

Abstract
Women with polycystic ovary syndrome (PCOS) are often presented with hyperandrogenemia along with vascular dysfunction and elevated blood pressure. In animal models of PCOS, anti-androgen treatment decreased blood pressure, indicating a key role for androgens in the development of hypertension. However, the underlying androgen-mediated mechanism that contributes to increased blood pressure is not known. This study determined whether elevated androgens affect endothelium-derived hyperpolarizing factor (EDHF)-mediated vascular relaxation responses through alteration in function of gap junctional proteins. Female rats were implanted with placebo or dihydrotestosterone (DHT) pellets (7.5 mg, 90-day release). After 12 weeks of DHT exposure, blood pressure was assessed through carotid arterial catheter and endothelium-dependent mesenteric arterial EDHF relaxation using wire myograph. Connexin expression in mesenteric arteries was also examined. Elevated DHT significantly increased mean arterial pressure and decreased endothelium-dependent EDHF-mediated acetylcholine relaxation. Inhibition of Cx40 did not have any effect, while inhibition of Cx37 decreased EDHF relaxation to a similar magnitude in both controls and DHT females. On the other hand, inhibition of Cx43 significantly attenuated EDHF relaxation in mesenteric arteries of controls but not DHT females. Elevated DHT did not alter Cx37 or Cx40, but decreased Cx43 mRNA and protein levels in mesenteric arteries. In vitro exposure of DHT to cultured mesenteric artery smooth muscle cells dose-dependently downregulated Cx43 expression. In conclusion, increased blood pressure in hyperandrogenic females is due, at least in part, to decreased EDHF-mediated vascular relaxation responses. Decreased Cx43 expression and activity may play a role in contributing to androgen-induced decrease in EDHF function.
Keywords: testosterone, connexin, blood pressure, vascular, EDHF, mesenteric arteries
Summary Sentence
Hyperandrogenism in female rats reduced EDHF function via decrease in connexin 43 expression and activity in mesenteric arteries, providing a molecular mechanism linking elevated androgens and increased blood pressure.
Introduction
The endothelium plays a major role in control of vascular tone. This function is achieved by the release of nitric oxide (NO), prostacyclin (PGI2), and the endothelium-derived hyperpolarizing factor (EDHF) [1]. Of these autacoids, NO predominates in large conducting arteries, whereas the importance of EDHF increases as the size of the arteries decreases [2–4]. NO and PGI2 are well characterized with respect to chemistry and signaling cascades, but the nature of EDHF remains obscure [5,6]. The endothelial hyperpolarization mediated by Ca2+-activated K+ (KCa) channels, such as small and intermediate conductance KCa channels, has been suggested to play a critical role in initiating EDHF-type relaxation responses in the arteries of many species, including humans [7–10]. Gap junctional proteins, such as connexins (Cxs), play an important role in spreading this K+-channel-initiated hyperpolarization along the blood vessels, thus propagating vasodilatation or vasoconstriction [11]. In blood vessels, gap junctions allow direct coupling between adjacent endothelial cells and adjacent smooth muscle cells and can even connect endothelial to smooth muscle cells in structures corresponding to myoendothelial gap junctions. At least three Cxs are involved in the formation of vascular gap junctions: Cx37, Cx40, and Cx43 [11,12]. A close correlation between the distribution of myoendothelial gap junctions along the mesenteric arterial tree and EDHF-mediated responses has been suggested [13]. The functional role of gap junctions in EDHF-mediated responses has been confirmed in rat mesenteric artery [14–16] and in other arteries (e.g. omental arteries) [17].
Decreased generation of EDHF, which is particularly critical in resistance arteries, has been shown to contribute to impaired endothelium-dependent vasodilation in hypertension [18–25]. The expression levels of arterial Cxs are reduced in animal models of hypertension [26–29]. Studies using transgenic mice showed that Cxs exert a profound hyperpolarizing influence in resistance arteries and that suppression of Cx expression induced hypertension [30–32]. Overexpression of Cx is also shown to reverse vascular dysfunction and hypertension [33]. These observations indicate the significance of Cxs in regulating vasomotor tone and blood pressure. However, the underlying mechanism that contributes to reduction in expression and function of Cxs during hypertension remains largely unclear.
Most studies have investigated the beneficial role of sex steroid hormones, especially estradiol, on cardiovascular function. Studies have demonstrated that estradiol has beneficial vascular effects because it increases EDHF function as well as expression and activity of Cxs [34–36]. However, despite both endothelial and vascular smooth muscle cells express androgen receptors [37,38], little is known about the role of androgens. The relationship between testosterone and female cardiovascular function deserves special consideration because plasma levels of testosterone are elevated approximately 2- to 3-fold in women with polycystic ovary syndrome (PCOS) [39–41], who are at increased risk for developing vascular dysfunction and hypertension [42–44]. In animal models of PCOS, anti-androgen treatment decreased blood pressure [45,46], indicating a key role for androgens in the development of hypertension in females. However, the underlying mechanism that contributes to androgen-mediated vascular dysfunction and increased blood pressure is not known. Since EDHF is more important for vasodilation in females than males [34,47,48], we hypothesized that elevated testosterone decreases EDHF function and induces hypertension in female rats. We tested this hypothesis by administering dihydrotestosterone (DHT, a nonaromatizable form of testosterone) into female rats, mimicking the 2-fold elevation in plasma DHT levels observed in PCOS, to investigate (1) whether the systemic arterial pressure is enhanced in DHT-treated compared with control rats; (2) whether endothelium-dependent EDHF-mediated vascular relaxation, particularly in the resistance mesenteric arteries, is inhibited in DHT–treated compared with control rats; and (3) whether the DHT-induced changes in EDHF relaxation involve alterations in Cx expression and function.
Materials and methods
All experimental procedures were performed in accordance with the National Institutes of Health guidelines (NIH Publication No. 85–23, revised 2011) with approval by the Animal Care and Use Committee at the University of Texas Medical Branch. Eight-week-old female Sprague-Dawley rats (Envigo, Indianapolis, IN) were divided into two groups, and one group was implanted subcutaneously with DHT pellets (7.5 mg, 90-day release, n = 8). The other group received placebo pellets (n = 8). At the end of a 12-week treatment period, this DHT treatment regimen resulted in rats with fewer estrus cycles and higher numbers of large cystic and atretic follicles in their ovaries, as previously reported [49,50]. In these rats, mean arterial pressure (MAP) was monitored using a carotid arterial catheter. Following blood pressure measurements, the animals were sacrificed, plasma was separated, and mesenteric arteries were isolated. A portion of the mesenteric arteries was used for vascular reactivity studies, and the remaining was quickly frozen for RNA/protein analysis. Plasma DHT levels in the samples were measured using radioimmunoassay (Biovendor, Asheville, NC) as per manufacturer's instructions. The intra- and interassay variations are 5.4% and 7.5%, respectively, and the detection limit is 6 pg/mL.
Experimental procedures
Mean arterial pressure
MAP in conscious free-moving control and DHT rats was determined using indwelling carotid arterial catheters as described in our previous publications [51]. Briefly, rats under anesthesia (Isoflurane, Henry Schein Animal Health, Dublin, OH) were surgically instrumented with flexible catheters (PE 50 tubing) in the right carotid artery. The catheters were tunneled to the nape of the neck and exteriorized. After a 24-h recovery period, when the animals were fully conscious and in a free-moving state, the catheter was connected to a pressure transducer, and arterial blood pressure was obtained using a data acquisition system (DBP001 direct blood pressure system and Workbench for Windows software; both from Kent Scientific, Litchfield, CT). Following a 30-min stabilization period, the arterial pressure was monitored continuously for 30 min and averaged to determine the baseline values.
Ex vivo vascular reactivity studies
Rats were sacrificed by CO2 inhalation, and the mesenteric arcade was removed. Resistance mesenteric arteries (2-mm segments of the third-order branch of the superior mesenteric artery, 150 to 200 μm diameter) were dissected free of fat and connective tissue and mounted in a Mulvany-style isometric wire myograph (Danish Myotechnology, Aarhus, Denmark) for vessel reactivity assessment. Vessels were maintained at 37°C in a physiologic Krebs buffer of the following composition (mM): NaCl, 120; NaHCO3, 25; KCl, 4.8; NaH2PO4, 1.2; MgSO4, 1.2; dextrose, 11.0; CaCl, 1.8. They were then aerated with 95% O2 and 5% CO2 (pH = 7.4). The rings were bathed in 6 mL Krebs buffer and allowed to equilibrate for 60 min before normalizing to an internal diameter of 0.9 of L13.3kPa by using a normalization software package (Danish Myotechnology). The rings were then assessed for vascular function. Data were captured using a Power Lab data acquisition system (AD Instruments, Colorado Springs, CO). The presence of intact endotheliums in the vascular preparations was confirmed by observing the relaxation response to acetylcholine (ACh, 10−6 M) in rings precontracted with phenylephrine (PE, 10−6 M) as described previously [52]. In our preliminary experiment, the concentration–response curves for the PE-induced contractions were not different between the two groups. After the PE-induced contraction (approximately the ED80 concentration) stabilized, relaxation responses to cumulative concentrations of ACh (10−10 to 10−5 M) were elicited. The EDHF-mediated component of ACh vasorelaxation was assessed after inhibiting NO production with NG-nitro-L-arginine methyl ester (L-NAME, 10−4 M) and PGI2 synthesis with indomethacin (10−5 M) for 30 min. In some experiments, the EDHF component of ACh relaxation was generated in the presence of Cx mimetic peptides (300 μM each, 90-min preincubation) to inhibit Cx37 (40,37GAP26, sequence VCYDQAFPISHIR), Cx40 (40GAP27, sequence SRPTEKNVFIV), or Cx43 (43,37GAP27, sequence SRPTEKTIFII) [53]. These peptides and their scrambled version were custom synthesized by Biopeptide Co. Inc. (San Diego, CA) or were purchased from AnaSpec (San Jose, CA). The purity of these peptides was >95% by HPLC. Endothelium-independent relaxation responses to levcromakalim (10−9 to 10−5 M) in PE precontracted rings were also determined.
Quantitative real-time reverse transcription polymerase chain reaction
Total RNA was extracted using RNeasy mini kit (QIAGEN, Valencia, CA) according to manufacturer's instructions. RNA concentration and integrity were determined using a DS-11 spectrophotometer (DeNovix, Wilmington, DE). One microgram of total RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). After dilution, cDNA corresponding to 100 ng of RNA was amplified by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) using FAM (Invitrogen; Thermo Scientific, Grand Island, NY) as the fluorophore in a CFX96 real-time thermal cycler (Bio-Rad). PCR conditions for TaqMan Gene Expression Assay were 2 min at 50°C and 10 min at 95°C for 1 cycle, then 15 s at 95°C and 1 min at 60°C for 50 cycles. Results were calculated using the 2–ΔΔCT method and expressed in fold change of the gene of interest in treated versus control samples. All reactions were performed in duplicate, and β-actin was used as an internal control. TaqMan assays were carried out in 10 μL volumes for real-time PCR at a final concentration of 250 nM TaqMan probe and 900 nM of each primer. Cx37 (Rn00572193_s1), Cx40 (Rn00570632_m1), Cx43 (Rn01433957_m1), and β-actin (Rn00667869_m1) assays were obtained by Assay-on-Demand (Applied Biosystems; Thermo Scientific).
Western blotting
Mesenteric arteries were homogenized in ice-cold radioimmunoprecipitation assay buffer (Cell Signaling Technology, Danvers, MA) containing a protease inhibitor tablet and phosphatase inhibitor cocktail-2 and -3 (Sigma-Aldrich, St. Louis, MO). Tissue lysates were centrifuged (14 000 × g for 10 min at 4°C), and the protein content was measured using the BCA protein assay kit (Pierce; Thermo Scientific). The supernatant was resuspended in NuPAGE lithium dodecyl sulfate sample buffer and reducing agent (Invitrogen; Thermo Scientific). Proteins (30 μg) alongside Precision Plus Standard (Kaleidoscope; Bio-Rad, Hercules, CA), and negative controls were resolved on 4%–12% gradient NuPAGE Bis-Tris gels (Invitrogen) at 100 V for 2 h at room temperature and then transferred onto Immobilon-P membranes (Millipore, Billerica, MA) at 100 V for 1 h. The membranes were blocked with 5% nonfat dry milk for 1 h and then incubated overnight at 4°C with primary antibodies of Cx37, Cx40, Cx43, and β-actin. Antibody details are in Supplementary Table S1. After being washed, the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase at 1:5000 dilution and detected with the ECL detection kit (Pierce; Thermo Scientific). Densitometric measurement was done using ImageJ software. Briefly, the film was scanned using gray scale mode at 300 DPI and saved file in a TIFF format. Images were opened with ImageJ program and under gel analyzer option checked the boxes for Label with percentages and Invert peaks. Using rectangle selection tool selected each band and assigned numbers. After selecting all the bands, histograms for all the bands were generated and using line tool enclosed each peak from equal distance from the base line. Using magic wand tool, each enclosed peak was selected and through analyze tool the area under each peak was obtained which was used for further calculation in spreadsheet program and data were represented normalized with housekeeping loading control. Results were expressed as ratios of the intensity of a specific band to that of β-actin.
Immunofluorescence microscopy
Freshly isolated rat mesenteric arteries were cryopreserved in OCT compound (Tissue-Tek; Sakura Finetek, Torrance, CA) cooled by liquid nitrogen. Cryosections of transverse rings (8–10 μm thick) were prepared and mounted onto poly-l-lysine-coated slides, air-dried, and stored at −20°C. Immediately before immunostaining, sections were fixed in −20°C methanol for 10 min and then rehydrated in PBS (120 mM NaCl and 2.7 mM Na2PO4 · 2H2O, pH 7.4) for 10 min. Sections were permeabilized with PBS containing 0.1% (vol/vol) Triton X-100 for 30 min followed by blocking with PBS containing 0.5% (wt/vol) BSA for 30 min at room temperature. After blocking, sections were stained with primary antibodies against Cx43 (details on Supplementary Table S1) overnight at 4°C and then washed for 30 min at room temperature in PBS. Sections were incubated with secondary antibody (Abcam, ab97050; 1:500 dilution) conjugated to fluorescein isothiocyanate for 1 h at 37°C and then washed to remove unbound antibody. All sections were counterstained with 4΄, 6-diamidino-2-phenylindole (DAPI, Thermo Fisher) per manufacturer's instruction to visualize cell nuclei. After washing, sections were air-dried and mounted under Dako fluorescence mounting medium (Agilent Technologies, Santa Clara, CA). After drying, immunofluorescent signals were viewed in an Olympus BX60 upright compound florescence microscope. The specificity of the immunostaining was evaluated by omission of the primary antibody and processed as described above.
Vascular smooth muscle cell isolation and culture
Mesenteric artery smooth muscle cells were isolated from adult female rats as described earlier [54]. Isolated cells were assessed for their purity by α-actin staining and were found to be >95% pure. Cells were routinely cultured in DMEM containing 4.5 g/L glucose supplemented with 25 mM HEPES, 2 mM L-glutamine, 10% FBS, and antibiotics without sodium pyruvate. Cells were changed to serum and phenol red-free media 48 h prior to treatments. Cells were treated with DHT (1–100 nM) for 5 days with fresh media changes every day. After treatments, cells were washed with PBS and lysed for RNA or protein preparation.
Statistical analysis
All values are given as mean ± SEM. Differences in tensions between PE contraction and basal tension were considered as maximal tension (100%), and relaxation to ACh was expressed as the percentage of relaxation from the maximal response induced by PE. Cumulative concentration–response curves were analyzed by computer fitting to a four-parameter sigmoid curve using the Prism 6 program (GraphPad, San Diego, CA) to evaluate pD2 value (negative logarithm of the molar concentration producing the half maximum response) and Emax (maximum asymptote of the curve). For statistical comparison of single parameters, an independent t test was used. Multiple comparisons in cultured vascular smooth muscle cells were made using ANOVA with post hoc Bonferroni test. Data from two vascular rings of the same rat were averaged and presented as the datum for one rat, with the n value representing the number of rats. Statistical significance was assumed if P < 0.05.
Results
Dihydrotestosterone exposure leads to hyperandrogenism and hypertension in females
Plasma DHT levels were significantly increased in DHT females (230.4 ± 16.2 pg/mL; n = 6; P < 0.05) compared with controls (110.8 ± 11.6 pg/mL; n = 6). However, estradiol levels were comparable between control (13.1 ± 0.5 pg/mL; n = 6) and DHT (13.7 ± 0.6 pg/mL; n = 6) groups. MAP was increased significantly in DHT rats (131 ± 9.2 mm Hg; n = 6, P < 0.05) compared with control rats (105 ± 2.5 mm Hg; n = 6) (Figure 1A). There was a proportional increase in systolic and diastolic pressures in DHT females compared to controls (Figure 1B and C). Heart rate was not significantly different between control (420 ± 18.5 beats per minute; n = 6) and DHT (408 ± 6.0 beats per minute; n = 6) females (Figure 1D).
Figure 1.

MAP and heart rate in control and DHT rats. MAP (A), systolic pressure (B), diastolic pressure (C), and heart rate (D) were continuously monitored via carotid arterial catheters in rats treated with placebo or DHT pellets (7.5 mg, 90-day release, subcutaneous). Values are presented as mean ± SEM of six animals in each group. *P < 0.05 vs control.
Endothelium-derived hyperpolarizing factor-mediated relaxation in mesenteric arteries is decreased in dihydrotestosterone females
To investigate the EDHF-mediated relaxation in the rat mesenteric artery, we examined ACh-induced relaxation in the presence of L-NAME and indomethacin. The ACh-induced EDHF-mediated relaxation was significantly lower in rings from DHT rats (n = 6, P < 0.05) than in those from controls (n = 6) (Figure 2 and Table 1).
Figure 2.

Endothelium-dependent EDHF-mediated vascular relaxation in mesenteric arteries of control and DHT rats. Arterial rings were isolated from control and DHT rats, contracted with PE, and examined for relaxation to ACh in the presence of NOS inhibitor L-NAME (10−4 M) and with PGI2 blocker indomethacin (10−5 M). Values are means ± SEM (n = 6 rats, two vessel segments/rat).
Table 1.
Vascular function (pD2 and Emax values) in control and DHT rats.
| Relaxation | pD2 | Emax | ||
|---|---|---|---|---|
| Control | DHT | Control | DHT | |
| EDHF mediated | 6.96 ± 0.05 | 6.36 ± 0.09* | 86.5 ± 3.42 | 66.2 ± 5.75* |
| Cx37 inhibition | 6.42 ± 0.09⁁ | 5.71 ± 0.08*⁁ | 75.9 ± 4.01⁁ | 54.65 ± 4.83*⁁ |
| Cx40 inhibition | 6.84 ± 0.06 | 6.24 ± 0.07* | 86.1 ± 4.17 | 61.5 ± 3.81* |
| Cx43 inhibition | 5.98 ± 0.12⁁ | 6.22 ± 0.04 | 66.9 ± 3.80⁁ | 62.6 ± 2.43 |
| Levcromakalim induced | 6.18 ± 0.04 | 6.11 ± 0.03 | 98.8 ± 0.35 | 97.5 ± 0.38 |
Values are expressed as mean ± SEM of 12 mesenteric arterial rings from six rats in each group. pD2 is presented as negative logarithm of the molar concentration producing the half maximum response. Emax is presented as a percentage of PE relaxation.
*P < 0.05 compared to control.
⁁ P < 0.05 compared to EDHF-mediated relaxation in their respective control and DHT groups.
Cx43-mediated vasodilator response was reduced in mesenteric arteries of dihydrotestosterone females
To address the involvement of Cx activities in DHT-impaired mesenteric EDHF vasodilation, we examined ACh-induced EDHF relaxation in the absence or presence of specific inhibitors. To examine the contribution of Cx37 in the EDHF-mediated relaxation, rings were incubated with 37,43Gap27, a Cx37 inhibitor. Preincubation with 37,43Gap27 significantly attenuated the ACh-induced EDHF relaxation to a similar magnitude in mesenteric arteries from control and DHT rats (n = 6 in each group, Figure 3A and Table 1).
Figure 3.

Role of Cxs in EDHF-mediated vascular relaxation in mesenteric arteries from control and DHT rats. Arterial rings were contracted with PE and examined for EDHF-mediated ACh relaxation in the presence of Cx mimetic peptides to inhibit (A) Cx37 (40,37GAP26, 300 μM), (B) Cx40 (40GAP27, 300 μM), or (C) Cx43 (43,37GAP27, 300 μM). Left panel is control and right panel is DHT group. Values are means ± SEM (n = 6 rats, two vessel segments/rat).
To examine the part played by Cx40 in EDHF-mediated relaxation, rings were incubated with 40Gap27, a Cx40 inhibitor. Presence of 40Gap27 did not alter EDHF relaxation responses in the mesenteric arteries of control and DHT rats (n = 6 in each group, Figure 3B and Table 1).
To examine the contribution of Cx43 in the EDHF-mediated relaxation, rings were incubated with 43Gap27, a Cx43 inhibitor. Preincubation with 43Gap27 did not affect EDHF relaxation in DHT rats, but it significantly attenuated the EDHF relaxation in control rats (n = 6 in each group, Figure 3C and Table 1, P < 0.05). Overall, these data imply that DHT treatment does not affect Cx37 or Cx40 components of EDHF relaxation, but only inhibit the Cx43 component of relaxation response to ACh.
Endothelium-independent levcromakalim-induced relaxation response was not altered in dihydrotestosterone females
Vascular relaxation to levcromakalim, an ATP-sensitive K+ channel opener used to determine the smooth muscle vasodilating capacity [55], was not significantly different between the control and DHT rats (n = 5 in each group, Table 1).
Cx43 expression is decreased in the mesenteric arteries of dihydrotestosterone females
We next determined the expression profile of Cxs in the mesenteric arteries. As shown in Figure 4, quantitative real-time PCR shows the expression of all three Cxs in rat mesenteric arteries. Compared to control rats, vessels in DHT rats showed significantly decreased expression levels of Cx43 mRNA only (↓48%, Figure 4, P < 0.05, n = 5 in each). The expression of the Cx37 and Cx40 mRNA was not different between control and DHT rats (n = 5, Figure 4).
Figure 4.

Cxs mRNA expression in mesenteric arteries of control and DHT rats. Cxs mRNA expressions were assessed using real-time reverse transcriptase PCR. Quantitation of vascular Cxs was normalized relative to β-actin levels. Values are given as means ± SEM of five rats in each group. *P < 0.05 vs control.
Western blotting showed that Cx37 and Cx40 protein levels were unaltered, while Cx43 protein levels were significantly decreased (↓46%) in the mesenteric arteries from DHT rats (full length western blot in supplementary Figure S1) than in those from the control (n = 5 in each) (Figure 5, P < 0.05). The immunofluorescent pattern of Cx43 was identified in endothelial and smooth muscle cells of mesenteric arteries of both control and DHT groups (Figure 6, n = 4). However, a local marked decrease in the number and intensity of fluorescent spots was observed significantly in the media than endothelium of DHT compared to control rats (Figure 6, n = 4).
Figure 5.
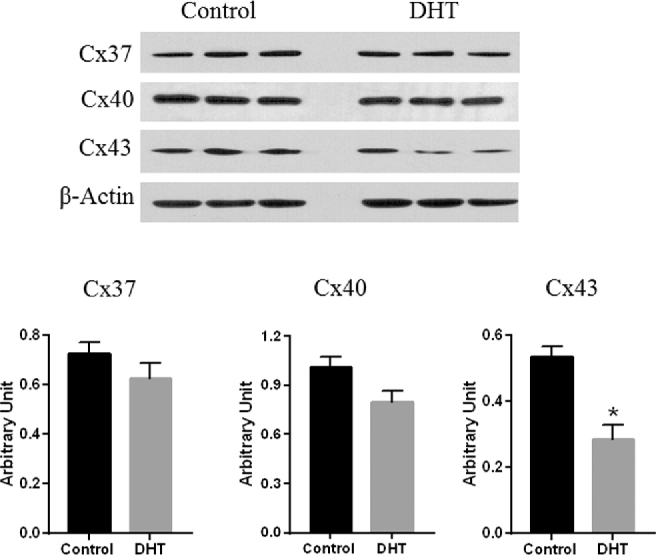
Cxs protein expression in mesenteric arteries of control and DHT rats. Cxs protein expressions were assessed using western blotting. Representative western blots for respective Cxs are shown at top; blot density obtained from densitometric scanning normalized to β-actin is shown at bottom. Values are given as means ± SEM of five rats in each group. *P < 0.05 vs control.
Figure 6.

Representative immunofluorescence of Cx43 in media and endothelium of the mesenteric artery of control and DHT rats revealed differences in the amount and intensity of immunolabeling. Original magnification of all pictures: ×200. Relative fluorescence intensity in the endothelium and vascular smooth muscle cell are presented. Values are given as means ± SEM of four rats in each group. *P < 0.05 vs control.
Dihydrotestosterone decreases Cx43 gene expression levels in cultured mesenteric artery smooth muscle cells
Based on the effects of hyperandrogenism on Cx43 expression and relaxation in mesenteric arteries, we next assessed the direct effects of DHT stimulation on Cx43 expression using an in vitro cell culture model. Cx43 mRNA levels in cultured mesenteric artery smooth muscle cells were dose-dependently decreased by DHT treatment (Figure 7A, P < 0.05, n = 4). DHT treatment also significantly decreased Cx43 protein levels (Figure 7B, P < 0.05, n = 4), indicating that androgens directly decrease Cx43 expression.
Figure 7.

DHT downregulates Cx43 expression in cultured mesenteric artery smooth muscle (MASM) cells. Concentration-dependent DHT-induced decrease in Cx43 (A) mRNA, and (B) protein in cultured MASM cells. The primary MASM cells were treated with vehicle or DHT for 5 days with fresh medium along with DHT replaced every day. Cell extracts were prepared and subjected to Cx43 expression analysis using qRT-PCR and western blot. Quantitation of vascular Cx43 mRNA and protein expression was normalized relative to β-actin. Data represent mean of four independent experiments. *P < 0.05 vs controls.
Discussion
The major finding of this study is the identification of a novel androgen-mediated mechanism that controls the expression of Cx43 in mesenteric artery smooth muscle cells by negatively regulating Cx43 transcript and protein levels. Regulation of Cx43 expression by androgens has significant functional consequences, as we determined that androgens exert significant control by blunting EDHF-mediated endothelial relaxation and inducing hypertension. Therefore, we suggest that decreases in vascular Cx43-stimulated responses may mediate the development and maintenance of hypertension induced by hyperandrogenism in adult females.
The model was designed to create a physiological state of hyperandrogenism that mimics androgen levels observed in women with PCOS. DHT, a nonaromatizable androgen, was selected to avoid the confounding effects of estrogen. DHT treatment increased plasma DHT levels by 2-fold compared to controls, which is consistent with the available data that DHT levels remain relatively stable across the menstrual cycle and are approximately 2-fold higher in women with PCOS compared with those without PCOS (140–160 pg/mL vs 70–80 pg/mL, respectively) [56,57].
The DHT-treated rats have higher blood pressure compared to control rats, which is consistent with the reports of increased blood pressure in hyperandrogenic PCOS women [42–44] and suggests that the mechanisms controlling blood pressure in adult females are perturbed by elevated androgens. The effect on arterial pressure increase is without any changes in heart rate, indicating that DHT does not affect the sympathetic activity.
Kidneys play an important role in long-term control of blood pressure [58]. To dissect out the effect of DHT on vascular function, we examined EDHF function in isolated mesenteric arteries. The current study shows that EDHF contributes substantially to mesenteric arterial relaxation, supporting earlier findings [59,60]. The most striking finding of our study is that EDHF-mediated mesenteric vasodilation was significantly decreased in DHT rats compared with controls, suggesting that elevated androgens blunt endothelial EDHF function. It has been shown that EDHF plays a more important role in vasodilation in females than in males [61–63], and thus suppression of such a critical regulator of vascular tone by androgens could contribute to increased blood pressure in DHT females. Mesenteric vasorelaxation to levcromakalim was not different between control and DHT females, suggesting that it is not the smooth muscle vasodilating capability that is reduced in DHT females, but some function related to the initiation and propagation of hyperpolarization signals.
In the systemic circulation, the principal EDHF components that mediate initiation and propagation of hyperpolarization responses and vasodilation are Cx37, Cx40, and Cx43. We analyzed the effect of DHT on the EDHF function with the specific role of Cx37, Cx40, and Cx43. The current study shows that Cx37 contributes substantially to mesenteric arterial relaxation as in earlier studies [64–66], but the presence of elevated DHT does not affect Cx37-mediated relaxation. Consistently, elevated DHT did not alter Cx37 mRNA and protein levels. Studies have shown that Cx40 plays an important role in mediating vascular relaxation in mesenteric artery of male Wistar rats [67]; however, its contribution in the mesenteric arterial relaxation in females, and in general to systemic blood pressure, is minimal [34,68.69], which is consistent with our findings. Furthermore, we find that the Cx40-mediated relaxation was not affected by the presence of elevated DHT. This observation is also supported by unchanged Cx40 mRNA and protein levels in the mesenteric arteries of control and DHT rats.
The Cx43 contributed to EDHF relaxation in controls, and its lack of effect on EDHF relaxation in mesenteric arteries of DHT rats suggests that androgen exposure resulted in a loss of the regulatory role of Cx43 in initiating or propagating hyperpolarization. It has been suggested that in females there is a relative predominance of Cx43-dependent vasodilation [34,47,48] that may increase its susceptibility to the effect of elevated androgens. Our data also suggest that the loss of Cx43’s regulatory role in mesenteric arteries of DHT rats resulted chiefly from reduced gap junctional activities due to suppressed expression of this Cx. At present, the mechanisms by which androgens downregulate expression and function of Cx43 in mesenteric arteries specifically in the vascular smooth muscle cells of DHT rats are not clear. The finding that DHT exposure to cultured mesenteric artery smooth muscle cells downregulated Cx43 mRNA suggests that Cx43 is a physiological target for DHT and that androgens can directly regulate Cx43 expression, possibly at the transcriptional level. This ability of androgens to suppress Cx43 expression and activity could contribute to lower levels of Cx43 in male than female organs, like in the heart [70]. This notion is supported by the fact that castration induced increased Cx43 mRNA and protein levels in rat prostate tissue which was abolished by androgen replacement [71]. Further studies are needed to examine the exact mechanism by which androgen regulates Cx43 transcription. It is important to emphasize the following cautionary remarks regarding the aforementioned interpretations. First, although we hypothesize that the decreased Cx43 vascular function in DHT rats could contribute to the observed increase in arterial pressure; further studies using genetic approaches such as the Cx43 knockout mice will be required to demonstrate a causal role for Cx43. Second, although widely used, the pharmacological specificity of Cx mimetic peptides should be carefully examined in future studies. The failure to detect a role for Cx37 and Cx40 could be due to the delivery or pharmokinetic issues of Cx37 and Cx40 mimetic peptides, pointing again for the need for genetic approaches.
In conclusion, this study demonstrates that elevated levels of androgens at concentrations found in clinical conditions induce reduced EDHF-mediated vasodilation and hypertension in females rats, possibly via downregulating Cx43-mediated signaling, providing a molecular mechanism linking elevated androgens and increased risk of hypertension in females. Although caution should be always observed in extrapolating the findings of animal studies directly to humans, the present finding has translational potential and provides a mechanistic understanding worthy of investigation in humans. Strategies targeting attenuation of excessive androgen or upregulation of Cx43 action in the systemic circulation could have important therapeutic potential in treatment of hypertension complicated by elevated androgens.
Supplementary data
Supplementary data are available at BIOLRE online.
Supplementary Table S1. Antibodies used for Western blot and immunofluorescence studies.
Supplementary Figure S1. Full-length western blot showing Cxs protein expression in mesenteric arteries of control and DHT rats.
Supplementary data are available at BIOLRE online.
References
- 1. Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev 2001; 81:1415–1459. [DOI] [PubMed] [Google Scholar]
- 2. Stankevicius E, Dalsgaard T, Kroigaard C, Beck L, Boedtkjer E, Misfeldt MW, Nielsen G, Schjorring O, Hughes A, Simonsen U. Opening of small and intermediate calcium-activated potassium channels induces relaxation mainly mediated by nitric-oxide release in large arteries and endothelium-derived hyperpolarizing factor in small arteries from rat. J Pharmacol Exp Ther 2011; 339:842–850. [DOI] [PubMed] [Google Scholar]
- 3. Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol 2006; 291:H985–1002. [DOI] [PubMed] [Google Scholar]
- 4. Hilgers RH, Todd J Jr, Webb RC. Regional heterogeneity in acetylcholine-induced relaxation in rat vascular bed: role of calcium-activated K+ channels. Am J Physiol Heart Circ Physiol 2006; 291:H216–H222. [DOI] [PubMed] [Google Scholar]
- 5. Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol 2004; 141:881–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de WC, Griffith TM. Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers Arch 2010; 459:897–914. [DOI] [PubMed] [Google Scholar]
- 7. Crane GJ, Gallagher N, Dora KA, Garland CJ. Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol 2003; 553:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, Hoyer J, Kohler R. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br J Pharmacol 2003; 138:594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feletou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond) 2009; 117:139–155. [DOI] [PubMed] [Google Scholar]
- 10. Hinton JM, Langton PD. Inhibition of EDHF by two new combinations of K+-channel inhibitors in rat isolated mesenteric arteries. Br J Pharmacol 2003; 138:1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Christ GJ, Spray DC, el-Sabban M, Moore LK, Brink PR. Gap junctions in vascular tissues. Evaluating the role of intercellular communication in the modulation of vasomotor tone. Circ Res 1996; 79:631–646. [DOI] [PubMed] [Google Scholar]
- 12. Hill CE, Rummery N, Hickey H, Sandow SL. Heterogeneity in the distribution of vascular gap junctions and connexins: implications for function. Clin Exp Pharmacol Physiol 2002; 29:620–625. [DOI] [PubMed] [Google Scholar]
- 13. Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res 2000; 86:341–346. [DOI] [PubMed] [Google Scholar]
- 14. Edwards G, Feletou M, Gardener MJ, Thollon C, Vanhoutte PM, Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br J Pharmacol 1999; 128:1788–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ Res 2002; 90:1108–1113. [DOI] [PubMed] [Google Scholar]
- 16. Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res 2005; 97:399–407. [DOI] [PubMed] [Google Scholar]
- 17. Urakami-Harasawa L, Shimokawa H, Nakashima M, Egashira K, Takeshita A. Importance of endothelium-derived hyperpolarizing factor in human arteries. J Clin Invest 1997; 100:2793–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gray C, Li M, Reynolds CM, Vickers MH. Pre-weaning growth hormone treatment reverses hypertension and endothelial dysfunction in adult male offspring of mothers undernourished during pregnancy. PLoS One 2013; 8:e53505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chinnathambi V, Yallampalli C, Sathishkumar K. Prenatal testosterone induces sex-specific dysfunction in endothelium-dependent relaxation pathways in adult male and female rats. Biol Reprod 2013; 89:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weston AH, Porter EL, Harno E, Edwards G. Impairment of endothelial SK(Ca) channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br J Pharmacol 2010; 160:836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goto K, Fujii K, Kansui Y, Iida M. Changes in endothelium-derived hyperpolarizing factor in hypertension and ageing: response to chronic treatment with renin-angiotensin system inhibitors. Clin Exp Pharmacol Physiol 2004; 31:650–655. [DOI] [PubMed] [Google Scholar]
- 22. Yang Z, Kaye DM. Endothelial dysfunction and impaired L-arginine transport in hypertension and genetically predisposed normotensive subjects. Trends Cardiovasc Med 2006; 16:118–124. [DOI] [PubMed] [Google Scholar]
- 23. Dal-Ros S, Bronner C, Schott C, Kane MO, Chataigneau M, Schini-Kerth VB, Chataigneau T. Angiotensin II-induced hypertension is associated with a selective inhibition of endothelium-derived hyperpolarizing factor-mediated responses in the rat mesenteric artery. J Pharmacol Exp Ther 2009; 328:478–486. [DOI] [PubMed] [Google Scholar]
- 24. Hilgers RH, Webb RC. Reduced expression of SKCa and IKCa channel proteins in rat small mesenteric arteries during angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 2007; 292:H2275–H2284. [DOI] [PubMed] [Google Scholar]
- 25. Torrens C, Brawley L, Anthony FW, Dance CS, Dunn R, Jackson AA, Poston L, Hanson MA. Folate supplementation during pregnancy improves offspring cardiovascular dysfunction induced by protein restriction. Hypertension 2006; 47:982–987. [DOI] [PubMed] [Google Scholar]
- 26. Yeh HI, Lee PY, Su CH, Tian TY, Ko YS, Tsai CH. Reduced expression of endothelial connexins 43 and 37 in hypertensive rats is rectified after 7-day carvedilol treatment. Am J Hypertens 2006; 19:129–135. [DOI] [PubMed] [Google Scholar]
- 27. Kansui Y, Fujii K, Nakamura K, Goto K, Oniki H, Abe I, Shibata Y, Iida M. Angiotensin II receptor blockade corrects altered expression of gap junctions in vascular endothelial cells from hypertensive rats. Am J Physiol Heart Circ Physiol 2004; 287:H216–H224. [DOI] [PubMed] [Google Scholar]
- 28. Haefliger JA, Castillo E, Waeber G, Aubert JF, Nicod P, Waeber B, Meda P. Hypertension differentially affects the expression of the gap junction protein connexin43 in cardiac myocytes and aortic smooth muscle cells. Adv Exp Med Biol 1997; 432:71–82. [DOI] [PubMed] [Google Scholar]
- 29. Haefliger JA, Meda P, Formenton A, Wiesel P, Zanchi A, Brunner HR, Nicod P, Hayoz D. Aortic connexin43 is decreased during hypertension induced by inhibition of nitric oxide synthase. Arterioscler Thromb Vasc Biol 1999; 19:1615–1622. [DOI] [PubMed] [Google Scholar]
- 30. de WC, Roos F, Bolz SS, Kirchhoff S, Kruger O, Willecke K, Pohl U. Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ Res 2000; 86:649–655. [DOI] [PubMed] [Google Scholar]
- 31. de WC, Roos F, Bolz SS, Pohl U. Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol Genomics 2003; 13:169–177. [DOI] [PubMed] [Google Scholar]
- 32. Gerl M, Vockl J, Kurt B, van Veen TA, Kurtz A, Wagner C. Inducible deletion of connexin 40 in adult mice causes hypertension and disrupts pressure control of renin secretion. Kidney Int 2015; 87:557–563. [DOI] [PubMed] [Google Scholar]
- 33. Tsang H, Leiper J, Hou LK, Dowsett L, Delahaye MW, Barnes G, Wharton J, Howard L, Iannone L, Lang NN, Wilkins MR, Wojciak-Stothard B. Role of asymmetric methylarginine and connexin 43 in the regulation of pulmonary endothelial function. Pulm Circ 2013; 3:675–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu MY, Hattori Y, Sato A, Ichikawa R, Zhang XH, Sakuma I. Ovariectomy attenuates hyperpolarization and relaxation mediated by endothelium-derived hyperpolarizing factor in female rat mesenteric artery: a concomitant decrease in connexin-43 expression. J Cardiovasc Pharmacol 2002; 40:938–948. [DOI] [PubMed] [Google Scholar]
- 35. Liu MY, Hattori Y, Fukao M, Sato A, Sakuma I, Kanno M. Alterations in EDHF-mediated hyperpolarization and relaxation in mesenteric arteries of female rats in long-term deficiency of oestrogen and during oestrus cycle. Br J Pharmacol 2001; 132:1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sakuma I, Liu MY, Sato A, Hayashi T, Iguchi A, Kitabatake A, Hattori Y. Endothelium-dependent hyperpolarization and relaxation in mesenteric arteries of middle-aged rats: influence of oestrogen. Br J Pharmacol 2002; 135:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Torres-Estay V, Carreno DV, San Francisco IF, Sotomayor P, Godoy AS, Smith GJ. Androgen receptor in human endothelial cells. J Endocrinol 2015; 224:R131–R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Higashiura K, Mathur RS, Halushka PV. Gender-related differences in androgen regulation of thromboxane A2 receptors in rat aortic smooth-muscle cells. J Cardiovasc Pharmacol 1997; 29:311–315. [DOI] [PubMed] [Google Scholar]
- 39. Legro RS, Schlaff WD, Diamond MP, Coutifaris C, Casson PR, Brzyski RG, Christman GM, Trussell JC, Krawetz SA, Snyder PJ, Ohl D, Carson SA et al. Total testosterone assays in women with polycystic ovary syndrome: precision and correlation with hirsutism. J Clin Endocrinol Metab 2010; 95:5305–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nelson VL, Legro RS, Strauss JF III, McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol 1999; 13:946–957. [DOI] [PubMed] [Google Scholar]
- 41. Yildizhan B, Anik IG, Pekin T. The impact of insulin resistance on clinical, hormonal and metabolic parameters in lean women with polycystic ovary syndrome. J Obstet Gynaecol 2016; 36:893–896. [DOI] [PubMed] [Google Scholar]
- 42. Reckelhoff JF. Polycystic ovary syndrome: androgens and hypertension. Hypertension 2007; 49:1220–1221. [DOI] [PubMed] [Google Scholar]
- 43. Chen MJ, Yang WS, Yang JH, Chen CL, Ho HN, Yang YS. Relationship between androgen levels and blood pressure in young women with polycystic ovary syndrome. Hypertension 2007; 49:1442–1447. [DOI] [PubMed] [Google Scholar]
- 44. Bentley-Lewis R, Seely E, Dunaif A. Ovarian hypertension: polycystic ovary syndrome. Endocrinol Metab Clin North Am 2011; 40:433–43x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blesson CS, Chinnathambi V, Hankins GD, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure induces hypertension in adult females via androgen receptor-dependent protein kinase Cdelta-mediated mechanism. Hypertension 2014; 65:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hurliman A, Keller BJ, Maille N, Mandala M, Casson P, Osol G. Hyperandrogenism and insulin resistance, not changes in body weight, mediate the development of endothelial dysfunction in a female rat model of polycystic ovary syndrome (PCOS). Endocrinology 2015; 156:4071–4080. [DOI] [PubMed] [Google Scholar]
- 47. Lang NN, Luksha L, Newby DE, Kublickiene K. Connexin 43 mediates endothelium-derived hyperpolarizing factor-induced vasodilatation in subcutaneous resistance arteries from healthy pregnant women. Am J Physiol Heart Circ Physiol 2007; 292:H1026–H1032. [DOI] [PubMed] [Google Scholar]
- 48. Tribulova N, Dupont E, Soukup T, Okruhlicova L, Severs NJ. Sex differences in connexin-43 expression in left ventricles of aging rats. Physiol Res 2005; 54:705–708. [PubMed] [Google Scholar]
- 49. Osuka S, Iwase A, Nakahara T, Kondo M, Saito A, Bayasula Nakamura T, Takikawa S, Goto M, Kotani T, Kikkawa F. Kisspeptin in the hypothalamus of two rat models of polycystic ovary syndrome. Endocrinology 2017; 158:367–377. [DOI] [PubMed] [Google Scholar]
- 50. Wang H, Wang X, Zhu Y, Chen F, Sun Y, Han X. Increased androgen levels in rats impair glucose-stimulated insulin secretion through disruption of pancreatic beta cell mitochondrial function. J Steroid Biochem Mol Biol 2015; 154:254–266. [DOI] [PubMed] [Google Scholar]
- 51. Sathishkumar K, Elkins R, Yallampalli U, Yallampalli C. Protein restriction during pregnancy induces hypertension in adult female rat offspring - influence of oestradiol. Br J Nutr 2012; 107:665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sathishkumar K, Elkins R, Yallampalli U, Balakrishnan M, Yallampalli C. Fetal programming of adult hypertension in female rat offspring exposed to androgens in utero. Early Hum Dev 2011; 87:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yi FX, Boeldt DS, Gifford SM, Sullivan JA, Grummer MA, Magness RR, Bird IM. Pregnancy enhances sustained Ca2+ bursts and endothelial nitric oxide synthase activation in ovine uterine artery endothelial cells through increased connexin 43 function. Biol Reprod 2010; 82:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sathishkumar K, Yallampalli U, Elkins R, Yallampalli C. Raf-1 kinase regulates smooth muscle contraction in the rat mesenteric arteries. J Vasc Res 2010; 47:384–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest 2000; 106:1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fassnacht M, Schlenz N, Schneider SB, Wudy SA, Allolio B, Arlt W. Beyond adrenal and ovarian androgen generation: increased peripheral 5 alpha-reductase activity in women with polycystic ovary syndrome. J Clin Endocrinol Metab 2003; 88:2760–2766. [DOI] [PubMed] [Google Scholar]
- 57. Silfen ME, Denburg MR, Manibo AM, Lobo RA, Jaffe R, Ferin M, Levine LS, Oberfield SE. Early endocrine, metabolic, and sonographic characteristics of polycystic ovary syndrome (PCOS): comparison between nonobese and obese adolescents. J Clin Endocrinol Metab 2003; 88:4682–4688. [DOI] [PubMed] [Google Scholar]
- 58. Crowley SD, Coffman TM. In hypertension, the kidney rules. Curr Hypertens Rep 2007; 9:148–153. [DOI] [PubMed] [Google Scholar]
- 59. Zygmunt PM, Ryman T, Hogestatt ED. Regional differences in endothelium-dependent relaxation in the rat: contribution of nitric oxide and nitric oxide-independent mechanisms. Acta Physiol Scand 1995; 155:257–266. [DOI] [PubMed] [Google Scholar]
- 60. Plane F, Holland M, Waldron GJ, Garland CJ, Boyle JP. Evidence that anandamide and EDHF act via different mechanisms in rat isolated mesenteric arteries. Br J Pharmacol 1997; 121:1509–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McCulloch AI, Randall MD. Sex differences in the relative contributions of nitric oxide and EDHF to agonist-stimulated endothelium-dependent relaxations in the rat isolated mesenteric arterial bed. Br J Pharmacol 1998; 123:1700–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wangensteen R, Moreno JM, Sainz J, Rodriguez-Gomez I, Chamorro V, Luna JD, Osuna A, Vargas F. Gender difference in the role of endothelium-derived relaxing factors modulating renal vascular reactivity. Eur J Pharmacol 2004; 486:281–288. [DOI] [PubMed] [Google Scholar]
- 63. White RM, Rivera CO, Davison CA. Nitric oxide-dependent and -independent mechanisms account for gender differences in vasodilation to acetylcholine. J Pharmacol Exp Ther 2000; 292:375–380. [PubMed] [Google Scholar]
- 64. Earley S, Resta TC, Walker BR. Disruption of smooth muscle gap junctions attenuates myogenic vasoconstriction of mesenteric resistance arteries. Am J Physiol Heart Circ Physiol 2004; 287:H2677–H2686. [DOI] [PubMed] [Google Scholar]
- 65. Chaytor AT, Bakker LM, Edwards DH, Griffith TM. Connexin-mimetic peptides dissociate electrotonic EDHF-type signalling via myoendothelial and smooth muscle gap junctions in the rabbit iliac artery. Br J Pharmacol 2005; 144:108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Haddock RE, Grayson TH, Brackenbury TD, Meaney KR, Neylon CB, Sandow SL, Hill CE. Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40. Am J Physiol Heart Circ Physiol 2006; 291:H2047–H2056. [DOI] [PubMed] [Google Scholar]
- 67. Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res 2005; 97:399–407. [DOI] [PubMed] [Google Scholar]
- 68. Halidi N, Alonso F, Burt JM, Beny JL, Haefliger JA, Meister JJ. Intercellular calcium waves in primary cultured rat mesenteric smooth muscle cells are mediated by connexin43. Cell Commun Adhes 2012; 19:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Alonso F, Krattinger N, Mazzolai L, Simon A, Waeber G, Meda P, Haefliger JA. An angiotensin II- and NF-kappaB-dependent mechanism increases connexin 43 in murine arteries targeted by renin-dependent hypertension. Cardiovasc Res 2010; 87:166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stauffer BL, Sobus RD, Sucharov CC. Sex differences in cardiomyocyte connexin43 expression. J Cardiovasc Pharmacol 2011; 58:32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Huynh HT, Alpert L, Laird DW, Batist G, Chalifour L, Alaoui-Jamali MA. Regulation of the gap junction connexin 43 gene by androgens in the prostate. J Mol Endocrinol 2001; 26:1–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available at BIOLRE online.