMRK trial [28]. We are unable to explore whether CD4 immune restoration continued to diverge after 48 weeks to larger, more clinically relevant differences [29] but plan to measure inflammatory biomarkers [30,31], T-cell subsets, and markers of T-cell turnover and activation in a subset of participants in whom peripheral blood mononuclear cells were stored. However, our results do suggest that quadruple ART is unlikely to provide any clinical benefit over standard triple-drug ART in patients with VL >100,000 copies/mL or advanced disease. Furthermore, the lack of evidence of clinical benefit does not support evaluation of adjunctive INSTIs in less immunocompromised patients, whose risk of clinical events is markedly lower [5].
With increasing moves towards replacing NNRTIs with INSTIs in first-line ART, findings from retrospective observational studies [32,33] have raised major concerns regarding the potential for increased IRIS events with faster VL declines, which may have particularly important consequences in severely immunocompromised patients and where CD4 counts may no longer be available, so that closer clinical monitoring cannot be instigated. Our randomised trial found no evidence of higher rates of clinical IRIS events associated with the significantly faster VL declines with raltegravir-intensified ART. This finding also has relevance for the increasing numbers of treatment-experienced patients returning after previous disengagement from care [34], for whom INSTIs are an attractive option given challenges in accessing resistance testing. Whilst unaffected by raltegravir intensification, IRIS events were significantly reduced by the enhanced-prophylaxis bundle also investigated in this trial [35].
One potential benefit from faster declines in VL is reduced potential for onward transmission, particularly because almost half of the trial participants had minimal symptoms (WHO stage 1 or 2). However, the differential suppression occurred between weeks 4 and 12, with overall only 15.8% less time spent with VL <50 c/mL in the standard ART group through week 24 (albeit predominantly at the time of greatest risk of clinical events). Therefore, benefits at a population level would likely be modest at best. However, such a strategy might have particular value in swiftly reducing VL in women identified as HIV-infected during pregnancy to reduce mother-to-child transmission [36].
Poorer compliance with raltegravir-intensified ART could explain the lack of clinical benefit, although the substantial differences in early VL suppression suggest that adherence to raltegravir-intensified ART was reasonable. However, the additional twice-daily raltegravir pill reduced self-reported adherence to ART in participants receiving other ART once daily by about 4% (S2B Fig). Association with twice-daily dosing, rather than pill burden, is supported by the lack of difference in self-reported ART adherence between participants randomised in this trial to enhanced-prophylaxis versus standard-of-care co-trimoxazole (one additional pill once daily from day 5 to 12 weeks) [17]. Although the induction of UGT1A1 by efavirenz (reducing raltegravir levels by about one third) and/or incomplete adherence in those on twice-daily regimens might have reduced efficacy, there was no evidence of heterogeneity in VL suppression at week 4, or mortality, according to initial NNRTI or according to whether other ART was once daily versus twice daily.
At 48 weeks, 20.0% of participants had a VL ≥50 copies/mL and 12.6% had ≥1,000 copies/mL; this may be unsurprising given advanced disease at ART initiation but limits power to investigate drug resistance. Despite similar rates of virological failure ≥1,000 copies/mL, raltegravir-intensified ART was associated with lower rates of the NRTI mutation K219E/Q and the NNRTI mutations K101E/P and P225H. A limitation is that we did not assay samples at ART initiation; however, randomising large numbers should provide balance between raltegravir-intensified ART and standard ART in transmitted drug resistance. This may suggest that these specific mutations arise early in treatment and, even if then suppressed, ultimately re-emerge at failure. However, differences in predicted NRTI/NNRTI resistance were at most marginal for tenofovir, abacavir, and rilpivirine, and raltegravir intensification did not substantially protect against developing clinically meaningful NRTI/NNRTI resistance. Major integrase mutations potentially compromising raltegravir occurred in very small numbers and did not preclude future use of dolutegravir.
The trial’s major strengths were the mortality primary endpoint in a well-defined high-risk population from eight HIV clinics across four African countries, increasing generalisability. Limitations include the fact that some WHO 3/4 and IRIS events were probably misclassified due to limited clinical, radiological, and/or microbiological information for some participants, reflecting available services at the centres, meaning we were also unable to accurately distinguish between IRIS-associated unmasking versus new active disease. Furthermore, the first post-randomisation CD4 was done at 4 weeks only and VL data were retrospective and not available for review. However, adjudications were made blinded to trial drugs, minimising bias in the overall findings, and the primary endpoint, 24-week mortality, was completely objective. IRIS rates were lower than some previous studies but likely included most clinically important events. Ultimately, the trial was well powered to detect differences in mortality, providing confidence that even if some IRIS events were missed, these were not leading to excess deaths given the relatively tight 95% CI around absolute mortality differences. We did not assess VL response before 4 weeks (first stored sample), when one third of the deaths had already occurred. We were unable to genotype Malawian samples (12.5% of the participants) and obtained integrase genotypes in only 73% of samples with VL >1,000 copies/mL at 12 weeks, possibly because of challenges with primers for non-clade-B viruses. We recruited fewer children than planned, limiting our ability to assess effects in this subgroup.
Overall, REALITY trial findings do not support intensification of ART with the INSTI, raltegravir, for the first 12 weeks in severely immunocompromised adults and children infected with HIV. In contrast, the enhanced prophylaxis bundle also tested in the REALITY trial provided substantial reduction in both mortality [17] and morbidity (including IRIS) [35] and could also be practically implemented at primary care level, provided CD4 counts were available to identify those at risk. Successful future interventions would need to improve both the rapidity and functional quality of immunological recovery and/or reduce inflammation and/or further improve prevention and management of IRIS. Earlier diagnosis and initiation of lifelong ART should continue to be a major focus, particularly in sub-Saharan Africa, as, despite recommendations for universal ART, the proportion of ‘late presenters’ appears to have plateaued in both low- and middle-income countries [37] and high-income countries [38], suggesting that substantial numbers will continue to present late for care in low- and middle-income countries for the foreseeable future. Finally and importantly, this study provides no evidence that moving to INSTIs as part of standard first-line therapy, as currently planned across many low- and middle-income countries, will increase rates of clinically important IRIS in such individuals.
Supporting information
(DOC)
The full trial protocol can be accessed at http://www.ctu.mrc.ac.uk/research/documents/hiv_protocols/reality_protocol [accessed 24 August 2018].
(PDF)
(DOC)
(DOC)
SAE, serious adverse event.
(DOC)
AE, adverse event.
(DOC)
IRIS, immune reconstitution inflammatory syndrome.
(DOC)
IRIS, immune reconstitution inflammatory syndrome.
(DOC)
CD4, cluster of differentiation 4.
(DOC)
(TIF)
Self-reported percentage reporting missing doses of any ART in the last 4 weeks (a) overall and (b) by backbone NRTI and NNRTI frequency. ART, antiretroviral therapy; NNRTI, non-nucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor.
(TIF)
VL suppression (a) <400 copies/mL and (b) <1,000 copies/mL. VL, viral load.
(TIF)
(TIF)
ART, antiretroviral therapy.
(TIF)
(TIF)
VL, viral load.
(TIF)
NNRTI, non-nucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; VL, viral load.
(TIF)
VL, viral load.
(TIF)
CD4, cluster of differentiation 4.
(TIF)
Changes in body composition, (a) fat mass and (b) muscle mass.
(TIF)
Changes in (a) CD4 cell count and (b) weight in children/adolescents (5–17 years) versus adults (18 years or older) at ART initiation. ART, antiretroviral therapy; CD4, cluster of differentiation 4.
(TIF)
CD8, cluster of differentiation 8.
(TIF)
IRIS, immune reconstitution inflammatory syndrome.
(TIF)
Acknowledgments
We thank all the participants and staff from all the centres participating in the REALITY trial.
The REALITY trial group consists of the following:
Participating centres: Joint Clinical Research Centre (JCRC), Kampala, Uganda (coordinating centre for Uganda): P. Mugyenyi, C. Kityo, V. Musiime, P. Wavamunno, E. Nambi, P. Ocitti, and M. Ndigendawani. JCRC, Fort Portal, Uganda: S. Kabahenda, M. Kemigisa, J. Acen, D. Olebo, G. Mpamize, A. Amone, D. Okweny, A. Mbonye, F. Nambaziira, A. Rweyora, M. Kangah, and V. Kabaswahili. JCRC, Gulu, Uganda: J. Abach, G. Abongomera, J. Omongin, I. Aciro, A. Philliam, B. Arach, E. Ocung, G. Amone, P. Miles, C. Adong, C. Tumsuiime, P. Kidega, B. Otto, and F. Apio. JCRC, Mbale, Uganda: K. Baleeta, A. Mukuye, M. Abwola, F. Ssennono, D. Baliruno, S. Tuhirwe, R. Namisi, F. Kigongo, D. Kikyonkyo, F. Mushahara, D. Okweny, J. Tusiime, A. Musiime, A. Nankya, D. Atwongyeire, S. Sirikye, S. Mula, and N. Noowe. JCRC, Mbarara, Uganda: A. Lugemwa, M. Kasozi, S. Mwebe, L. Atwine, T. Senkindu, T. Natuhurira, C. Katemba, E. Ninsiima, M. Acaku, J. Kyomuhangi, R. Ankunda, D. Tukwasibwe, and L. Ayesiga. University of Zimbabwe Clinical Research Centre, Harare, Zimbabwe: J. Hakim, K. Nathoo, M. Bwakura-Dangarembizi, A. Reid, E. Chidziva, T. Mhute, G.C. Tinago, J. Bhiri, S. Mudzingwa, M. Phiri, J. Steamer, R. Nhema, C. Warambwa, G. Musoro, S. Mutsai, B. Nemasango, C. Moyo, S. Chitongo, K. Rashirai, S. Vhembo, B. Mlambo, S. Nkomani, B. Ndemera, M. Willard, C. Berejena, Y. Musodza, P. Matiza, B. Mudenge, and V. Guti. KEMRI Wellcome Trust Research Programme, Kilifi, Kenya: A. Etyang, C. Agutu, J. Berkley, K. Maitland, P. Njuguna, S. Mwaringa, T. Etyang, K. Awuondo, S. Wale, J. Shangala, J. Kithunga, S. Mwarumba, S. Said Maitha, R. Mutai, M. Lozi Lewa, G. Mwambingu, A. Mwanzu, C. Kalama, H. Latham, J. Shikuku, A. Fondo, A. Njogu, C. Khadenge, and B. Mwakisha. Moi University Clinical Research Centre, Eldoret, Kenya: A. Siika, K. Wools-Kaloustian, W. Nyandiko, P. Cheruiyot, A. Sudoi, S. Wachira, B. Meli, M. Karoney, A. Nzioka, M. Tanui, M. Mokaya, W. Ekiru, C. Mboya, D. Mwimali, C. Mengich, J. Choge, W. Injera, K. Njenga, S. Cherutich, M. Anyango Orido, G. Omondi Lwande, P. Rutto, A. Mudogo, I. Kutto, A. Shali, L. Jaika, H. Jerotich, and M. Pierre. Department of Medicine and Malawi-Liverpool Wellcome Trust Clinical Research Programme, College of Medicine, Blantyre, Malawi: J. Mallewa, S. Kaunda, J. Van Oosterhout, B. O'Hare, R. Heydermann, C. Gonzalez, N. Dzabala, C. Kelly, B. Denis, G. Selemani, L. Nyondo Mipando, E. Chirwa, P. Banda, L. Mvula, H. Msuku, M. Ziwoya, Y. Manda, S. Nicholas, C. Masesa, T. Mwalukomo, L. Makhaza, I. Sheha, J. Bwanali, and M. Limbuni.
Trial Coordination and Oversight: MRC Clinical Trials Unit at UCL, London, UK: D. Gibb, M. Thomason, A.S. Walker, S. Pett, A. Szubert, A. Griffiths, H. Wilkes, C. Rajapakse, M. Spyer, A. Prendergast, and N. Klein. Data Management Systems: M. Rauchenberger, N. Van Looy, E. Little, and K. Fairbrother.
Social Science Group: F. Cowan, J. Seeley, S. Bernays, R. Kawuma, and Z. Mupambireyi.
Independent REALITY Trial Monitors: F. Kyomuhendo, S. Nakalanzi, J. Peshu, S. Ndaa, J. Chabuka, N. Mkandawire, L. Matandika, and C. Kapuya.
Trial Steering Committee: I. Weller (Chair), E. Malianga, C. Mwansambo, F. Miiro, P. Elyanu, E. Bukusi, E. Katabira, O. Mugurungi, D. Gibb, J. Hakim, A. Etyang, P. Mugyenyi, and J. Mallewa.
Data Monitoring Committee: T. Peto (Chair), P. Musoke, J. Matenga, and S. Phiri.
Endpoint Review Committee (independent members): H. Lyall (Cochair), V. Johnston (Cochair), F. Fitzgerald, F. Post, F. Ssali, A. Prendergast, A. Arenas-Pinto, A. Turkova, and A. Bamford. Members from MRC CTU, independent of day-to-day patient management: S.L. Pett and D.M. Gibb.
Abbreviations
- AE
adverse event
- aHR
adjusted hazard ratio
- ART
antiretroviral therapy
- CD4
cluster of differentiation 4
- ERC
Endpoint Review Committee
- GEE
generalised estimating equation
- IL-6
interleukin 6
- INSTI
integrase strand-transfer inhibitor
- IRIS
immune reconstitution inflammatory syndrome
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- NRTI
nucleoside reverse transcriptase inhibitor
- REALITY
Reduction of EArly mortaLITY
- RUSF
ready-to-use supplementary food
- SAE
serious adverse event
- SE
standard error
- VL
viral load
Data Availability
The REALITY trial data are held at MRC CTU at UCL, which encourages optimal use of data by employing a controlled access approach to data sharing, incorporating a transparent and robust system to review requests and provide secure data access consistent with the relevant ethics committee approvals. All requests for data are considered and can be initiated by contacting mrcctu.ctuenquiries@ucl.ac.uk.
Funding Statement
REALITY was funded by Joint Global Health Trials Scheme (JGHTS) of the UK Department for International Development (DFID), the Wellcome Trust, and Medical Research Council (MRC) (G1100693). Additional funding support was provided by the PENTA foundation and core support to the MRC Clinical Trials Unit at UCL (MC_UU_12023/23) (MC_UU_12023/26). Cipla, Gilead Sciences, ViiV Healthcare/GlaxoSmithKline, and Merck Sharp & Dohme donated drugs for REALITY, and Ready-to-Use-Supplementary-Food (RUSF) was purchased from Valid International. The Malawi–Liverpool–Wellcome Trust Clinical Research Programme, University of Malawi College of Medicine (101113/Z/13/Z), and the KEMRI/Wellcome Trust Research Programme, Kilifi (203077/Z/16/Z), are supported by strategic awards from the Wellcome Trust, United Kingdom. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.World Health Organisation. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach (second edition) (http://www.who.int/hiv/pub/arv/arv-2016/en/). Geneva: 2016. [PubMed]
- 2.IeDea ART Cohort Collaborations, Avila D, Althoff KN, Mugglin C, Wools-Kaloustian K, et al. Immunodeficiency at the start of combination antiretroviral therapy in low-, middle-, and high-income countries. J Acquir Immune Defic Syndr. 2014;65(1):e8–16. 10.1097/QAI.0b013e3182a39979 ; PubMed Central PMCID: PMC3894575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulle A, Schomaker M, May MT, Hogg RS, Shepherd BE, Monge S, et al. Mortality in patients with HIV-1 infection starting antiretroviral therapy in South Africa, Europe, or North America: a collaborative analysis of prospective studies. PLoS Med. 2014;11(9):e1001718 10.1371/journal.pmed.1001718 ; PubMed Central PMCID: PMC4159124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornell M, Grimsrud A, Fairall L, Fox MP, van Cutsem G, Giddy J, et al. Temporal changes in programme outcomes among adult patients initiating antiretroviral therapy across South Africa, 2002–2007. AIDS. 2010;24(14):2263–70. 10.1097/QAD.0b013e32833d45c5 ; PubMed Central PMCID: PMC2948209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker AS, Prendergast AJ, Mugyenyi P, Munderi P, Hakim J, Kekitiinwa A, et al. Mortality in the year following antiretroviral therapy initiation in HIV-infected adults and children in Uganda and Zimbabwe. Clin Infect Dis. 2012;55(12):1707–18. 10.1093/cid/cis797 ; PubMed Central PMCID: PMC3501336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta A, Nadkarni G, Yang WT, Chandrasekhar A, Gupte N, Bisson GP, et al. Early mortality in adults initiating antiretroviral therapy (ART) in low- and middle-income countries (LMIC): a systematic review and meta-analysis. PLoS ONE. 2011;6(12):e28691 10.1371/journal.pone.0028691 ; PubMed Central PMCID: PMC3248405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Churchill D, Waters L, Ahmed N, Angus B, Boffito M, Bower M, et al. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2015. HIV Med. 2016;17 Suppl 4:s2–s104. 10.1111/hiv.12426 . [DOI] [PubMed] [Google Scholar]
- 8.Department of Health and Human Services Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed 11 November 2018.
- 9.Asmuth DM, Ma ZM, Mann S, Knight TH, Yotter T, Albanese A, et al. Gastrointestinal-associated lymphoid tissue immune reconstitution in a randomized clinical trial of raltegravir versus non-nucleoside reverse transcriptase inhibitor-based regimens. AIDS. 2012;26(13):1625–34. 10.1097/QAD.0b013e3283546595 . [DOI] [PubMed] [Google Scholar]
- 10.Patterson KB, Prince HA, Stevens T, Shaheen NJ, Dellon ES, Madanick RD, et al. Differential penetration of raltegravir throughout gastrointestinal tissue: implications for eradication and cure. AIDS. 2013;27(9):1413–9. 10.1097/QAD.0b013e32835f2b49 ; PubMed Central PMCID: PMC4016763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buzon MJ, Massanella M, Llibre JM, Esteve A, Dahl V, Puertas MC, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med. 2010;16(4):460–5. 10.1038/nm.2111 . [DOI] [PubMed] [Google Scholar]
- 12.Hatano H, Strain MC, Scherzer R, Bacchetti P, Wentworth D, Hoh R, et al. Increase in 2-long terminal repeat circles and decrease in D-dimer after raltegravir intensification in patients with treated HIV infection: a randomized, placebo-controlled trial. J Infect Dis. 2013;208(9):1436–42. 10.1093/infdis/jit453 ; PubMed Central PMCID: PMC3789577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vallejo A, Gutierrez C, Hernandez-Novoa B, Diaz L, Madrid N, Abad-Fernandez M, et al. The effect of intensification with raltegravir on the HIV-1 reservoir of latently infected memory CD4 T cells in suppressed patients. AIDS. 2012;26(15):1885–94. 10.1097/QAD.0b013e3283584521 . [DOI] [PubMed] [Google Scholar]
- 14.Lake JE, McComsey GA, Hulgan T, Wanke CA, Mangili A, Walmsley SL, et al. Switch to raltegravir decreases soluble CD14 in virologically suppressed overweight women: the Women, Integrase and Fat Accumulation Trial. HIV Med. 2014;15(7):431–41. 10.1111/hiv.12128 ; PubMed Central PMCID: PMC4107004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelesidis T, Tran TT, Stein JH, Brown TT, Moser C, Ribaudo HJ, et al. Changes in Inflammation and Immune Activation With Atazanavir-, Raltegravir-, Darunavir-Based Initial Antiviral Therapy: ACTG 5260s. Clin Infect Dis. 2015;61(4):651–60. 10.1093/cid/civ327 ; PubMed Central PMCID: PMC4542595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merck Sharp & Dohme. Raltegravir Summary of Product Characteristics (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000860/WC500037405.pdf, accessed 12 October 2018) 2015.
- 17.Hakim J, Musiime V, Szubert AJ, Mallewa J, Siika A, Agutu C, et al. Enhanced Prophylaxis plus Antiretroviral Therapy for Advanced HIV Infection in Africa. N Engl J Med. 2017;377(3):233–45. 10.1056/NEJMoa1615822 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallewa J, Szubert AJ, Mugyenyi P, Chidziva E, Thomason MJ, Chepkorir P, et al. Effect of ready-to-use supplementary food on mortality in severely immunocompromised HIV-infected individuals in Africa initiating antiretroviral therapy (REALITY): an open-label, parallel-group, randomised controlled trial. Lancet HIV. 2018. 10.1016/S2352-3018(18)30038-9 ; PubMed Central PMCID: PMC5932190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.World Health Organisation. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach (http://www.who.int/hiv/pub/guidelines/arv2013/download/en/index.html). Geneva: 2013. [PubMed]
- 20.Division of AIDS of the US National Institutes of Health. Table for Grading the Severity of Adult and Pediatric Adverse Events v1.0 (https://rsc.tech-res.com/docs/default-source/safety/table_for_grading_severity_of_adult_pediatric_adverse_events.pdf?sfvrsn=6, accessed 12 October 2018). 2004.
- 21.National Institutes of Health Division of Microbiology and Infectious Diseases (DMID). Toxicity tables (https://www.niaid.nih.gov/sites/default/files/dmidpedtox.pdf, accessed 10 March 2017). 2007.
- 22.French MA, Price P, Stone SF. Immune restoration disease after antiretroviral therapy. AIDS. 2004;18(12):1615–27. [DOI] [PubMed] [Google Scholar]
- 23.Meintjes G, Lawn SD, Scano F, Maartens G, French MA, Worodria W, et al. Tuberculosis-associated immune reconstitution inflammatory syndrome: case definitions for use in resource-limited settings. Lancet Infect Dis. 2008;8:516–23. 10.1016/S1473-3099(08)70184-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raffi F, Rachlis A, Stellbrink HJ, Hardy WD, Torti C, Orkin C, et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet. 2013;381(9868):735–43. 10.1016/S0140-6736(12)61853-4 . [DOI] [PubMed] [Google Scholar]
- 25.Lawn SD, Myer L, Orrell C, Bekker LG, Wood R. Early mortality among adults accessing a community-based antiretroviral service in South Africa: implications for programme design. AIDS. 2005;19(18):2141–8. . [DOI] [PubMed] [Google Scholar]
- 26.Wilson EM, Sereti I. Immune restoration after antiretroviral therapy: the pitfalls of hasty or incomplete repairs. Immunol Rev. 2013;254(1):343–54. 10.1111/imr.12064 ; PubMed Central PMCID: PMC3694599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stirrup OT, Copas AJ, Phillips AN, Gill MJ, Geskus RB, Touloumi G, et al. Predictors of CD4 cell recovery following initiation of antiretroviral therapy among HIV-1 positive patients with well-estimated dates of seroconversion. HIV Med. 2018;19(3):184–94. 10.1111/hiv.12567 ; PubMed Central PMCID: PMC5836945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rockstroh JK, Lennox JL, Dejesus E, Saag MS, Lazzarin A, Wan H, et al. Long-term treatment with raltegravir or efavirenz combined with tenofovir/emtricitabine for treatment-naive human immunodeficiency virus-1-infected patients: 156-week results from STARTMRK. Clin Infect Dis. 2011;53(8):807–16. 10.1093/cid/cir510 . [DOI] [PubMed] [Google Scholar]
- 29.Serrano-Villar S, Zhou Y, Rodgers AJ, Moreno S. Different impact of raltegravir versus efavirenz on CD4/CD8 ratio recovery in HIV-infected patients. J Antimicrob Chemother. 2017;72(1):235–9. 10.1093/jac/dkw375 . [DOI] [PubMed] [Google Scholar]
- 30.Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5(10):e203 10.1371/journal.pmed.0050203 ; PubMed Central PMCID: PMC2570418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prendergast AJ, Szubert AJ, Berejena C, Pimundu G, Pala P, Shonhai A, et al. Baseline Inflammatory Biomarkers Identify Subgroups of HIV-Infected African Children With Differing Responses to Antiretroviral Therapy. J Infect Dis. 2016;214(2):226–36. 10.1093/infdis/jiw148 ; PubMed Central PMCID: PMC4918830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wijting IEA, Rokx C, Wit FWNM, Postma AM, Hoepelman AIM, van der Ende ME, et al. Integrase Inhibitors are an Independent Risk Factor for IRIS: an ATHENA-Cohort Study. Conference on Retroviruses and Opportunistic Infections; Seattle, WA. 13–16 February, 2017.
- 33.Dutertre M, Cuzin L, Pugliese P, Joly V, Valantin MA, Cotte L, et al. Initiation of ART based on Integrase Inhibitors increases the risk of IRIS. Conference on Retroviruses and Opportunistic Infections; Seattle, WA. 13–16 February, 2017.
- 34.Kaplan SR, Oosthuizen C, Stinson K, Little F, Euvrard J, Schomaker M, et al. Contemporary disengagement from antiretroviral therapy in Khayelitsha, South Africa: A cohort study. PLoS Med. 2017;14(11):e1002407 10.1371/journal.pmed.1002407 ; PubMed Central PMCID: PMC5675399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Post FA, Szubert AJ, Prendergast AJ, Johnston V, Lyall H, Fitzgerald F, et al. Causes and Timing of Mortality and Morbidity Among Late Presenters Starting Antiretroviral Therapy in the REALITY Trial. Clin Infect Dis. 2018;66(suppl_2):S132–S9. 10.1093/cid/cix1141 ; PubMed Central PMCID: PMC5850430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahangdale L, Cates J, Potter J, Badell ML, Seidman D, Miller ES, et al. Integrase inhibitors in late pregnancy and rapid HIV viral load reduction. Am J Obstet Gynecol. 2016;214(3):385 e1–7. 10.1016/j.ajog.2015.12.052 ; PubMed Central PMCID: PMC4995881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmona S, Bor J, Nattey C, Maughan-Brown B, Maskew M, Fox MP, et al. Persistent High Burden of Advanced HIV Disease Among Patients Seeking Care in South Africa's National HIV Program: Data From a Nationwide Laboratory Cohort. Clin Infect Dis. 2018;66(suppl_2):S111–S7. 10.1093/cid/ciy045 ; PubMed Central PMCID: PMC5850436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Late presenters working group in Cohere in EuroCoord, Mocroft A, Lundgren J, Antinori A, Monforte A, Brannstrom J, et al. Late presentation for HIV care across Europe: update from the Collaboration of Observational HIV Epidemiological Research Europe (COHERE) study, 2010 to 2013. Euro Surveill. 2015;20(47). 10.2807/1560-7917.ES.2015.20.47.30070 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)
The full trial protocol can be accessed at http://www.ctu.mrc.ac.uk/research/documents/hiv_protocols/reality_protocol [accessed 24 August 2018].
(PDF)
(DOC)
(DOC)
SAE, serious adverse event.
(DOC)
AE, adverse event.
(DOC)
IRIS, immune reconstitution inflammatory syndrome.
(DOC)
IRIS, immune reconstitution inflammatory syndrome.
(DOC)
CD4, cluster of differentiation 4.
(DOC)
(TIF)
Self-reported percentage reporting missing doses of any ART in the last 4 weeks (a) overall and (b) by backbone NRTI and NNRTI frequency. ART, antiretroviral therapy; NNRTI, non-nucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor.
(TIF)
VL suppression (a) <400 copies/mL and (b) <1,000 copies/mL. VL, viral load.
(TIF)
(TIF)
ART, antiretroviral therapy.
(TIF)
(TIF)
VL, viral load.
(TIF)
NNRTI, non-nucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; VL, viral load.
(TIF)
VL, viral load.
(TIF)
CD4, cluster of differentiation 4.
(TIF)
Changes in body composition, (a) fat mass and (b) muscle mass.
(TIF)
Changes in (a) CD4 cell count and (b) weight in children/adolescents (5–17 years) versus adults (18 years or older) at ART initiation. ART, antiretroviral therapy; CD4, cluster of differentiation 4.
(TIF)
CD8, cluster of differentiation 8.
(TIF)
IRIS, immune reconstitution inflammatory syndrome.
(TIF)
Data Availability Statement
The REALITY trial data are held at MRC CTU at UCL, which encourages optimal use of data by employing a controlled access approach to data sharing, incorporating a transparent and robust system to review requests and provide secure data access consistent with the relevant ethics committee approvals. All requests for data are considered and can be initiated by contacting mrcctu.ctuenquiries@ucl.ac.uk.