

Abstract
Aims
Dickkopf-3 (DKK3), a secreted protein in the Dickkopf family, is expressed in various tissues, including the heart, and has been shown to play an important role in tissue development. However, the biological function of DKK3 in the heart remains largely unexplored. This study aimed to examine the role of DKK3 in pathological cardiac hypertrophy.
Methods and results
We performed gain-of-function and loss-of-function studies using DKK3 cardiac-specific transgenic (TG) mice and DKK3 knockout (KO) mice (C57BL/6J background). Cardiac hypertrophy was induced by aortic banding. Cardiac hypertrophy was evaluated by echocardiographic, haemodynamic, pathological, and molecular analyses. Our results demonstrated that the loss of DKK3 exaggerated pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction, whereas the overexpression of DKK3 protected the heart against pressure overload-induced cardiac remodelling. These beneficial effects were associated with the inhibition of the ASK1-JNK/p38 (apoptosis signal-regulating kinase 1-c-Jun N-terminal kinase/p38) signalling cascade. Parallel in vitro experiments confirmed these in vivo observations. Co-immunoprecipitation experiments suggested that physical interactions occurred between DKK3 and ASK1. Moreover, rescue experiments indicated that, in DKK3 TG mice, the activation of ASK1 using a cardiac-specific conditional ASK1 transgene reduced the functionality of DKK3 in response to pressure overload; furthermore, the inactivation of ASK1 by dominant-negative ASK1 rescued pressure overload-induced cardiac abnormalities in DKK3 KO mice.
Conclusion
Taken together, our findings indicate that DKK3 acts as a cardioprotective regulator of pathological cardiac hypertrophy and that this function largely occurs via the regulation of ASK1-JNK/p38 signalling.
Keywords: Cardiac hypertrophy, Fibrosis, DKK3, ASK1
1. Introduction
Heart failure, which is increasing in prevalence, is a debilitating disease with high morbidity and mortality.1 At present, it is widely recognized that arterial hypertension is a major risk factor for the development of heart failure.2 In response to pressure overload, the heart initially activates an adaptive physiological response to its increased workload in the form of cardiac hypertrophy; however, over time, the increased cardiac workload and the burden of continuous mechanical stress ultimately promote heart failure, arrhythmias, and sudden death.3,4 Thus, the suppression of cardiac hypertrophy has emerged as a possible strategy for mitigating hypertension-induced end-organ damage. At the cellular level, cardiac hypertrophy is characterized by increased cardiomyocyte size, enhanced protein synthesis, and foetal gene programming reactivation.5–7 Although the multiple signalling mechanisms that control cardiomyocyte growth have been extensively studied, the molecular mechanisms that mediate the development of cardiac hypertrophy and the transition to heart failure are incompletely understood.
The Dickkopf (DKK) proteins, including not only proteins DKK1–DKK4 but also a unique DKK3-related protein that is known as Soggy (DKKL1 or SGY-1), are secreted Wnt signalling regulators.8,9 All the DKK proteins except for SGY-1 share two conserved cysteine-rich domains that are separated by a variable linker region.9 DKK3, which is also known as REIC (Reduced Expansion in Immortalized Cells), is a divergent member of the DKK family.8 DKK3 is expressed during embryonic development in many tissues, including bone tissue, the neural epithelium, limb buds, and the heart.10 The available evidence suggests that DKK3 plays an important role in regulating tissue development, apoptosis, proliferation, and immunity.11–14 DKK3 is also widely expressed in various adult tissues, including the heart,9 suggesting that this protein may play a role in heart physiology and pathology. However, the role of DKK3 in cardiac disease has not previously been investigated. DKK3 is a potential tumour suppressor gene for a range of tumours,15–17 and several studies suggest that tumour suppressors negatively modulate cardiac hypertrophy;18,19 thus, DKK3 may be an attractive target for therapeutic intervention to prevent cardiac hypertrophy and heart failure. The current study featured the following objectives: (i) to determine whether DKK3 is altered in dilated cardiomyopathy (DCM) patients and a model of pressure overload-induced cardiac hypertrophy; (ii) to determine whether DKK3 expression affects cardiac hypertrophy; and (iii) to identify the mechanisms that would be involved in any such effects that are observed.
2. Methods
The animal protocol was approved by the Animal Care and Use Committee of the Renmin Hospital of Wuhan University, China. Sodium pentobarbital (50 mg/kg, ip) was used to anaesthetize mice, and the adequacy of anaesthesia was confirmed by the absence of reflex response to foot squeeze. All surgeries and subsequent analyses were performed in a blinded fashion. All procedures that involved human samples conformed to the principles that have been outlined in the Declaration of Helsinki and were approved by the Renmin Hospital of Wuhan University Review Board, Wuhan, China. An expanded method section is available in Supplementary material online, Supplementary Methods, which includes detailed methods on the following: reagents and animals, aortic banding model,6,7,20,21 echocardiography and haemodynamic measurements,6,7,20–23 histological analysis, cultured neonatal rat cardiac myocytes (NRCM) and recombinant adenoviral vectors,6,7,20 immunofluorescence, quantitative real-time PCR and western blotting, immunoprecipitation, in vitro kinase assay,24,25 human heart samples,20 and statistical analysis.
3. Results
3.1. DKK3 expression is decreased in DCM human hearts and in hypertrophic murine hearts
To study the potential role of DKK3 in cardiac hypertrophy and heart failure, we first determined whether DKK3 expression is altered in hearts with these pathologies. Western blot and real-time PCR analyses demonstrated that in left ventricle (LV) myocardial samples, compared with healthy donors, end-stage heart failure DCM patients who were undergoing heart transplantation demonstrated dramatically decreased DKK3 protein and mRNA expression (Figure 1A and D), but markedly elevated levels of the hypertrophic markers β-myosin heavy chain (MHC) and atrial natriuretic factor (ANP; Figure 1B and D). The immunofluorescence results showed that DKK3 was distributed in cytoplasm and was extracellular, and its expression was down-regulated in DCM hearts (Figure 1C). Similarly, in a murine model of aortic binding (AB)-induced cardiac hypertrophy, AB-treated mice exhibited increased β-MHC and ANP relative to sham-operated mice (Figure 1E), and cardiac DKK3 was ∼57% lower in 4-week AB-treated hearts compared with sham-operated hearts (Figure 1E, n = 3 independent experiments, P < 0.01 vs. sham). Furthermore, DKK3 levels were significantly down-regulated in in vitro cultured neonatal cardiomyocytes that had been treated with either angiotensin II (Ang II) or phenylephrine for 48 h to induce cardiomyocyte hypertrophy (Figure 1F, n = 3 independent experiments, P < 0.01 vs. phosphate buffer solution (PBS)). These findings suggest that DKK3 deficiency is associated with the development of cardiac hypertrophy and heart failure.
Figure 1.
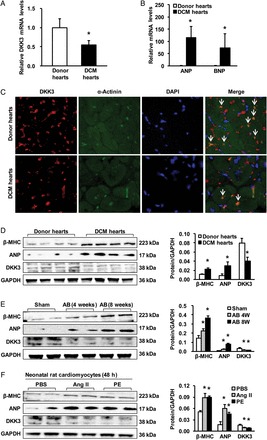
DKK3 expression is decreased in DCM human hearts and in hypertrophic murine hearts. (A) The relative levels of DKK3 mRNA in donor hearts and DCM hearts. (B) Relative ANP and brain natriuretic peptide mRNA levels in donor hearts and DCM hearts. (C) The immunostaining results showed that DKK3 both is distributed in cytoplasm and is extracellular and decreased in DCM hearts (n = 6 hearts per experimental group; *P < 0.05 vs. donor hearts). (D–F) β-MHC, ANP, and DKK3 protein levels in samples from (D) donor hearts and DCM hearts; (E) mice at the indicated times after sham or aortic binding (AB) surgery; and (F) neonatal rat cardiomyocytes that have been treated with angiotensin II (Ang II) or phenylephrine for 48 h (n = 3 independent experiments, *P < 0.05 vs. donor or sham or phosphate buffer solution (PBS)). (left) Representative blots and (right) quantitative results.
3.2. DKK3 modulates Ang II-induced cardiomyocyte hypertrophy in vitro
The observation of decreased DKK3 expression in response to hypertrophic stimuli suggests that DKK3 may regulate pathological cardiac hypertrophy. To test this conjecture, we performed controlled gain- and loss-of-function studies utilizing cultured isolated cardiomyocytes. Cells were infected with AdshDKK3 to knockdown DKK3 expression or with AdDKK3 to induce DKK3 overexpression (see Supplementary material online, Figure S1); these cells were then treated with either 1 μM Ang II or PBS control for 48 h and immunostained with α-actinin to allow for measurements of cardiomyocyte size. The experimental results with respect to the cell surface area of cardiomyocytes indicated that AdshDKK3-mediated DKK3 deficiency exacerbated Ang II-mediated cardiomyocyte hypertrophy; in contrast, AdDKK3-mediated DKK3 overexpression attenuated cardiomyocyte hypertrophy (Figure 2A and B). Consistent with these results, compared with controls, the Ang II-induced expression of the hypertrophic markers ANP and β-MHC was markedly increased in AdshDKK3-infected cardiomyocytes (Figure 2C, left panel), but decreased in AdDKK3-infected cardiomyocytes (Figure 2C, right panel) after Ang II stimulation. These in vitro data suggest that DKK3 exerts an inhibitory effect upon cardiac hypertrophy.
Figure 2.
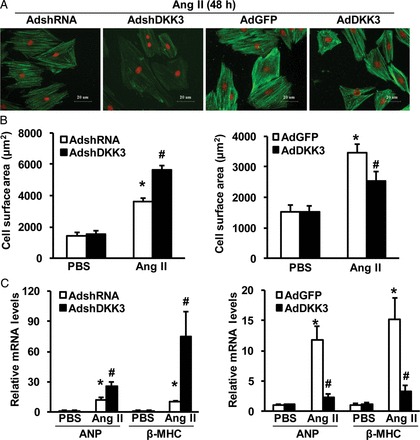
DKK3 modulates Ang II-induced cardiomyocyte hypertrophy in vitro. (A) Representative images of neonatal rat cardiomyocytes that have been infected with AdshDKK3 or AdDKK3 and treated with Ang II (1 μM) for 48 h. (B) Cell surface area of neonatal rat cardiomyocytes that have been infected with (left) AdshDKK3 or (right) AdDKK3 and treated with Ang II (1 μM) for 48 h (n = 40+ cells per experimental group). (C) The relative levels of ANP and β-MHC mRNAs in neonatal rat cardiomyocytes that have been infected with (left) AdshDKK3 or (right) AdDKK3 and treated with Ang II (1 μM) for 48 h (n = 3 independent experiments). *P < 0.05 vs. AdshRNA or AdGFP/PBS; #P < 0.05 vs. AdshRNA or AdGFP/Ang II.
3.3. Loss of DKK3 exacerbates pressure overload-induced hypertrophy
Our in vitro experimental results strongly suggest that DKK3 plays a critical regulatory role in hypertrophic responses to Ang II. To investigate the potential involvement of DKK3 in suppressing in vivo cardiac hypertrophy in response to stimuli that are more pathologically relevant than Ang II exposure, DKK3 knockout (KO; DKK3−/−) mice (see Supplementary material online, Figure S2A) were examined at 4 weeks after an AB or sham operation. Under basal conditions, DKK3−/− mice manifested no pathological abnormalities in cardiac structure or cardiac function (see Supplementary material online, Table). However, relative to AB-treated wild-type (WT) mice (DKK3+/+ mice), DKK3−/− mice exhibited markedly increased cardiac hypertrophy after 4 weeks of AB, as evidenced by increased heart weight (HW)/body weight (BW), lung weight (LW)/BW, and HW/tibia length (TL) ratios compared with AB-treated DKK3+/+ mice (Figure 3A–C). Histological analyses revealed an increased cardiomyocyte cross-sectional area (CSA) in DKK3−/− hearts than in DKK3+/+ hearts after 4 weeks of AB (Figure 3D and E). Echocardiography and haemodynamic measurements indicated that these changes were accompanied by increased cardiac dilation and dysfunction in DKK3−/− than in DKK3+/+ mice (Figure 3F and G). In addition, after 4 weeks of AB, increased levels of the hypertrophic markers ANP, brain natriuretic peptide (BNP), and β-MHC were observed in DKK3−/− than in DKK3+/+ mice (see Supplementary material online, Figure S2B).
Figure 3.
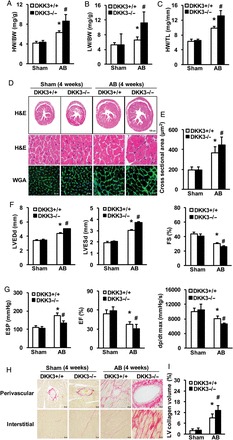
Loss of DKK3 exacerbates pressure overload-induced hypertrophy. (A–C) Statistical results for the following ratios in the indicated groups (n = 12–13 mice per experimental group): (A) HW/body weight (BW); (B) LW/BW; and (C) HW/TL. (D) Images of heart sections from WT and KO mice at 4 weeks after sham or AB surgery (n = 5–8 mice per experimental group) that have been stained with H&E or FITC-conjugated wheat germ agglutinin (WGA). (E) Statistical results for cross-sectional area (CSA) (n = 100+ cells per experimental group). (F) Parameters of the echocardiographic results for WT and KO mice (n = 7–8 mice per experimental group). (G) Parameters of haemodynamic measurement results for WT and KO mice (n = 7–8 mice per experimental group). (H) Images of picrosirius red (PSR)-stained heart sections from WT and KO mice at 4 weeks after sham or AB surgery (n = 5–8 mice per experimental group). (I) Statistical results for left ventricle (LV) collagen volume (%) (n = 25+ fields per experimental group). *P < 0.05 vs. WT/sham; #P < 0.05 vs. WT/AB.
Fibrosis constitutes a major hypertrophic pathological development. To further characterize the effects of DKK3 on maladaptive cardiac remodelling, cardiac fibrosis was measured. Paraffin-embedded tissue sections were stained with picrosirius red (PSR) to assess cardiac fibrotic extent. Marked perivascular and interstitial fibrosis was observed in AB-treated DKK3+/+ mice, but this phenomenon was more prominently observed in AB-treated DKK3−/− mice (Figure 3H). Subsequent analyses of LV collagen content and the mRNA expression levels of various fibrotic markers, such as connective tissue growth factor (CTGF), collagen I, and collagen III, consistently demonstrated that the fibrotic response was greater in DKK3−/− than in DKK3+/+ mice (Figure 3I and see Supplementary material online, Figure S2C). Collectively, these loss-of-function data indicate that DKK3 deficiency exacerbates cardiac hypertrophy in response to pressure overload.
3.4. DKK3 overexpression mitigates pressure overload-induced hypertrophy
We then addressed whether elevated levels of DKK3 in the heart attenuate cardiac hypertrophy. To examine this issue, transgenic (TG) mice with the cardiac-specific overexpression of mouse DKK3 were generated using the α-MHC promoter (see Supplementary material online, Figure S3A). Four germ lines of DKK3 TG mice were established; the successful creation of these lines was verified by western blotting (see Supplementary material online, Figure S3B). At baseline, none of the DKK3 TG mice manifested any apparent morphological or pathological cardiac abnormalities (see Supplementary material online, Table). The TG line (Tg4) that exhibited the highest DKK3 levels was selected for the following experiments. DKK3 TG mice and their WT littermates [i.e. non-transgenic (NTG) mice] were examined at 8 weeks after either AB treatment or a sham operation. As expected, the myocardial hypertrophic response was significantly blocked in TG mice, as indicated by lower ratios of HW/BW, LW/BW, and HW/TL in TG than in NTG mice (Figure 4A–C). Similarly, the CSA of cardiomyocytes was much smaller in AB-treated DKK3 TG than in AB-treated NTG mice (Figure 4D and E). Consistent with these morphological alterations, the AB-induced expression of markers of cardiac hypertrophy (ANP, BNP, and β-MHC) was greatly reduced in TG than in NTG mice (see Supplementary material online, Figure S3C). After 8 weeks of AB, profoundly better cardiac structure and cardiac function were exhibited by TG than by NTG mice, as measured by echocardiography and haemodynamic measurements (see Supplementary material online, Figure S3E and F).
Figure 4.
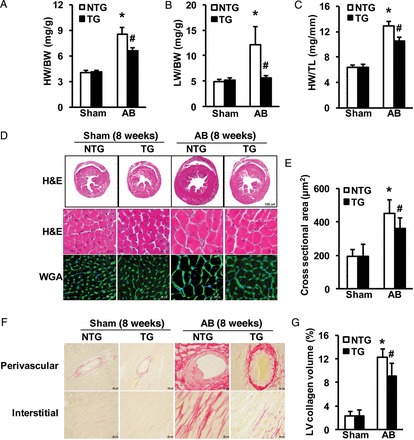
DKK3 overexpression mitigates pressure overload-induced hypertrophy. (A–C) Statistical results for the following ratios in the indicated groups (n = 12–15 mice per experimental group): (A) HW/BW; (B) LW/BW; and (C) HW/TL. (D) Images of heart sections from NTG and transgenic (TG) mice at 8 weeks after sham or AB surgery (n = 5–6 mice per experimental group) that have been stained with H&E or FITC-conjugated WGA. (E) Statistical results for CSA (n = 100+ cells per experimental group). (F) Images of PSR-stained heart sections from NTG and TG mice at 8 weeks after sham or AB surgery (n = 5–6 mice per experimental group). (G) Statistical results for LV collagen volume (%) (n = 25+ fields per experimental group). *P < 0.05 vs. NTG/sham; #P < 0.05 vs. NTG/AB.
We next assessed cardiac fibrosis by examining the PSR staining of collagen within the interstitial and perivascular spaces and by measuring the collagen volume and the expression of the fibrotic markers collagen I, collagen III, and CTGF in the LV. In contrast to DKK3 KO mice, DKK3 TG mice exhibited remarkably attenuated cardiac fibrosis compared with NTG animals (Figure 4F and G and see Supplementary material online, Figure S3D). Taken together, the results of these gain-of-function investigations suggest that the increased expression of DKK3 in the heart protects against pressure overload-induced cardiac hypertrophy.
3.5. DKK3 regulates ASK1 signalling in the heart
Given the aforementioned results, which demonstrate that the down-regulation of DKK3 expression in failing hearts plays a causative role in cardiomyocyte hypertrophy, we attempted to identify the molecules that are involved in DKK3 signalling. Because mitogen-activated protein kinase (MAPK) signalling pathways in heart tissue and in isolated cardiomyocytes are activated by various hypertrophic stresses, including haemodynamic overload and neurohormonal stimuli,26 the possible involvement of MAPK pathways in DKK3 signalling was investigated. Western blot analyses demonstrated that 4 weeks of AB caused significant increases in the levels of phosphorylated MEK1/2, extracellular signal-regulated kinases (ERK) 1/2, c-Jun N-terminal kinase (JNK)1/2, and p38 in both DKK3−/− and DKK3+/+ hearts (Figure 5A). The degree of JNK1/2 and p38 activation was significantly greater in DKK3−/− than in DKK3+/+ hearts, whereas similar levels of MEK1/2 and ERK1/2 activation were observed in these two groups of animals (Figure 5A). In gain-of-function experiments, we observed that the AB-induced activation of JNK1/2 and p38 was almost completely blocked in DKK3 TG hearts (Figure 5B). To confirm these findings, we altered DKK3 expression in vitro by transfecting cultured neonatal rat cardiomyocytes with AdshDKK3 or AdDKK3 and then subjecting the transfected cardiomyocytes to 60 min of 1 μM Ang II treatment. In accordance with the in vivo findings, greater levels of phosphorylated JNK1/2 and p38 were observed in AdshDKK3-transfected cells compared with control AdshRNA-transfected cells (Figure 5C). In contrast, DKK3 overexpression reduced the phosphorylated JNK1/2 and p38 in Ang II-treated cells to levels that were comparable with the levels of phosphorylated JNK1/2 and p38 that were observed in control cells (Figure 5D). Collectively, these data suggest that in hearts that have been subjected to hypertrophic stimulation, DKK3 suppresses the activation of JNK1/2 and p38, but has no effect on MEK1/2 and ERK1/2 activation.
Figure 5.
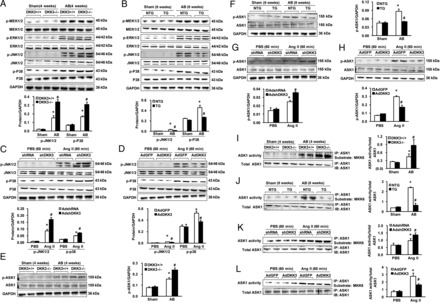
DKK3 regulates ASK1 signalling in the heart. (A and B) The levels of phosphorylated and total MEK1/2, extracellular signal-regulated kinases 1/2, JNK1/2, and P38 proteins in samples from (A) WT and KO mice and (B) NTG and TG mice at the indicated times after a sham or AB operation (n = 3, *P < 0.05 vs. WT or NTG/sham; #P < 0.05 vs. WT or NTG/AB). (C and D) Levels of phosphorylated and total JNK1/2 and p38 proteins in samples of neonatal rat cardiomyocytes that were first infected with (C) AdshDKK3 or (D) AdDKK3 and then treated with Ang II for 60 min (n = 3, *P < 0.05 vs. AdshRNA or AdGFP/PBS; #P < 0.05 vs. AdshRNA or AdGFP/Ang II). (E and F) The levels of phosphorylated and total ASK1 proteins in samples from (E) WT and KO mice and (F) NTG and TG mice at the indicated times after a sham or AB operation (n = 3, *P < 0.05 vs. WT or NTG/sham; #P < 0.05 vs. WT or NTG/AB). (G and H) The levels of phosphorylated and total ASK1 proteins in samples of neonatal rat cardiomyocytes that were infected with (G) AdshDKK3 or (H) AdDKK3 and then treated with Ang II for 60 min (n = 3, *P < 0.05 vs. AdshRNA or AdGFP/PBS; #P < 0.05 vs. AdshRNA or AdGFP/Ang II). (top) Representative blots and (bottom) quantitative results. (I and J) ASK1 kinase activity in samples from (I) WT and KO mice and (J) NTG and TG mice at the indicated times after a sham or AB operation (n = 3, *P < 0.05 vs. WT or NTG/sham; #P < 0.05 vs. WT or NTG/AB). (K and L) ASK1 kinase activity in neonatal rat cardiomyocytes that have been infected with (K) AdshDKK3 or (L) AdDKK3 and treated with Ang II for 60 min (n = 3, *P < 0.05 vs. AdshRNA or AdGFP/PBS; #P < 0.05 vs. AdshRNA or AdGFP/Ang II). (left) Representative blots and (right) quantitative results. For this figure, n indicates the number of independent experiments.
Considerable evidence exists to indicate that the activation of JNK1/2 and p38 plays a critical role in apoptotic cell death.27,28 Apoptosis signal-regulating kinase 1 (ASK1) is an MAPK kinase kinase that is activated by various stresses. ASK1 activation causes the activation of MKK3/6 and MKK4/7, which activate JNK1/2 and p38, respectively.29 To determine whether DKK3 directly inhibits JNK1/2 and p38 activation or indirectly by inhibiting upstream components of the signalling pathway, the effects of DKK3 on ASK1 activation in DKK3−/− and DKK3+/+ hearts that have been subjected to pressure overload were assessed through immunoblotting. Although the immunoreactive signals that were associated with ASK1 protein content/phosphorylation were weak, quantitative immunoblotting indicated that chronic pressure overload induced a greater increase in cardiac phosphorylated ASK1 levels in DKK3−/− than in DKK3+/+ animals (Figure 5E). In vitro kinase activity assays further demonstrated significantly greater ASK1 activity in AB-treated DKK3−/− than in AB-treated DKK3+/+ hearts (Figure 5I). Although AB significantly increased ASK1 phosphorylation in NTG mice, this increase in ASK1 phosphorylation was almost completely eliminated in TG hearts (Figure 5F). Importantly, DKK3 overexpression also mitigated AB-induced increases in ASK1 activity (Figure 5J). To further determine whether DKK3 directly inhibits ASK1 in cardiomyocytes, cultured neonatal rat cardiomyocytes were subjected to either AdshDKK3-mediated DKK3 knockdown or AdDKK3-mediated DKK3 overexpression followed by treatment with 1 μM Ang II for 60 min. As expected, Ang II-induced ASK1 phosphorylation was markedly increased by AdshDKK3, but dramatically decreased by AdDKK3 (Figure 5G and H); moreover, Ang II-stimulated ASK1 activity was increased in AdshDKK3-infected cardiomyocytes (Figure 5K), but significantly attenuated in AdDKK3-infected cardiomyocytes (Figure 5L). Notably, co-immunoprecipitation experiments in HEK293T cells transfected with FLAG-tagged ASK1 and hemagglutinin-tagged DKK3 showed that DKK3 could co-immunoprecipitate along with ASK1 and vice versa (Figure 6A and B), which was consistent with the results of endogenous co-immunoprecipitation in NRCMs (see Supplementary material online, Figure S4A and B). Taken together, these results indicate that the overexpression of DKK3 blocks the activation of ASK1 in hypertrophic hearts; in contrast, a loss of DKK3 promotes the hypertrophic stress-induced activation of ASK1.
Figure 6.
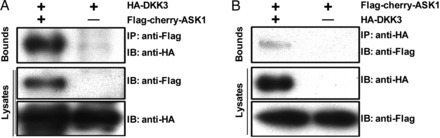
DKK3 interacts with ASK1. (A) Western blots with Flag or hemagglutinin antibodies after the use of a Flag antibody to co-immunoprecipitate ASK1 from HEK293T whole cell lysates. (B) Western blots with Flag or HA antibodies after the use of an HA antibody to co-immunoprecipitate DKK3 from HEK293T whole cell lysates. Three independent experiments were performed.
3.6. DKK3-mediated anti-hypertrophic effects are largely dependent on the inhibition of ASK1 signalling
We and others have previously demonstrated that the activation of the ASK1 signalling cascade is associated with cardiac hypertrophy.30–34 To determine whether DKK3 exerts anti-hypertrophic effects in an ASK1 inhibition-dependent fashion, we co-infected neonatal cardiomyocytes with both AdASK1 and AdDKK3; these cardiomyocytes were then treated with Ang II. The results of analyses of cardiomyocte surface area revealed that the overexpression of ASK1 significantly attenuated DKK3-elicited inhibitory effects on Ang II-induced cell hypertrophy (see Supplementary material online, Figure S5A), suggesting that DKK3-induced anti-hypertrophy may depend on the inhibition of ASK1.
To confirm the above in vitro findings in an in vivo model that exhibits greater pathological relevance, ASK1-inducible transgenes were introduced into mice with DKK3 TG backgrounds. Conditional ASK1 TG mice (ASK1-Cre-TG mice) were generated by crossing ASK1 flox mice with TG α-MHC-MerCreMer (MEM) mice, and DKK3 TG mice and ASK1-Cre-TG mice were crossed to create DKK3/ASK1-Cre DTG animals (see Supplementary material online, Figure S5B and C). To induce Cre-dependent recombination, 6-week-old ASK1-Cre-TG, and DTG mice were treated with tamoxifen (80 mg/kg/day, ip) for 5 consecutive days. The mice were identified by western blot analysis (see Supplementary material online, Figure S5D). After 4 weeks of AB, ASK1-Cre-TG mice exhibited significantly increased cardiac hypertrophy compared with MEM-Cre controls (see Supplementary material online, Figure S5E–L). Importantly, in the ASK1 TG background, DKK3 overexpression no longer protected against the hypertrophic effects that were induced by 4 weeks of AB, as evidenced by the fact that AB treatment caused increases in the HW/BW, LW/BW, and HW/TL ratios in these animals (see Supplementary material online, Figure S5E–G). Histological analyses also revealed that, as indicated by relative cardiomyocyte CSA, the protective effect of DKK3 overexpression on cardiac hypertrophy was compromised during cardiac ASK1 activation (see Supplementary material online, Figure S5H and I). In DKK3 TG mice with an ASK1 TG background, DKK3 overexpression did not attenuate myocardial fibrosis after 4 weeks of AB (see Supplementary material online, Figure S5H and J). Furthermore, in the cardiac ASK1 TG background, DKK3 overexpression did not blunt either the foetal cardiac gene or fibrotic marker expression after chronic pressure overload (see Supplementary material online, Figure S5K and L). Thus, the experimental results generated for both in vitro cardiomyocytes and in vivo animal hearts consistently demonstrate that restored cardiac ASK1 expression negates DKK3-induced anti-hypertrophic effects. These data suggest that the protective role of DKK3 in hypertrophic hearts is at least partially dependent on the inactivation of ASK1.
3.7. Inactivation of cardiac ASK1 rescues the adverse effect of DKK3 deficiency on pressure overload-induced cardiac remodelling
As shown in Supplementary material online, Figure S5, ASK1 activation could, at least partially, negate the anti-hypertrophic effect of DKK3 overexpression in the heart. Thus, we were interested in examining whether the inactivation of ASK1 could have therapeutic potential for the treatment of cardiac hypertrophy. To conduct this examination, we infected neonatal rat cardiomyocytes with both AddnASK1 (a catalytically inactive mutant form of ASK1 in which Lys-709 has been replaced by Arg) and AdshDKK3. We then treated the infected cells with Ang II to induce cell hypertrophy. The results of cell surface area analyses revealed that increases in cell hypertrophy caused by DKK3 knockdown were significantly mitigated by the prevention of ASK1 activation (see Supplementary material online, Figure S6A). To test whether ASK1 inactivation could rescue the abnormalities that have been observed in AB-treated DKK3−/− mice, we generated a cross model to produce animals with dnASK1 overexpression in DKK3-null hearts (see Supplementary material online, Figure S6B and C). In these genetic models, the inactivation of ASK1 by its dominant-negative mutant was confirmed by western blotting (ASK1-MKK6-JNK/P38; see Supplementary material online, Figure S6D). After 4 weeks of AB, cardiac-specific dnASK1-TG mice exhibited significant attenuation of cardiac hypertrophy and fibrosis compared with NTG controls (see Supplementary material online, Figure S6E–L). Interestingly, after DKK3-deficient mice were crossed into the dnASK1-TG mice background, DKK3 deficiency no longer promoted AB-triggered increases in the HW/BW, LW/BW, and HW/TL ratios (see Supplementary material online, Figure S6E–G), the CSA of cardiomyocytes, or volumes of LV fibrosis (see Supplementary material online, Figure S6H–L). Taken together, these data suggest that the inhibition of ASK1 can prevent DKK3 reduction-induced cardiac remodelling in response to hypertrophic stimuli.
4. Discussion
It has been well documented that cardiac hypertrophy is an important cause of heart failure that reciprocally reinforces the development of heart failure. However, our understanding of the pathogenesis of cardiac hypertrophy at the molecular level remains extremely limited. In the present study, we identify DKK3 as an intrinsic negative regulator of cardiac hypertrophy and demonstrate that DKK3 prevents maladaptive remodelling and a transition to heart failure in the context of chronic pressure overload. The major novel findings of this study are as follows: (i) DKK3 expression is significantly decreased in both failing human hearts and hypertrophic murine hearts; (ii) DKK3 protects against cardiac hypertrophy in response to hypertrophic stimuli both in vitro and in vivo; and (iii) DKK3 functions as a negative regulator of pressure overload-induced cardiac hypertrophy, at least partly, through the regulation of ASK1-JNK/p38 signalling. Thus, we provide the first indications that DKK3 plays a critical role in attenuating pressure overload-induced cardiac remodelling.
The mechanisms by which DKK3 attenuates cardiac hypertrophy are likely mediated by the inactivation of the MAPK signalling cascade. Numerous studies have determined that the activation of the MAPK signalling pathway contributes to the development of cardiac hypertrophy and heart failure.26,35,36 The MAPK cascade, which is initiated in cardiomyocytes by stress stimuli, involves a sequence of successively acting kinases, including p38, JNKs, and ERKs.36 Once this cascade has been activated, p38, JNKs, and ERKs phosphorylate a wide array of intracellular targets, including numerous transcription factors; this phosphorylation results in the reprogramming of cardiac gene expression.26 To elucidate the molecular mechanisms that are involved in DKK3-mediated anti-hypertrophic effects, we examined the status of the MAPK signalling cascade in our hypertrophic models. Interestingly, we found that the activation of both p38 and JNK1/2 in response to chronic pressure overload or Ang II stimulation was almost completely blocked by the cardiac-specific overexpression of DKK3, but greatly enhanced by DKK3 deficiency. These findings suggest that DKK3 exerts its anti-hypertrophic effects through the inhibition of p38 and JNK1/2 signalling.
Given that ASK1 is a major upstream regulator of p38 and JNK,27,34 DKK3 inhibition of p38/JNK suggests that DKK3 may affect the activation of ASK1. Indeed, the results presented in this study reveal that ASK1 activity was significantly reduced in AB-treated DKK3 hearts, and that the overexpression of ASK1 offsets the protective effects of DKK3 against AB-induced cardiac remodelling. Moreover, our data indicate that ASK1 is required for pressure overload-induced cardiac hypertrophy and dysfunction in DKK3-null hearts; these findings are consistent with previous reports that identify ASK1 as an essential factor for the development of cardiac hypertrophy and failure.30–34 Thus, we believe that the mechanisms that underlie DKK3-induced anti-hypertrophic effects could plausibly be dependent on the inhibition of the ASK1-p38/JNK cascade. In co-immunoprecipitation experiments that addressed how DKK3 negatively regulates the activation of ASK1, we observed that DKK3 can physically bind to ASK1, which was confirmed by endogenous co-immunoprecipitation (Figure 6A and B and see Supplementary material online, Figure S4A and B), suggesting that DKK3 may cause ASK1 to become inaccessible to activation. Future studies will be required to map the interactive domains of these two proteins.
There is general agreement that DKK3 is a secreted protein.9,10 However, in recent years, numerous studies have demonstrated that DKK3 not only functions as a secreted protein, but also plays an important role as a cytosolic protein. Hsieh et al.37 showed that DKK3 has two isoforms and the 55 kDa in the cytosol while the 50-kDa isoform was secreted into the medium. Krupnik et al.9 showed that soluble human DKK3 protein displayed increase in mobility after N-glycanase treatment, and the major form (45–65 kDa) of soluble human DKK3 was observed but two splices of 45–55 and 40 kDa following deglycosylation. In our study, we also found that DKK3 also runs in a double band all the time in the western blot, which indicated that DKK3 was deglycosylation in the heart. Sakaguchi et al.38 showed that IL-7 mRNA was induced by intracellular production of DKK3 but not secreted DKK3 protein. Ochiai et al.39 demonstrated that DKK3 was co-localized with Tctex-1 around the endoplasmic reticulum of human fibroblasts. Overexpression of DKK3 in HEK293 cells showed that DKK3 was located throughout the cytoplasm.40 Immunohistochemical staining showed that DKK3 was punctuated in the cellular cytoplasm in the brain and liver tissue.41 All these studies have indicated that DKK3 both was distributed in cytoplasm and was extracellular. Notably, in our study, the results of immunostaining (Figure 1C), endogenous Co-IP (see Supplementary material online, Figure S4A and B), and western blot of DKK3 showed that DKK3 also located intracellular and functions as a regulator to interact with ASK1, and then to suppress cardiac hypertrophy. Even so, further experiments will be needed to be carried out to elucidate how DKK3 could escape from the exocytosis and locate and function intracellularly in the heart.
In conclusion, this investigation is the first study to define the role of DKK3 in cardiac remodelling. Our findings indicate that DKK3 functions as a novel negative modulator of cardiac hypertrophy and failure in response to pressure overload. The underlying mechanism of the protective role of DKK3 against the development of cardiac hypertrophy appears to involve the inhibition of ASK1-JNK/p38 signalling (Figure 7). Therefore, the present study also provides novel insights into the molecular mechanisms of pathological cardiac remodelling. Based on these findings, DKK3 may represent a new therapeutic target for suppressing the onset of heart failure.
Figure 7.
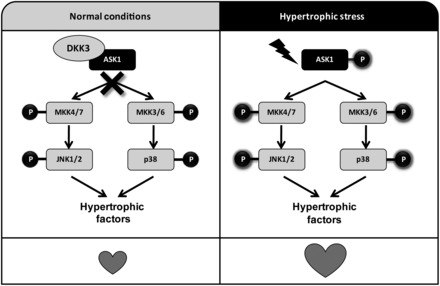
The proposed mechanism of DKK3 regulation of cardiac hypertrophy. On the basis of our in vitro and in vivo data, we propose that DKK3 physically interacts with ASK1 to inhibit ASK1 activity under normal conditions (left panel). Under conditions of aortic banding or other hypertrophic stress, DKK3 is down-regulated and dissociates from ASK1 (right panel); as a result, ASK1 is phosphorylated and activates MKK4/7 and MKK3/6, which then sequentially phosphorylate downstream JNK1/2 and P38, which activate multiple transcription factors that relate to cardiac hypertrophy.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by grants from the National Natural Science Foundation of China (nos 81100230, 81070089, 81200071, 81270306, and 81370365), National Science and Technology Support Project (nos 2011BAI15B02, 2012BAI39B05, 2013YQ030923-05, and 2014BAI02B01), the National Basic Research Program of China (no 2011CB503902), and the Key Project of the National Natural Science Foundation (no 81330005).
Supplementary Material
Acknowledgements
We thank Dr Takahisa Furukawa (Osaka Bioscience Institute, Japan) for providing DKK3 KO mice and Prof. Hidenori Ichijo (University of Tokyo, Tokyo, Japan) for providing the dnASK1 plasmid. We also acknowledge the valuable technological assistance that Li-Hua Gan, Rui Zhang, Ya-Fen Lin, Xue-Yong Zhu, Li Yang, and Xin Zhang provided for this study.
Conflict of interest: none declared.
References
- 1.Chen J, Normand SL, Wang Y, Krumholz HM. National and regional trends in heart failure hospitalization and mortality rates for Medicare beneficiaries 1998–2008. JAMA. 2011;306:1669–1678. doi: 10.1001/jama.2011.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–334. doi: 10.1161/CIRCULATIONAHA.108.845792. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Tang QZ, Liu C, Moon M, Chen M, Yan L, et al. Cellular FLICE-inhibitory protein protects against cardiac remodeling induced by angiotensin II in mice. Hypertension. 2010;56:1109–1117. doi: 10.1161/HYPERTENSIONAHA.110.157412. [DOI] [PubMed] [Google Scholar]
- 4.Kang YJ. Cardiac hypertrophy: a risk factor for QT-prolongation and cardiac sudden death. Toxicol Pathol. 2006;34:58–66. doi: 10.1080/01926230500419421. [DOI] [PubMed] [Google Scholar]
- 5.Li H, He C, Feng J, Zhang Y, Tang Q, Bian Z, et al. Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc Natl Acad Sci USA. 2010;107:13818–13823. doi: 10.1073/pnas.1008397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang DS, Bian ZY, Zhang Y, Zhang SM, Liu Y, Zhang R, et al. Role of interferon regulatory factor 4 in the regulation of pathological cardiac hypertrophy. Hypertension. 2013;61:1193–1202. doi: 10.1161/HYPERTENSIONAHA.111.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang DS, Luo YX, Zhang R, Zhang XD, Chen HZ, Zhang Y, et al. Interferon regulatory factor 9 protects against cardiac hypertrophy by targeting myocardin. Hypertension. 2013;63:119–127. doi: 10.1161/HYPERTENSIONAHA.113.02083. [DOI] [PubMed] [Google Scholar]
- 8.Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25:7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- 9.Krupnik VE, Sharp JD, Jiang C, Robison K, Chickering TW, Amaravadi L, et al. Functional and structural diversity of the human Dickkopf gene family. Gene. 1999;238:301–313. doi: 10.1016/s0378-1119(99)00365-0. [DOI] [PubMed] [Google Scholar]
- 10.Monaghan AP, Kioschis P, Wu W, Zuniga A, Bock D, Poustka A, et al. Dickkopf genes are co-ordinately expressed in mesodermal lineages. Mech Dev. 1999;87:45–56. doi: 10.1016/s0925-4773(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 11.Mallarino R, Campas O, Fritz JA, Burns KJ, Weeks OG, Brenner MP, et al. Closely related bird species demonstrate flexibility between beak morphology and underlying developmental programs. Proc Natl Acad Sci USA. 2012;109:16222–16227. doi: 10.1073/pnas.1206205109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abarzua F, Sakaguchi M, Takaishi M, Nasu Y, Kurose K, Ebara S, et al. Adenovirus-mediated overexpression of REIC/Dkk-3 selectively induces apoptosis in human prostate cancer cells through activation of c-Jun-NH2-kinase. Cancer Res. 2005;65:9617–9622. doi: 10.1158/0008-5472.CAN-05-0829. [DOI] [PubMed] [Google Scholar]
- 13.Kawano Y, Kitaoka M, Hamada Y, Walker MM, Waxman J, Kypta RM. Regulation of prostate cell growth and morphogenesis by Dickkopf-3. Oncogene. 2006;25:6528–6537. doi: 10.1038/sj.onc.1209661. [DOI] [PubMed] [Google Scholar]
- 14.Papatriantafyllou M, Moldenhauer G, Ludwig J, Tafuri A, Garbi N, Hollmann G, et al. Dickkopf-3, an immune modulator in peripheral CD8 T-cell tolerance. Proc Natl Acad Sci USA. 2012;109:1631–1636. doi: 10.1073/pnas.1115980109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee EJ, Jo M, Rho SB, Park K, Yoo YN, Park J, et al. Dkk3, downregulated in cervical cancer, functions as a negative regulator of beta-catenin. Int J Cancer. 2009;124:287–297. doi: 10.1002/ijc.23913. [DOI] [PubMed] [Google Scholar]
- 16.Mizobuchi Y, Matsuzaki K, Kuwayama K, Kitazato K, Mure H, Kageji T, et al. REIC/Dkk-3 induces cell death in human malignant glioma. Neuro Oncol. 2008;10:244–253. doi: 10.1215/15228517-2008-016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koppen A, Ait-Aissa R, Koster J, Ora I, Bras J, van Sluis PG, et al. Dickkopf-3 expression is a marker for neuroblastic tumor maturation and is down-regulated by MYCN. Int J Cancer. 2008;122:1455–1464. doi: 10.1002/ijc.23180. [DOI] [PubMed] [Google Scholar]
- 18.Oceandy D, Pickard A, Prehar S, Zi M, Mohamed TM, Stanley PJ, et al. Tumor suppressor Ras-association domain family 1 isoform A is a novel regulator of cardiac hypertrophy. Circulation. 2009;120:607–616. doi: 10.1161/CIRCULATIONAHA.109.868554. [DOI] [PubMed] [Google Scholar]
- 19.Huang H, Tang QZ, Wang AB, Chen M, Yan L, Liu C, et al. Tumor suppressor A20 protects against cardiac hypertrophy and fibrosis by blocking transforming growth factor-beta-activated kinase 1-dependent signaling. Hypertension. 2010;56:232–239. doi: 10.1161/HYPERTENSIONAHA.110.149963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang DS, Zhang XF, Gao L, Zong J, Zhou H, Liu Y, et al. Signal regulatory protein-alpha protects against cardiac hypertrophy via the disruption of Toll-like receptor 4 signaling. Hypertension. 2013;63:96–104. doi: 10.1161/HYPERTENSIONAHA.113.01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu J, Bian ZY, Zhang R, Zhang Y, Liu C, Yan L, et al. Interferon regulatory factor 3 is a negative regulator of pathological cardiac hypertrophy. Basic Res Cardiol. 2013;108:326. doi: 10.1007/s00395-012-0326-9. [DOI] [PubMed] [Google Scholar]
- 22.Li HL, Zhuo ML, Wang D, Wang AB, Cai H, Sun LH, et al. Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation. 2007;115:1885–1894. doi: 10.1161/CIRCULATIONAHA.106.656835. [DOI] [PubMed] [Google Scholar]
- 23.Yi W, Sun Y, Yuan Y, Lau WB, Zheng Q, Wang X, et al. C1q/tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation. 2012;125:3159–3169. doi: 10.1161/CIRCULATIONAHA.112.099937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung H, Seong HA, Manoharan R, Ha H. Serine-threonine kinase receptor-associated protein inhibits apoptosis signal-regulating kinase 1 function through direct interaction. J Biol Chem. 2010;285:54–70. doi: 10.1074/jbc.M109.045229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park HS, Cho SG, Kim CK, Hwang HS, Noh KT, Kim MS, et al. Heat shock protein hsp72 is a negative regulator of apoptosis signal-regulating kinase 1. Mol Cell Biol. 2002;22:7721–7730. doi: 10.1128/MCB.22.22.7721-7730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 27.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 28.Xiao J, Moon M, Yan L, Nian M, Zhang Y, Liu C, et al. Cellular FLICE-inhibitory protein protects against cardiac remodelling after myocardial infarction. Basic Res Cardiol. 2012;107:239. doi: 10.1007/s00395-011-0239-z. [DOI] [PubMed] [Google Scholar]
- 29.Takeda K, Matsuzawa A, Nishitoh H, Ichijo H. Roles of MAPKKK ASK1 in stress-induced cell death. Cell Struct Funct. 2003;28:23–29. doi: 10.1247/csf.28.23. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Tao L, Jiao X, Gao E, Lopez BL, Christopher TA, et al. Nitrative thioredoxin inactivation as a cause of enhanced myocardial ischemia/reperfusion injury in the aging heart. Free Radic Biol Med. 2007;43:39–47. doi: 10.1016/j.freeradbiomed.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao L, Jiao X, Gao E, Lau WB, Yuan Y, Lopez B, et al. Nitrative inactivation of thioredoxin-1 and its role in postischemic myocardial apoptosis. Circulation. 2006;114:1395–1402. doi: 10.1161/CIRCULATIONAHA.106.625061. [DOI] [PubMed] [Google Scholar]
- 32.Izumiya Y, Kim S, Izumi Y, Yoshida K, Yoshiyama M, Matsuzawa A, et al. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II-induced cardiac hypertrophy and remodeling. Circ Res. 2003;93:874–883. doi: 10.1161/01.RES.0000100665.67510.F5. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi O, Higuchi Y, Hirotani S, Kashiwase K, Nakayama H, Hikoso S, et al. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc Natl Acad Sci USA. 2003;100:15883–15888. doi: 10.1073/pnas.2136717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taniike M, Yamaguchi O, Tsujimoto I, Hikoso S, Takeda T, Nakai A, et al. Apoptosis signal-regulating kinase 1/p38 signaling pathway negatively regulates physiological hypertrophy. Circulation. 2008;117:545–552. doi: 10.1161/CIRCULATIONAHA.107.710434. [DOI] [PubMed] [Google Scholar]
- 35.Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh SY, Hsieh PS, Chiu CT, Chen WY. Dickkopf-3/REIC functions as a suppressor gene of tumor growth. Oncogene. 2004;23:9183–9189. doi: 10.1038/sj.onc.1208138. [DOI] [PubMed] [Google Scholar]
- 38.Sakaguchi M, Kataoka K, Abarzua F, Tanimoto R, Watanabe M, Murata H, et al. Overexpression of REIC/Dkk-3 in normal fibroblasts suppresses tumor growth via induction of interleukin-7. J Biol Chem. 2009;284:14236–14244. doi: 10.1074/jbc.M808002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ochiai K, Watanabe M, Ueki H, Huang P, Fujii Y, Nasu Y, et al. Tumor suppressor REIC/Dkk-3 interacts with the dynein light chain, Tctex-1. Biochem Biophys Res Commun. 2011;412:391–395. doi: 10.1016/j.bbrc.2011.07.109. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura RE, Hackam AS. Analysis of Dickkopf3 interactions with Wnt signaling receptors. Growth Factors. 2010;28:232–242. doi: 10.3109/08977191003738832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang K, Watanabe M, Kashiwakura Y, Li SA, Edamura K, Huang P, et al. Expression pattern of REIC/Dkk-3 in various cell types and the implications of the soluble form in prostatic acinar development. Int J Oncol. 2010;37:1495–1501. doi: 10.3892/ijo_00000802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.