

Abstract
Aims
TMEM100 was previously identified as a downstream target of activin receptor-like kinase 1 (ALK1; ACVRL1) signalling. Mutations on ALK1 cause hereditary haemorrhagic telangiectasia (HHT), a vascular disorder characterized by mucocutaneous telangiectases and visceral arteriovenous malformations (AVMs). The aims of this study are to investigate the in vivo role of TMEM100 at various developmental and adult stages and to determine the extent to which TMEM100 contributed to the development of AVMs as a key downstream effector of ALK1.
Methods and results
Blood vasculature in Tmem100-null embryos and inducible Tmem100-null neonatal and adult mice was examined. We found that TMEM100 deficiency resulted in cardiovascular defects at embryonic stage; dilated vessels, hyperbranching, and increased number of filopodia in the retinal vasculature at neonatal stage; and various vascular abnormalities, including internal haemorrhage, arteriovenous shunts, and weakening of vasculature with abnormal elastin layers at adult stage. However, arteriovenous shunts in adult mutant mice appeared to be underdeveloped without typical tortuosity of vessels associated with AVMs. We uncovered that the expression of genes encoding cell adhesion and extracellular matrix proteins was significantly affected in lungs of adult mutant mice. Especially Mfap4, which is associated with elastin fibre formation, was mostly down-regulated.
Conclusion
These results demonstrate that TMEM100 has essential functions for the maintenance of vascular integrity as well as the formation of blood vessels. Our results also indicate that down-regulation of Tmem100 is not the central mechanism of HHT pathogenesis, but it may contribute to the development of vascular pathology of HHT by weakening vascular integrity.
Keywords: ALK1 (ACVRL1), Hereditary haemorrhagic telangiectasia, MFAP4, TMEM100, Vascular integrity
1. Introduction
Transmembrane protein 100 (TMEM100) contains two predicted transmembrane domains and is well conserved in vertebrates.1 We have previously proposed a hypothesis that TMEM100 is a potential downstream effector of activin receptor-like kinase 1 (ALK1; ACVRL1) signalling pertinent to hereditary haemorrhagic telangiectasia (HHT) pathogenesis,1 as Tmem100 mRNA was decreased in ALK1-deficient lungs, and Alk1 and Tmem100 expressions were largely overlapping in endothelial cells of major arteries during embryogenesis.1–3 Recently, Somekawa et al.4 further substantiated this hypothesis by demonstrating that TMEM100 is regulated by bone morphogenetic protein 9 (BMP9)-ALK1 signalling, and Tmem100-null embryos die at around E11.5 with a phenocopy of Alk1-null embryos.5,6 Mutations in components of TGF-β signalling, ALK1, endoglin (ENG), and SMAD4 cause HHT, characterized by recurrent epistaxis, mucocutaneous telangiectases, and arteriovenous malformations (AVMs) in various organs, such as the brain, lung, and liver.7,8 The fundamental pathology of this malady is direct connections between arteries and veins without intervening capillaries. More than 90% of HHT patients develop telangiectasia (small AVMs) in mucocutaneous layers of skin and in gastrointestinal tract. These telangiectases are mostly acquired during post-developmental stages, and the frequency and severity increase by age.9
We have previously shown that de novo mucocutaneous AVM development requires a secondary event such as wounding in addition to ALK1 deficiency.10 Currently available therapeutic approaches for nasal telangiectases are anti-angiogenic drugs such as bevacizumab, which directs to inhibiting the ‘secondary events’, e.g. angiogenesis.11,12 However, no therapy has ever developed to overcome the ALK1- or ENG deficiency. This is largely due to the paucity of data regarding the downstream effectors of BMP9-ALK1 signalling. Although the evidence has been clearly presented that TMEM100 is a downstream target of ALK1 signalling in vitro and in vivo at embryonic stage, it remains unknown whether down-regulation of TMEM100 is the central mechanism of AVM development caused by ALK1 deficiency. If proved, TMEM100 can be a potential target for developing drugs for HHT. To further test this hypothesis and to elucidate the role of TMEM100 in maintenance of adult vasculature, we examined the effects of Tmem100 deletion in neonatal and adult mice using inducible knockout (iKO) approach and compared vascular phenotypes of Tmem100-iKO with those of Alk1-iKO.
2. Methods
2.1. Mouse strains
Generation of Tmem100- and Alk1-conditional knockout and reporter mice was described previously.1,13 ROSA26CreER mice were purchased from The Jackson Laboratory. Deletion of Tmem100 using ROSA26CreER was achieved by three consecutive intragastric injections of 50 µg of tamoxifen (Sigma) at postnatal day (P)1, P2, and P3 or two consecutive intraperitoneal injections of tamoxifen at 0.1 mg/g of body weight to 6-week-old mice. Deletion of Alk1 in adult mice was achieved by one time tamoxifen injection (0.1 mg/g b.w., i.p.). Mice were maintained under standard specific pathogen-free conditions, and all animal procedures performed were reviewed and approved by the Institutional Animal Care and Use Committees of Gachon University (approval reference number, LCDI-2012-0074; LCDI-2014-0019) and University of Florida (UF 201101417). All animal procedures conformed to the NIH guidelines (Guide for the care and use of laboratory animals). Mice were on a mixed genetic background of C57BL/6J and 129SvJ.
2.2. X-gal staining and histology
Pregnant female mice for the isolation of the embryos were euthanized by CO2 asphyxiation. A full-thickness skin wound was made on the shaved back of an anaesthetized mouse by inhalation of isoflurane (4%) using a disposable biopsy punch 7 days before X-gal staining. The mouse was euthanized by CO2 asphyxiation, and back skin was removed. X-gal staining was carried out to detect LacZ reporter activity as described previously.14 Briefly, collected embryos, skin, and organs were fixed in fixing solution [1% formaldehyde, 0.2% glutaraldehyde, 2 mM MgCl2, 5 mM EGTA, 0.02% NP-40 in PBS], rinsed three times with PBS, and stained in X-gal staining solution [5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl2, 0.01% deoxycholate sodium salt, 0.02% NP-40, 0.75 mg/mL X-gal in 100 mM phosphate buffer (pH 7.3)] at 37°C overnight. Stained organs were post-fixed in post-fixing solution (4% paraformaldehyde, 0.1% Tween 20 in PBS) at 4°C overnight. X-gal stained organs were either cleared in benzyl benzoate : benzyl alcohol (1 : 1) solution for whole-mount imaging or embedded in paraffin. The embedded organs were cut into 7 µm slices and subjected to haematoxylin or nuclear fast red staining. Masson's trichrome staining was performed on paraffin-embedded sections with a Trichrome Stain (Masson) Kit (Sigma) according to the manufacturer's instruction. Elastin layers were stained by Hart's stain method.
2.3. Immunolabelling and quantitative analyses of retina vasculature
Retinas of tamoxifen-treated Tmem1002loxP/2loxP (control) and Tmem1002loxP/2loxP;ROSA26+/CreER mutant (Tmem100-iKO) newborn mice at postnatal day (P)8 were isolated as described.15 Mice were euthanized by anaesthesia with ketamine/xylazine (100/15 mg/kg b.w., i.p.) followed by bilateral thoracotomy. Isolated retina was divided into four quadrants by deep radial incisions and then fixed in 4% paraformaldehyde/PBS for 2 h at room temperature. After fixation, retinas were blocked with ice-cold blocking buffer (1% BSA, 0.5% Triton X-100 in PBS) overnight at 4°C and washed with Pblec solution (1 mM MgCl2, 1 mM CaCl2, 0.1 mM MnCl2, 1% TritonX-100 in PBS) three times. Blood vessels in the retina were labelled with FITC-conjugated isolectin B4 (L2895; Sigma, 20 µg/mL) in Pblec buffer overnight at 4°C. The samples were washed three times with PBS for 30 min and then flat-mounted with Simpo-Mount (GBI Labs). Fluorescence images were captured using an SZX7 fluorescent stereomicroscope (Olympus) and an LSM 710 confocal microscope (Zeiss). The progression of the vascular plexus towards the edge of a quadrant of the retina was measured by the ratio of the vascular radius and the quadrant's radius. The numbers of branch points (capillary junction numbers) and filopodial protrusions (sprout numbers) were counted using Image J program within a microscopic field at ×200 and ×100 magnification, respectively. The covered area by blood vessels was determined with VesSeg-Tool software (http://www.isip.uni-luebeck.de/index.php?id=150&L=0). Vascular density was represented by the ratio of the covered area by blood vessels and the area of the vascular plexus region.
2.4. India ink and latex dye injections
India ink and latex dye injections are routinely used to visualize vasculature and detect vascular leakage. For India ink injection, pregnant females for the isolation of the embryos were euthanized by CO2 asphyxiation, and E10.5 embryos were collected and anaesthetized on ice, and a 50% solution of India ink in PBS was injected intracardially using drawn glass pipettes. The latex dye is especially useful to distinguish AV shunts, because the latex dye cannot pass through the capillary network and retained within arteries in the normal vasculature. After tamoxifen treatment, control and Tmem100-iKO mutant mice were subjected to the latex dye injection when the mutant mice were curled up sick. Adult mice were anaesthetized with intraperitoneal injection of ketamine/xylazine (100/15 mg per 1 kg body weight), and the thoracic and abdominal cavities were opened. For pulmonary injection of the latex dye, the left atrium was cut and 10 mL of heparin/vasodilator mix (200 U/mL of heparin, 40 ng/mL of papaverine, 100 µM of sodium nitroprusside in PBS) was injected into the right ventricle using syringe pump at a rate of 0.5 mL/min. Pulmonary vessels were fixed with 10 mL of 10% formalin and then the latex dye (Blue latex, catalogue BR80B; Connecticut Valley Biological Supply Co.) was injected slowly and steadily with a 1 mL syringe. For systemic injection of the latex dye, the right atrium was cut and latex dye was injected into the left ventricle. The dye-injected mice were washed briefly with PBS and fixed in 10% formalin overnight. Organs and tissues corrected from the dye-injected mice were dehydrated in methanol series, cleared in benzyl benzoate : benzyl alcohol (1 : 1), and photographed. A skin wound was made on the shaved back of an anaesthetized mouse by inhalation of isoflurane (4%) using a 4 mm biopsy punch 7 days before latex dye injection.
2.5. Determination of haemoglobin level
Haemoglobin level was measured in a single drop of tail blood from control and Tmem100-iKO mutant mice 7 days after tamoxifen injection using an optical absorption photometric analyzer (HemoPoint H2, Standbio Laboratory) according to the manufacturer's instruction.
2.6. Immunohistochemistry
Primary antibodies used in this study were as follows: anti-α-smooth muscle actin (A5228; Sigma, 1 : 400), anti-von Willebrand factor (A008229; Dako, 1 : 200), anti-F4/80 (14-4801-85; eBioscience, 1 : 100), and anti-MFAP4 (AB103925; Abcam, 1 : 100). Control and Tmem100-iKO mutant mice were euthanized by CO2 asphyxiation. Lungs were embedded and cut into 7 µm slices. Deparaffinized sections were hybridized with a primary antibody. For colorimetric staining, the secondary antibody reaction and colour development were carried out with a VECTASTAIN Elite ABC Kit or a VECTOR M.O.M. staining kit (Vector Laboratories) according to the manufacturer's instruction. Diameters of MFAP4-positive vessels, which were surrounded >75% of the vessel circumstance by anti-MFAP4 staining, were measured in a microscopic field at ×100 magnification to access the expression of MFAP4 in the pulmonary vessels. Lung sections prepared from four control and eight Tmem100-iKO mice were immunostained with anti-MFAP4 antibody. Three microscopic fields without large vessels (>300 µm diameter) of each lung section were analysed. Blood vessels were divided into three groups according to their diameters (Group I: <50 µm diameter; Group II: 50–100 µm diameter; Group III: >100 µm diameter).
2.7. Microarray
Mice were euthanized by CO2 asphyxiation. Total RNAs were isolated from lungs of three control mice and three Tmem100-iKO mutant mice 7 days after tamoxifen injection. Microarray analyses were performed using the Illumina MouseRef-8 v2.0 Expression BeadChip (Illumina), which covered ∼25 000 well-annotated mouse RefSeq transcripts. Array data were processed and analysed using Illumina GenomeStudio v1.5.4 (Gene Expression Module v2009.2) (Illumina). Genes showing |fold change| ≥ 2 and adjusted P-value of <0.05 were considered significantly different. Identified genes were divided into groups sharing the same biological process or molecular function by panther classification.16,17 Gene Set Enrichment Analysis was performed using the DAVID Bioinformatic Resources.18,19 Microarray data are available at the NCBI's Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57256).
2.8. Quantitative real-time PCR
Tamoxifen-treated Tmem1002loxP/2loxP control and Tmem1002loxP/2loxP;ROSA26+/CreER mutant mice were used for quantitative real-time PCR. Mice were euthanized by CO2 asphyxiation. Total RNAs were isolated from lungs of each genotype 3 and 7 days after tamoxifen injection using a NucleoSpin RNA II kit (BD Biosciences), and then first-strand cDNA was synthesized with a PrimeScript RT Master Mix (TAKARA). Real-time PCR was performed using a SYBR Premix DimerEraser Kit (TAKARA) and CFX384 Real Time System (Bio-Rad). A mouse cyclophilin primer set (5′-TGGAGAGCACCAAGACAGACA-3′; 5′-TGCCGGAGTCGACAATGAT-3′) was used to normalize the amount of total cDNA in each sample. Predesigned PCR primer sets20–22 for microfibrillar-associated protein 4 (Mfap4) (5′-ATGGCTATACCCTCTACGTGG-3′; 5′-GGTCCCGATCAAAGGTGGAG-3′, PrimerBank ID 118130625c3) and Tmem 100 (5′-GACAATGGAGAAAAACCCCAAGA-3′; 5′-GGTAGCAGGAGAGTTCGGC-3′, PrimerBank ID 13385928a1) were used to determine the amount of each cDNA in control and mutant samples. Each sample was run in triplicate. After 40 cycles of PCR reaction, the relative amount of Mfap4 and Tmem100 transcripts was determined using the ΔΔCt method.
2.9. Statistics
Two-tailed Student's t-test was performed for statistical analysis; differences were considered significant if P < 0.05.
3. Results
3.1. Tmem100 expression was found in newly forming blood vessels of developing organs
Previously, we reported that Tmem100 was expressed in arterial endothelial cells, notochord, mammary glands, endocardium, and floor plate of early-stage embryos using Tmem100-reporter allele (Tmem100SIBN), in which the LacZ expression is controlled by endogenous Tmem100 promoter.1 Using this reporter line, we further analysed Tmem100 transcriptional activity at late gestational, neonatal, and adult stages. Expression in coronary arteries was first found at E17.5. It was the highest at newborn stage and disappeared at weaning stage (Figure 1A). In the lung, Tmem100 expression was first detected in the main pulmonary arteries at mid-gestation stage. As development proceeded, the expression extended to capillaries and persisted at adult stages (Figure 1B). In addition, Tmem100 expression also observed in blood vessels of other developing organs, such as eye, spleen, thymus, and skin (Figure 1C), but diminished at juvenile stages and disappeared after weaning stage. The spatiotemporal expression pattern of Tmem100 is largely overlapping with that of ALK1.2,3
Figure 1.
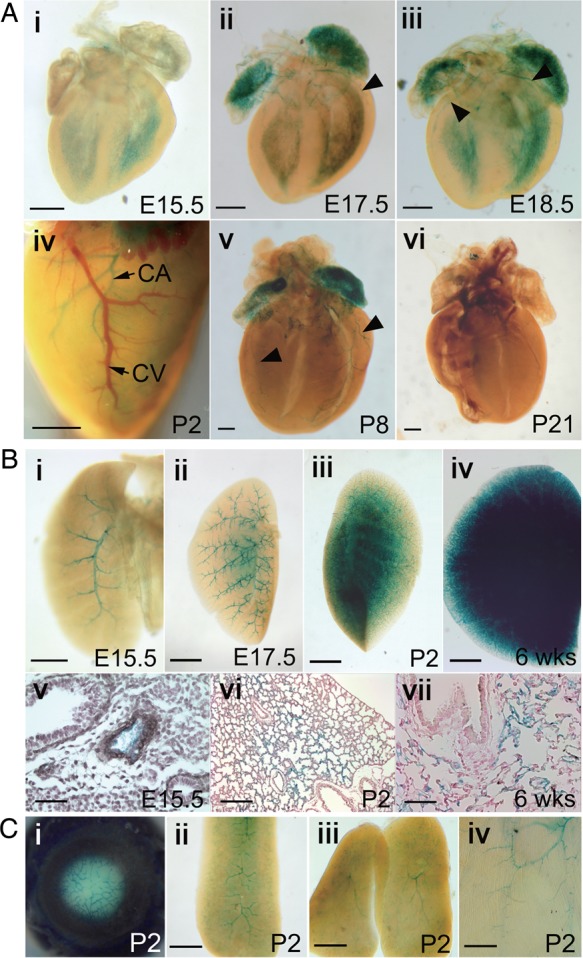
Expression of Tmem100 in developing organs. (A) Whole-mount X-gal-stained hearts of Tmem100+/SIBN mice at various embryonic, postnatal, and adult stages followed by clearing. X-gal-positive coronary arteries were indicated by arrowheads. LacZ expression was first detected at E17.5 (ii) and disappeared at weaning age (vi). Note X-gal-positive coronary artery (CA) and X-gal-negative coronary vein (CV) in the heart before clearing (iv). (B) Whole-mount X-gal-stained lungs followed by clearing (i–iv) and sections of X-gal-stained lungs counterstained with haematoxylin (v) or nuclear fast red (vi–vii). Only main pulmonary arteries were X-gal positive at E15.5 (i,v); however, LacZ expression spread as development proceeded (ii–iv and vi–vii). (C) X-gal-stained eye (i), spleen (ii), thymus (iii), and skin (iv) at P2 stage. Scale bars: A, Bi, and C, 500 μm; Bii–iv, 1 mm; Bv,vii, 50 μm; Bvi, 200 μm. More than three samples of each experiment were subjected to X-gal staining.
3.2. Tmem100-null mice at embryonic and neonatal stages phenocopy Alk1-null mice
Tmem100-null embryos die around E11.5 showing growth retardation, pericardial oedema, the absence of large branching vitelline vessels in the yolk sac, and abnormal connections between dorsal aorta and cardinal vein (see Supplementary material online, Figure S1). The phenotype of Tmem100-null embryos closely mimics that of Alk1-null embryos as reported by another group4 and indicates important roles of TMEM100 in angiogenesis during embryonic development. To further investigate the role of TMEM100 at neonatal stages, effects of TMEM100 deficiency in retinal vessel development were determined using tamoxifen-treated Tmem1002loxP/2loxP;ROSA26+/CreER newborn mice (Tmem100-iKO here after), in which inducible Cre recombinase (CreER) was ubiquitously expressed by ROSA26 promoter.23 Development of the retinal vasculature occurs postnatally in a tightly regulated spatiotemporal pattern and forms well-organized vascular network.24 Endothelial cell-specific disruption of Alk1 (Alk1-iKOec) results in impaired retinal angiogenesis, including increased retinal vascular density, failed vascular remodelling, and hyperbranching.25 Injection of neutralizing anti-BMP10 antibody into Bmp9-KO or neutralizing ALK1-Fc into WT newborns also showed similar vascular defects.26–28 Retinal vasculature of control (Tmem1002loxP/2loxP) and Tmem100-iKO mice was examined using FITC-conjugated isolectin IB4 at P8 when angiogenic front of superficial vascular plexus from the optic nerve reaches the retinal periphery.24 Retinal vasculature of Tmem100-iKO showed significantly reduced progression of angiogenic front (Figure 2A and D) in company with hyperbranched and dilated superficial vascular plexus, and increased vascular density, especially in angiogenic front region (Figure 2B, E, and F). The increased vascular branching and vascular density might be associated with dramatically increased number of filopodial protrusions in the tip cells. Number of filopodial protrusions in the frontal region and even in the central region of the plexus where filopodia formation is normally suppressed was shown to be increased (Figure 2C and G). AV shunt formation in the Tmem100-iKO retina was not obvious, but abnormally dilated vessels (indicated by arrows in Figure 2B) were detected. The retinal vascular phenotype of Tmem100-iKOs is remarkably similar to that of mice deficient in ALK1 signalling,25–28 further supporting the hypothesis that TMEM100 is an important downstream effector of ALK1 signalling for vascular development.
Figure 2.
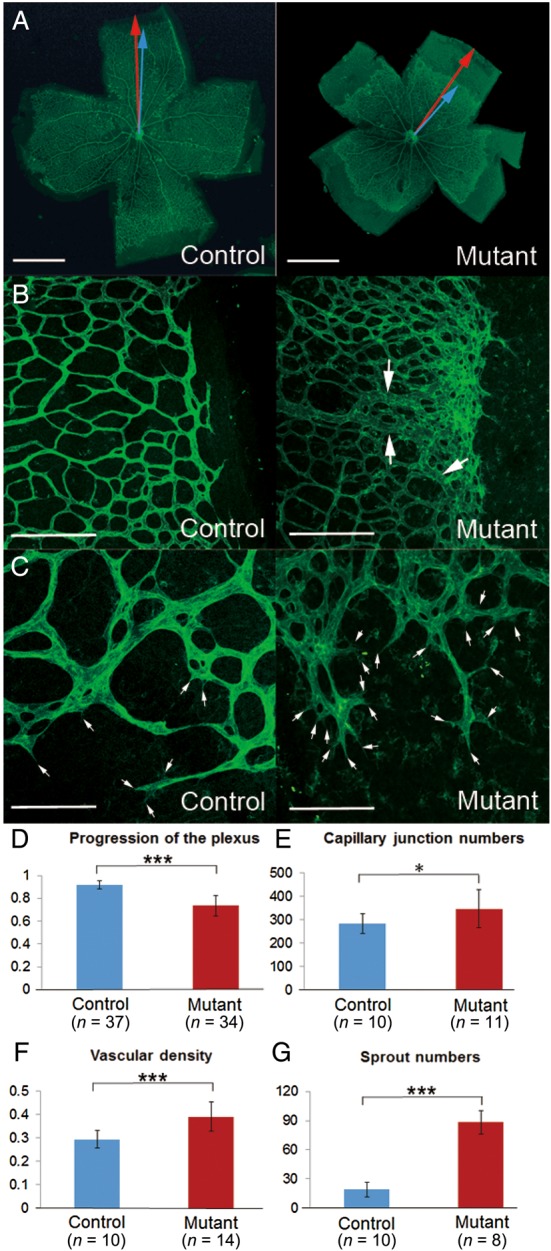
Abnormal development of retinal vasculature in Tmem100-iKO newborn mice. (A) FITC-conjugated isolectin B4 labelling showed complete formation of superficial vascular plexus in the control mice but delayed progression of the angiogenic front in the Tmem100-iKO mutant mice. The progression of the vascular plexus towards the edge of the retina was measured by the ratio of the vascular radius (blue arrow) and the retina's radius (red arrow). (B) Dilated and hyperbranched vascular plexus were observed in the mutants. Note that these defects were severer in the angiogenic front region. Arrows indicate abnormally dilated vessels resembling AV shunts. (C) High magnified image of the angiogenic front region represented increased number of filopodial protrusions (arrows) in the tip cells. (D–G), Comparison of several angiogenic parameters between the control and mutant retinas; progression of the vascular plexus towards the edge of the retina (D), number of capillary junctions within a field of view (×200) (E), vascular density (the covered area by blood vessels in the vascular plexus region) within a field of view (×25) (F), number of sprouts (filopodial protrusions in the tip cells) within a field of view (×100) (G). *P < 0.05, ***P < 0.001. Scale bars: A, 1 mm; B, 200 μm; C, 100 μm.
3.3. TMEM100 deficiency at adult stage caused mild AV shunts
Next, we investigated vascular defects in adult Tmem100-iKO mice. TMEM100 deficiency at adult stage was achieved by intraperitoneal tamoxifen injection. Most Tmem100-iKOs died ∼7–14 days after tamoxifen injection. The mutant mice showed reduced haemoglobin level (Figure 3A) and a wide degree of internal haemorrhages in visceral organs (Figure 3B–D), including the lung (8/16), liver (6/6), and intestine (8/16). To identify AV shunts, the latex dye was injected into the left ventricle of control and Tmem100-iKO mice at the moribund stage. Focal AV shunts were observed in the intestine (4/5) and liver (4/8) (Figure 3E and F). However, severity of intestinal and liver AV shunts in Tmem100-iKO was much milder compared with that in Alk1-iKOs10 (see Supplementary material online, Figure S2). Previously, we found that AVMs with a dilated and tortuous morphology developed in subdermal vessels in response to wound in Alk1-iKO.10 To test whether down-regulation of TMEM100 is a central mechanism of AVM development caused by ALK1 deficiency, wound-induced AVM formation was analysed in Tmem100-iKO. As in Figure 3G, no such AVMs were detected in Tmem100-iKO (n = 4). Alk1 expression in subdermal arteries is diminished at adult stage but induced in feeding arteries in response to wound (see Supplementary material online, Figure S3A).2 Lack of wound-induced Tmem100 expression (see Supplementary material online, Figure S3B) may explain the absence of wound-induced AVMs in Tmem100-iKOs. In the mutant lung, we found the leakage of the latex dye (Figure 3H), which is a typical phenotype observed in the Alk1-iKO lungs.10
Figure 3.
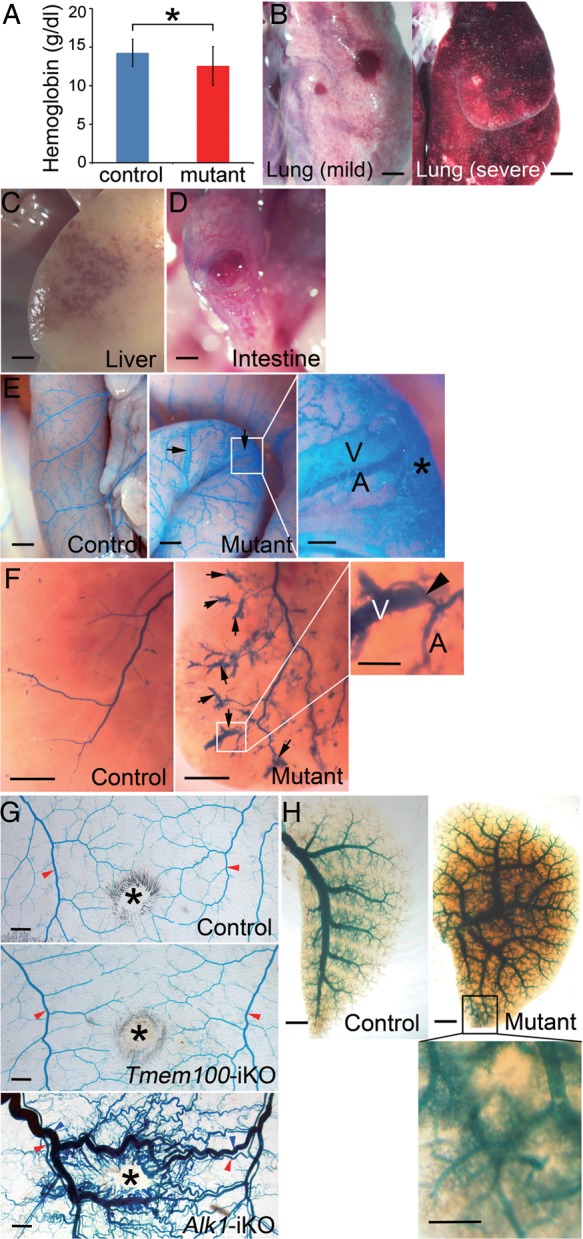
Vascular defects of Tmem100-iKO adult mice. (A–D) Reduced haemoglobin level (Control, n = 11; Mutant, n = 11, *P < 0.05) (A) and signs of internal haemorrhages in the lungs (B: left panel, mild haemorrhage; right panel, severe haemorrhage; 8/16), liver (C; 6/6), and intestine (D; 8/16) of Tmem100-iKO mutants. Two-tailed Student's t-test was performed for statistical analysis. (E–H) Blood vessels in the small intestine (E), liver (F), back skin (G), and lung (H) visualized by latex dye perfusion. Presence of the latex dye in both arteries (A) and veins (V) would indicate AV shunts (arrows). (E) The focal AV shunts in the intestine were detected close to haemorrhagic areas indicated by an asterisk. Control, n = 7; Mutant, n = 5 (four of them showed AV shunts). (F) Note direct connection (arrowhead) between arteries (A) and veins (V) in the AV shunts of the liver. Control, n = 7; Mutant, n = 8 (four of them showed AV shunts). (G) No sign of AV shunts was observed in the wounded skin area of control (n = 4) and Tmem100-iKOs (n = 4), compared with wounded skin of tamoxifen-treated Alk12loxP/2loxP;ROSA26+/CreER mice (Alk1-iKO; n = 20). *, wound; red arrowheads, arteries; blue arrowheads, veins. (H) Leakage of the latex dye was detected in the pulmonary vasculature. Scale bars: B–H, 1 mm; enlarged images, 0.25 mm.
3.4. Mfap4 was down-regulated in the lungs of TMEM100-deficient mice
To identify affected genes in TMEM100-deficiect mice, microarray analysis was performed using lungs isolated from control and Tmem100-iKO mice 7 days after tamoxifen injection, because Tmem100 was most intensely expressed in lungs at adult stage (Figure 1Biv and Bvii). Expression of genes encoding cell adhesion and extracellular matrix (ECM) proteins was most significantly affected (Figure 4; Table 1). Cell adhesion and ECM proteins as well as cytokine signalling proteins are crucial for blood vessel formation and maintenance. Abnormal cell–cell adhesion such as uneven and discontinuous smooth muscle layers and affected collagen and elastin layers were observed in HHT patients and animal models.10,29,30 Among genes in these categories, Mfap4 was the most highly affected gene (6.12-fold down) in the Tmem100-iKO lungs. In the lung, MFAP4 is mainly localized to elastic fibres of the arterioles and alveolar walls (Figure 5A).31,32 MFAP4 is known to directly interact with fibrillin-1, a component of elastin microfibrils, and plays an essential role in the assembly of elastic fibres.33 Elastin layers maintain elasticity of blood vessels, and disruption of elastin homeostasis causes various vascular disease.34 Transcriptional down-regulation of Mfap4 in the Tmem100-iKO lungs was confirmed by quantitative RT–PCR analysis, representing a 5.5-fold decrease (Figure 5B). Immunostaining using the MFAP4 antibody also showed that the number of small vessels (<100 µm diameter) expressing MFAP4 was markedly diminished in the Tmem100-iKO lungs (Figure 5C). Reduced number of elastin-positive vessels and elastin density in small vessels of the Tmem100-iKO lungs were confirmed by Hart's elastin staining (Figure 5D). To examine whether the down-regulation of Mfap4 was a cause or consequence of the reduced number of elastic small arteries, the expression level of Mfap4 and integrity of elastin layers were analysed in the lungs isolated from control and Tmem100-iKO mice 3 days after tamoxifen injection when no discernible abnormality was detected (see Supplementary material online, Figure S4A). The expression level of Mfap4 was significantly down-regulated in the mutant lungs at Day 3, but no significant defects of the integrity of elastin layers were detected at this stage (see Supplementary material online, Figure S4B). This result suggests that the down-regulation of Mfap4 is rather a cause than a consequence of elastin defects in the mutant lungs. We also found reduced number of organized vascular smooth muscle cell-associated vessels and irregular formation of endothelial and smooth muscle layers in the Tmem100-iKO lungs, especially in the haemorrhagic regions (Figure 5E and F), as in the Alk1-iKO lungs.10 These phenotypes are consistent with impaired vascular integrity and haemorrhage in Tmem100-iKOs (see Supplementary material online, Figure S5). We observed increased number of F4/80-positive macrophages in Tmem100-iKOs at the terminal stage but not 4 days after tamoxifen injection (see Supplementary material online, Figure S6), indicating that macrophages infiltration is not a cause but a consequence of vascular abnormality in the mutant lungs.
Figure 4.

Gene Set Enrichment Analysis (GSEA) of biological processes and molecular functions for differentially expressed genes in the lungs between control and Tmem100-iKO mice. Significantly enriched biological processes (A) and molecular functions (B) are represented with their Fisher P-value (<0.01). Expression of genes encoding cell adhesion proteins (biological process) and ECM proteins (molecular function) is most significantly affected in the lungs of tamoxifen-treated Tmem100-iKO mutant mice.
Table 1.
Differentially regulated cell adhesion and ECM genes in Tmem100-iKO mice
| Cell adhesion |
ECM |
||
|---|---|---|---|
| Gene | Fold change | Gene | Fold change |
| Mfap4 | −6.12 | Mfap4 | −6.12 |
| Lgals1 | −3.89 | Spon2 | −5.05 |
| Col1a1 | −3.35 | Fbln1 | −4.59 |
| Pcolce | −3.25 | Col1a1 | −3.35 |
| Loxl1 | −3.23 | Pcolce | −3.25 |
| Pcolce2 | −2.97 | Mfap5 | −2.98 |
| Ptprd | −2.44 | Pcolce2 | −2.97 |
| Vcam1 | −2.39 | Mmp2 | −2.50 |
| Tinag | −2.36 | Col17a1 | −2.23 |
| Ptprs | −2.21 | Col6a2 | −2.17 |
| Col6a2 | −2.17 | Scube2 | −2.15 |
| Itga1 | −2.17 | Matn4 | −2.07 |
| Emcn | −2.12 | Hspg2 | −2.04 |
| Matn4 | −2.07 | Lamc3 | 2.49 |
| Hspg2 | −2.04 | Spp1 | 2.69 |
| Cdh16 | −2.04 | Angptl4 | 3.17 |
| Pcdh20 | 2.10 | ||
| Tinagl | 2.51 | ||
| Spp1 | 2.69 | ||
| Lox | 3.81 | ||
Figure 5.
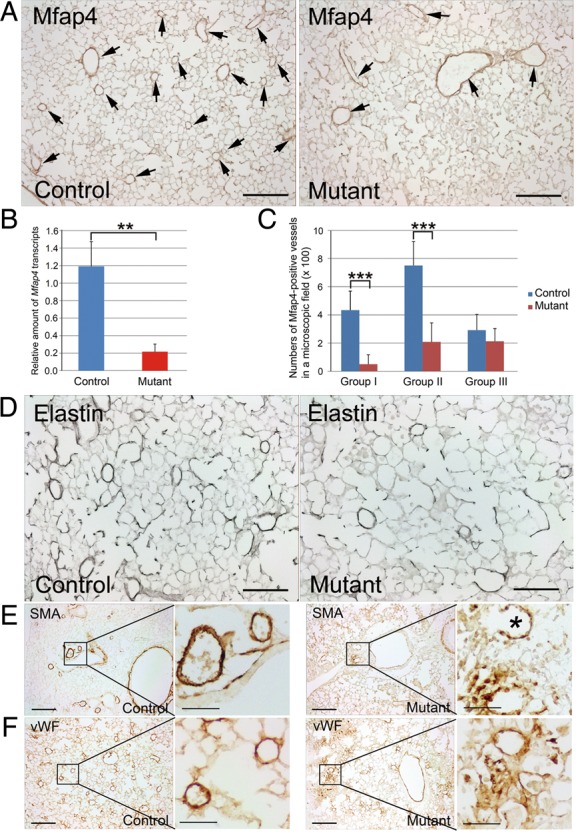
Mfap4 is down-regulated in the lungs of Tmem100-iKO mice. (A) Immunohistochemical staining of control (n = 4) and Tmem100-iKO mutant (n = 8) lungs with anti-MFAP4 antibody. The MFAP4-positive staining was predominantly detected in the pulmonary vessels with various sizes and alveolar walls in the control mice. However, the MFAP4 expression was diminished in small vessels and alveolar walls of Tmem100-iKO mice. Arrows indicate MFAP4-positive vessels. (B) Quantitative real-time PCR showing 5.5-fold down-regulation of Mfap4 in the lungs of Tmem100-iKO mice. **P < 0.01. Control, n = 3; Mutant, n = 3. (C) Number of vessels positive for MFAP4. Pulmonary vessels were divided into three groups according to their diameters (Group I: <50 µm diameter; Group II: 50–100 µm diameter; Group III: >100 µm diameter). Note that the numbers of MFAP4-positive vessels with diameter <100 µm were markedly reduced in the Tmem100-iKO lungs. However, the numbers of MFAP4-positive vessels with diameter larger than 100 µm were not affected. Two-tailed Student's t-test was performed for statistical analysis. ***P < 0.001. Control, n = 4; Mutant, n = 8. Three microscopic fields of each lung section were analysed. (D) Hart's stain showing reduced number of small vessels possessing elastin layers in the Tmem100-iKO lung. Control, n = 4; Mutant, n = 8. (E, F) Reduced number of organized blood vessels in the lung of Tmem100-iKO mice. Smooth muscle cells (SMCs) and vascular SMC-associated endothelial cells were immunostained by anti-α-smooth muscle actin (SMA) and anti-von Willebrand factor (vWF) antibodies, respectively. Note disarrayed vascular structure in the affected region and a ruptured blood vessel (indicated by an asterisk) of the Tmem100-iKO lung. Control, n = 4; Mutant, n = 8. Scale bars: A and D, 100 μm; E and F, 200 μm; enlarged images, 50 μm.
4. Discussion
Here, we showed that Tmem100 expression was detected in blood vessels of developing organs, but it diminished at adult stage except in the lung. The spatiotemporal expression pattern was similar with that of Alk1.2,3 Phenotypes of TMEM100 deficiency in embryonic and neonatal stages are strikingly similar with those of Alk1-null or -iKO mice,5,6,26,27 suggesting that TMEM100 could be an important downstream effector of ALK1 signalling in embryonic and neonatal angiogenesis. TMEM100 deficiency at adult stage resulted in haemorrhage in various organs, mild AV shunts in the intestine and liver, and increased vascular permeability in the lung, indicating that TMEM100 is essential for maintenance of adult vasculature. Overlapping features of vascular phenotypes between Tmem100- and Alk1-iKOs exist, but AV shunts found in adult Tmem100-iKOs appeared to be underdeveloped without typical tortuosity of vessels associated with AVMs. Absence of wound-induced AVMs in Tmem100-iKOs might be related with lack of wound-induced Tmem100 expression.
Symptoms in HHT patients are caused by both congenital and postnatal vascular malformations. Mostly, the vascular malformations are due to ALK1- or ENG deficiency.7,8Tmem100-null embryos phenocopied Alk1-null embryos including AV shunts between dorsal aorta and cardinal vein as well as hyperbranching of developing retinal vessels; however, severity of AV shunts in adult Tmem100-iKO was much milder compared with that in Alk1-iKOs. Even wound-induced skin AVMs were not detected in Tmem100-iKO. It suggests that down-regulation of TMEM100 may contribute to congenital vascular pathogenesis of HHT, but it is not the central mechanism of HHT at adult stage such as nasal telangiectases, which cause recurrent epistaxis. Therefore, TMEM100 does not appear to be the ideal target for developing therapies for epistaxis.
Microarray analysis showed that expression of genes encoding cell adhesion and ECM proteins was significantly affected in the lungs of adult Tmem100-iKOs. Some marker genes of arterial endothelial cells (Gja4, Gja5, and Efnb2) and Notch- and Akt-downstream genes (Hrt1, Hrt2, Hrt3, Hes1, Hes5, Klf2, and eNos) are reported to be down-regulated in E9.5 Tmem100-null embryos.4 The expression of these genes, however, was not significantly affected in the lungs of adult Tmem100-iKOs except Gja5 (4.92-fold down). It might be due to the different role of TMEM100 in embryonic angiogenesis and maintenance of adult vasculature. MFAP4, an important protein for elastic fibre assembly,33 was most highly down-regulated, and pulmonary vascular walls were disrupted in Tmem100-iKOs. Taken together, these data demonstrate that TMEM100 is essential for maintaining pulmonary vascular integrity by regulating cell adhesion and ECM proteins such as MFAP4.
Bone marrow (BM)-derived circulating haematopoietic cells and BM mesenchymal stromal cells are important for vascular homeostasis. We cannot exclude the possibility that dysfunction of these cells contributes to vascular abnormality and haemorrhage in Tmem100-iKO mice. Further study will be performed to clarify this issue. It would be also interesting to study whether the loss or reduced TMEM100 is associated with various pathological conditions that lead to pulmonary haemorrhages.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This study was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2011-0009321) to Y.J.L., in part by a NRF grant funded by the Korea government (MSIP) (NRF-2014R1A2A2A01007604) to Y.J.L., and a grant from the Kwang-dong pharmaceutical Cooperation (2011-5019) to Y.J.L., and by World Class University program funded by the Ministry of Education, Science and Technology through NRF (R32-10215) to S.P.O.
Supplementary Material
Acknowledgements
We thank C. Song, S.O. Park, and K.S. Ko for technical assistance in animal experiments.
Conflict of interest: none declared.
References
- 1.Moon EH, Kim MJ, Ko KS, Kim YS, Seo J, Oh SP, Lee YJ. Generation of mice with a conditional and reporter allele for Tmem100. Genesis. 2010;48:673–678. doi: 10.1002/dvg.20674. [DOI] [PubMed] [Google Scholar]
- 2.Seki T, Yun J, Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ Res. 2003;93:682–689. doi: 10.1161/01.RES.0000095246.40391.3B. [DOI] [PubMed] [Google Scholar]
- 3.Seki T, Hong KH, Yun J, Kim SJ, Oh SP. Isolation of a regulatory region of activin receptor-like kinase 1 gene sufficient for arterial endothelium-specific expression. Circ Res. 2004;94:e72–e77. doi: 10.1161/01.RES.0000127048.81744.31. [DOI] [PubMed] [Google Scholar]
- 4.Somekawa S, Imagawa K, Hayashi H, Sakabe M, Ioka T, Sato GE, Inada K, Iwamoto T, Mori T, Uemura S. Tmem100, an ALK1 receptor signaling-dependent gene essential for arterial endothelium differentiation and vascular morphogenesis. Proc Natl Acad Sci USA. 2012;109:12064–12069. doi: 10.1073/pnas.1207210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet. 2000;26:328–331. doi: 10.1038/81634. [DOI] [PubMed] [Google Scholar]
- 7.Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43:97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous MEM, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 9.Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24:203–219. doi: 10.1016/j.blre.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS, Oh SP. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119:3487–3496. doi: 10.1172/JCI39482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karnezis TT, Davidson TM. Treatment of hereditary hemorrhagic telangiectasia with submucosal and topical bevacizumab therapy. Laryngoscope. 2012;122:495–497. doi: 10.1002/lary.22501. [DOI] [PubMed] [Google Scholar]
- 12.Han C, Choe S-W, Kim YH, Acharya AP, Keselowsky BG, Sorg BS, Lee Y-J, Oh SP. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis. 2014;17:823–830. doi: 10.1007/s10456-014-9436-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL, Oh SP. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008;111:633–642. doi: 10.1182/blood-2007-08-107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joo JH, Lee YJ, Munguba GC, Park S, Taxter TJ, Elsagga MY, Jackson MR, Oh SP, Sugrue SP. Role of Pinin in neural crest, dorsal dermis, and axial skeleton development and its involvement in the regulation of Tcf/Lef activity in mice. Dev Dyn. 2007;236:2147–2158. doi: 10.1002/dvdy.21243. [DOI] [PubMed] [Google Scholar]
- 15.Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protocols. 2010;5:1518–1534. doi: 10.1038/nprot.2010.113. [DOI] [PubMed] [Google Scholar]
- 16.Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, Guo N, Muruganujan A, Doremieux O, Campbell MJ, Kitano H, Thomas PD. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33:D284–D288. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–D799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spandidos A, Wang X, Wang H, Dragnev S, Thurber T, Seed B. A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics. 2008;9:633. doi: 10.1186/1471-2164-9-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badea TC, Wang Y, Nathans J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci. 2003;23:2314–2322. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, Krah NM, Seaward MR, Willett KL, Aderman CM, Guerin KI, Hua J, Löfqvist C, Hellström A, Smith LEH. The mouse retina as an angiogenesis model. IOVS. 2010;51:2813–2826. doi: 10.1167/iovs.10-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tual-Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, Fruttiger M, Arthur HM. Endothelial depletion of acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS ONE. 2014;9:e98646. doi: 10.1371/journal.pone.0098646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niessen K, Zhang G, Ridgway JB, Chen H, Yan M. ALK1 signaling regulates early postnatal lymphatic vessel development. Blood. 2010;115:1654–1661. doi: 10.1182/blood-2009-07-235655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larrivée B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A. ALK1 signaling inhibits angiogenesis by cooperating with the notch pathway. Dev Cell. 2012;22:489–500. doi: 10.1016/j.devcel.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricard N, Ciais D, Levet S, Subileau M, Mallet C, Zimmers TA, Lee S-J, Bidart M, Feige J-J, Bailly S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood. 2012;119:6162–6171. doi: 10.1182/blood-2012-01-407593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torsney E, Charlton R, Diamond AG, Burn J, Soames JV, Arthur HM. Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation. 2003;107:1653–1657. doi: 10.1161/01.CIR.0000058170.92267.00. [DOI] [PubMed] [Google Scholar]
- 30.Brace LD. Qualitative disorders of platelets and vasculature. In: Rodak BF, Fritsma GA, Keohane EM, eds. Hematology: Clinical Principles and Applications. 4th ed. St. Louis: Elsevier; 2012. p. 718–733. [Google Scholar]
- 31.Wulf-Johansson H, Lock Johansson S, Schlosser A, Trommelholt Holm A, Melholt Rasmussen L, Mickley H, Diederichsen ACP, Munkholm H, Poulsen TS, Tornøe I, Nielsen V, Marcussen N, Vestbo J, Sækmose SG, Holmskov U, Sorensen GL. Localization of microfibrillar-associated protein 4 (MFAP4) in human tissues: clinical evaluation of serum MFAP4 and its association with various cardiovascular conditions. PLoS ONE. 2013;8:e82243. doi: 10.1371/journal.pone.0082243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlosser A, Thomsen T, Shipley JM, Hein PW, Brasch F, Tornøe I, Nielsen O, Skjødt K, Palaniyar N, Steinhilber W, McCormack FX, Holmskov U. Microfibril-associated protein 4 binds to surfactant protein A (SP-A) and colocalizes with SP-A in the extracellular matrix of the lung. Scand J Immunol. 2006;64:104–116. doi: 10.1111/j.1365-3083.2006.01778.x. [DOI] [PubMed] [Google Scholar]
- 33.Kasamatsu S, Hachiya A, Fujimura T, Sriwiriyanont P, Haketa K, Visscher MO, Kitzmiller WJ, Bello A, Kitahara T, Kobinger GP, Takema Y. Essential role of microfibrillar-associated protein 4 in human cutaneous homeostasis and in its photoprotection. Sci Rep. 2011;1:164. doi: 10.1038/srep00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsamis A, Krawiec JT, Vorp DA. Elastin and collagen fibre microstructure of the human aorta in ageing and disease: a review. J R Soc Interface. 2013;10:20121004. doi: 10.1098/rsif.2012.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.