

Abstract
Individual mutations in mice can slow aging: They extend life span by retarding a wide range of harmful, age-dependent changes in multiple cells and tissues. Evolutionary changes—by definition, changes in DNA sequence—can lead to even more dramatic postponement of age-dependent deterioration. Genetic variation within a species, for example among breeds of dogs, can also lead to major changes in aging rate, although there is not yet any strong evidence for similar genetic variation that modulates aging in rodents or humans. This essay compares different strategies for using genetic information to clarify questions in biogerontology, suggesting an emphasis on genes that can retard multiple forms of age-dependent dysfunction in parallel.
Keywords: Longevity, Mice, Genetics, Comparative biology
The goal of this essay is to provide a framework for thinking about how genetic data—DNA sequences and their variation—can help clarify the biology of aging, the process that leads to multiple forms of decline in the last third of the adult life span. The concepts of aging and its relationship to disease are slippery enough that expert opinion varies widely on the most productive approach to using genetic methods in biological gerontology. Many millions of dollars, and hundreds of research careers, have been committed based, too often implicitly, on such strategic branch points. Some of those who work on “the genetics of aging” focus on questions about segregating polymorphic alleles that modulate life span or risks of specific late-life diseases. Others study rare mutations that appear to accelerate the aging process or some segment of it. Some workers concentrate on mutations, in model organisms, that extend life span by deceleration of aging and its effects. A small contingent of comparative biologists seeks insight from differences in physiology and cell biology among related species, or breeds, that age at different rates. This article, which is intended to be provocative rather than to portray a consensus viewpoint, develops the position that some kinds of genetic research are more likely than others to produce important insights into how aging is regulated and how research on aging might someday lead to useful clinical maneuvers. More specifically, I want to argue that the principal goal of genetic research on aging should be to discover how specific genes can and do postpone, in parallel, the many varieties of age-related senescent change in dividing cells, nondividing cells, cell parts, and extracellular materials. The key question here is not “Are there genes for aging?” but rather “How do genes postpone aging?”
Asking the question “What is aging?” at a biogerontology meeting elicits an unsettling mixture of puzzlement and contempt. To neophytes, the question is too easy—they know full well what aging is—and to cognoscenti, the question is too besmirched by sad memories of prior debates to deserve any further dyspeptic rumination. This essay starts from the perspective that not all answers to this chestnut are equally persuasive and that picking the wrong answer can doom a scientist to wasting a lifetime of experimental attention on tangential problems. The issues are not merely semantic, merely philosophical; they are not trivial but pivotal. Finding “genes against aging” requires careful consideration of what aging is, how one might measure it, and how one might recognize such genes. This essay will present a series of interconnected questions, none of which can be answered persuasively without careful consideration of how aging, longevity, and diseases are distinct from one another. Box 1 provides one nosology of the questions at hand.
Box 1. Questions that might be of interest to biogerontologists who want to work with geneticists and vice versa.
(a) Are there polymorphisms present in natural populations of mice and people that modulate aging rate (as distinct from age at death)? What kind of evidence would be relevant to addressing this question? Are such polymorphisms present among breeds of dogs?
(b) What are the assumptions of studies that use age at death as a surrogate for aging rate and the problems with these assumptions? What needs to be done to improve the ability of gene association studies to find alleles (if indeed any exist) that modulate aging rate in humans?
(c) What can single gene mutations, whether naturally occurring or engineered by recombinant methods, tell us about the biology of aging as it affects normal mice and people?
(d) Are mutations that appear to mimic some features of aging, the so-called progeroid disease mutations, at all relevant to questions about the aging process in normal individuals?
(e) Do studies of animals that are prone to specific diseases, such as animal models of Alzheimer’s disease, give any insights into the biology of aging and its relationship to late-life diseases?
(f) Can studies of differences in physiology and cell biology among species that differ in aging rate guide evaluation of interspecies genetic changes relevant to aging?
Interest in the biology of aging is sparked mostly by the realization that aging leads to impairments of health and function, mediated by a mixture of diseases, disabilities, and troublesome loss of cognitive and physical function: a coordinated semisynchronous decline. Semisynchrony is not lockstep progression: It is rare for someone to develop cancer, Alzheimer’s disease, and a broken hip on the same day. But a person, or other mammal, who has reached an age where he/she already shows some loss of hearing, some osteoarthritis, and somewhat diminished cognitive function is also far more likely to show other manifestations of aging than someone just reaching mature adulthood. Members of differing mammalian species age in similar ways, but at very different rates. If a physical examination reveals mild cataracts, reduced cardiovascular reserves, diminished expiratory flow rates, poor immune response to vaccines, diminished olfaction, joint erosion, and a nascent malignancy, it is a very good bet that the examinee is old. But it is not possible to guess the age of the old subject unless you know which species you are dealing with, because the clinical features apply about equally well to a 2-year-old mouse, 15-year-old dog, 25-year-old horse, 30-year-old chimpanzee, or 80-year-old human. It is uncommon to find a 2-year-old mouse or 15-year-old dog who has avoided all such “age-dependent” changes and equally uncommon to encounter a 20-year-old human who exhibits serious decline in any of these listed systems. What biological process, or processes, can effectively postpone age-related changes in dividing and nonmitotic cells, in extracellular macromolecules, and in neuroendocrine control networks for 50 years in some mammals, 20 years in others, and a mere 3 years in still others, despite exposure to the same endogenous and external challenges that produce the signs and symptoms of aging?
It is a standard practice to refer to “the aging process” as though aging were a factory that transformed raw materials—healthy young adults—into the ailing and vulnerable forms we recognize in our old friends and old pets. This “process of aging” analogy invokes, sometimes explicitly, the success of developmental biologists in piecing together molecular programs of cause and effect by which the cells in a blastocyst build neonates and then adults. This mode of thinking prompts questions about which of the many proposed causes—oxidation, or glycation, or loss of stem cell function, or inflammatory circuits, or somatic mutation, or some combination of the above—give rise to the diseases and malfunctions seen in old people. This kind of metaphor is very familiar, and has been very productive, in experimental medicine: Helicobacter pylori causes ulcers, atherosclerosis causes heart attacks, so what causes aging? The “process of aging” metaphor is that of a blueprint or recipe or algorithm, a series of steps which, when executed, can turn a blastocyst into an adult and then turn an adult into an old person. From this viewpoint, the genes for aging, like genes that cause or promote specific developmental abnormalities or late-life illnesses, are imagined to work by blocking or speeding specific steps in the developmental progression that converts a healthy adult into someone aged.
The fundamental, and important, flaw in this metaphor is that aging and development have dissimilar, and in critical ways opposite, relationships to evolutionary pressures. Genetic alleles that affect the development of mature adults are under strong, direct, selective pressure. Mutants that interfere with healthy development are lost from the germline, and those rare mutations that improve Darwinian fitness can replace the normal alleles from which they arise. In contrast, the phenotypic changes that make up the biology and pathology of aging, that is, the cosmetic changes and immune dysfunction and hip fractures, etc., occur largely at ages beyond those of maximum reproductive output (1–3). A genetic allele that can prevent strokes in an 80-year-old woman, or prevent infection in a 20-year-old dog, or diminish cataract risk in a 4-year-old mouse, does not have any selective advantage because nearly all human, dog, or mouse reproduction occurs at earlier ages.
From this perspective, the “aging process” reflects the conjoined actions of many different forms of threat, including tenacious cross-linking of diverse macromolecules, neoplastic transformation, loss of irreplacable cells and organs (neurons, teeth, and pancreatic β-cells), decay of intracellular and extracellular homeostatic circuits, somatic mutations, and unrestrained accumulation of cells and cell products that have beneficial effects in young adults but pile up, to bad effect, beyond reproductive ages. Postponement of aging may indeed have evolutionary value in specific niches, that is, those in which reproductive success depends not so much on rapid production of initial litters (as in mice) but instead on production of high-quality offspring, repeatedly, over long time intervals (as in bats, humans, and many bird species).
But—and here is the crux of the argument—how is it possible for genetic alleles to protect against such a very wide range of deleterious molecular and cellular changes? Imagine a short-lived species, like a mouse or opossum, in which the multiple pathologies of aging all occur within 2 years, which finds itself in an ecological niche in which postponed aging would be advantageous. A mutant allele that delays cataract development would have no selective advantage because the exponential age-dependent increases in risks of cancer, sarcopenia, immune senescence, bone fragility, etc., would rapidly impair fitness even if cataracts were efficiently delayed. Similarly, an allele that protects against neoplasia would have little fitness value in organisms that continue to show rapid age-dependent increases of cataracts, bone fragility, sarcopenia, and immune senescence. Alleles that delay sarcopenia would be useless in an organism still vulnerable to neoplasia, cataracts, immune decline, and so forth. The implication here is that evolution into niches in which delayed aging conveys improved fitness must depend on mutations that can retard, in parallel, most or all of the signs and symptoms that afflict older animals.
It is clear that genetic mutation and selection can accomplish such a feat: The differences in aging rate and longevity between dogs and mice are due to differences in genetic constitution and cannot be replicated by environmental modulations. Evolution has created long-lived species from shorter-lived progenitors repeatedly in multiple branches of the mammalian radiation, from multiple starting points. Evidence on the speed with which longevity evolves is limited, but there are reasons to suspect that slow aging opossums have been created within a few thousand generations (4), and artificial selection on body size within dog breeds has created subgroups with radically different maximal life span within a few hundreds of generations (see later).
THERE ARE GENES AGAINST AGING
The idea that single genetic mutations, or small sets of mutations, could delay multiple distinct aspects of aging has seemed intuitively unlikely, so much so as have been declared impossible on theoretical grounds by distinguished biologists (5). These theoretical proofs of impossibility had to be revised (6) to deal with evidence that single gene mutations in Caenorhabditis elegans could have potent effects on maximal life span; the revision took the form of an argument that the dauer pathways used by C. elegans to adjust to resource-poor environments made them a special nonrepresentative case. Evidence that single gene mutations can delay aging and extend life span in mice (7–11) has now established convincingly that multiple aspects of aging can indeed be delayed, in parallel, by surprisingly modest changes in genetic sequence and has even begun to suggest molecular pathways (12–14) that could mediate such broad-spectrum resistance to threats as distinct as DNA damage, protein cross-linking, and disruption of cellular control pathways. It is, however, by no means clear that the genetic loci whose mutation can slow aging in mice, let alone in flies or worms, overlap to any extent with the set of loci responsible for differences in aging rate among mammalian species. Nor is it clear that the physiological processes that postpone aging in chimps are the same as those that slow aging in naked mole rats, elephants, and ostriches. Elucidating the physiology of slow aging in multiple clades and evaluating genetic variants that account for exceptional longevity is potentially a major theme for new discovery in biological gerontology.
How do genes postpone aging, that is, postpone all aspects of aging in parallel? Figuring out good strategies to attack this question requires that we both distinguish among and note the intimate connections among, aging, life span, and disease.
Aging is not the same as disease, even though it increases the incidence and severity of disease. Failing to make this distinction confuses cause with effect and process with product. The key point is that aging increases the risk of multiple diseases in parallel and at the same time also increases the incidence and severity of impairments that are not traditionally thought of as diseases (such as wrinkled skin, impairments of smell, and many others). Interventions that slow aging, such as single gene mutations, evolutionary divergence, and caloric restriction, have as a hallmark a parallel deceleration of multiple lethal diseases, multiple nonlethal diseases, and multiple forms of age-associated dysfunction whose impact on health and mortality may be either worrisome or trivial. An intervention (or genetic allele) that protects against wrinkled skin, or heart attacks, or hearing loss, but does not show parallel effectiveness against a wide range of age-dependent outcomes cannot sensibly be said to have an antiaging effect.
Although differences in the rate of aging alter life expectancy of adults, life expectancy of adults can also be modulated by pathways that do not involve aging. A strain of mice in which hemophilia is a common cause of death will benefit from injections of clotting factor, even though a bleeding dyscrasia is not related to the biology of aging. Similarly, environmental changes that reduce the incidence of smoking, improve dental hygiene, immunize against influenza, mandate seat belts, and provide insulin to diabetics may improve survival of older adults, but through mechanisms that do not modulate aging per se.
So: Is aging under genetic control? Can genetic studies tell us about the molecular and cellular biology of aging? How many human polymorphisms modulate aging? Can genetic alleles accelerate aging? The notion of aging as a process that leads to multisystem failure, at a rate which is itself subject to genetic modulation, leads to answers for each of these questions, answers that in some ways cast doubt on conventional wisdom and on consensus opinion about optimal deployment of resources for research on aging and age-associated disease.
ARE THERE POLYMORPHISMS PRESENTIN NATURAL POPULATIONSOF MICEAND PEOPLE THAT MODULATE AGING RATE (AS DISTINCT FROM AGEAT DEATH)?
I do not believe that there is any convincing evidence, yet, for polymorphisms that modulate rate of aging in mice or people. Worse yet, I do not believe that studies currently under way in either species are well designed to produce evidence on this question. Human gene mapping efforts, starting with candidate gene surveys dating back at least to 1993 (15), and extending to recent genome-wide association surveys (reviewed in [16]), have almost always sought associations between polymorphisms and age at death, with emphasis on contrasts between exceptionally old individuals, that is, centenarians or near-centenarians and suitable control populations. The design flaw here is in the choice of phenotype—age at death. In countries with sufficiently good health systems to produce very old citizens, most deaths are caused by complications of cardiovascular disease, and so alleles that have even modest propensity to speed or slow atherosclerosis have a major effect on survival to extreme old age. Alleles that modulate risks of the most common cancers would similarly be enriched or depleted in the very old. Surveys based only on age at death, if sufficiently powered, would thus be expected to collect loci whose effects on survival are mediated by modulation of these two common lethal diseases, whether or not the loci work through an alteration of the aging process per se.
A study to look for human loci that modify aging would need to use a different phenotype, a phenotype that incorporates age-sensitive traits from many tissues and organ systems, including many traits which seldom lead to death. The test battery would focus on traits that often show age-related change at ages where few adults have died. An allele that is seen mostly in those 50-year-olds that retain excellent hearing, are free of cataracts, respond vigorously to immunization, retain high levels of lung elasticity, have above average glucose tolerance, and have no signs of incipient chronic illness would have a decent prima facie case as an antiaging allele.
It is not yet clear if any such loci exist in humans, but there is a strong positive precedent from another familiar species, the dog. It is amply documented (17) that dog breeds generated by selective breeding for large body size are shorter-lived than average, and small breeds are longer-lived. In one survey (18,19), 56% of the variance among dog breeds in mean life span could be attributed by regression analysis to variance in mean breed body size, with similarly strong associations seen in many other reports on purebred (20) and mixed-breed (21) dogs. Breed size, in turn, is in many cases known to be related to genetically controlled differences in growth hormone–induced production of IGF-1 (insulin-like growth factor-1) (22). The key point is that these differences in breed-specific life span are accompanied by, and almost certainly due to, parallel differences in rate of occurrence of multiple age-dependent diseases. Data from dog insurance databases have shown that smaller breeds differ systematically from larger breeds in the risks of tumors, lethal trauma, locomotor dysfunction, heart disease, neurological disease, and a set of miscellaneous other lethal conditions (23). Risk of cataract, too, correlates well with breed longevity (24). As reviewed elsewhere (17), the connection of IGF-1 and body size to aging and longevity is consistent with data showing extended life span in many stocks of mice with abnormally low-growth hormone/IGF-1 tonicity, with connections of small size with healthspan among horse breeds (25), and with the amply documented resistance of shorter humans to multiple forms of neoplasia (26–32) and cataract (33). Recent work on a set of families with genetic defects in the receptor for growth hormone (34), who seem to be resistant to neoplasia and diabetes, is also consistent with the implication that polymorphisms that modulate growth hormone/IGF-1 in dogs may be able to delay multiple forms of disease in parallel, the hallmark of genes that affect aging. Whether human populations do indeed have polymorphic loci that modulate aging and whether these are related to IGF-1 signals are separate questions still to be answered.
HOW DO STUDIESOF SINGLE GENE MUTATIONS,IN MICEOR INVERTEBRATES, HELP ADDRESS QUESTIONSINTHE BIOLOGYAND GENETICSOF AGING?
There are now several hundred loci at which mutations have been shown to extend life span in worms or flies and more than 20 loci with at least initial claims of life-span extension in mice, though few of the latter have been confirmed, tested on multiple backgrounds, or evaluated for effects on age-dependent end points other than death. These mutant collections are exceptionally helpful in some respects, and much less so in others. Many of the antiaging mutations, such as the Snell and Ames dwarf models, produce animals that are infertile or subfertile and which have weaknesses (small body size and poor resistance to infection and to cold temperatures), which would greatly impair vigor and fitness in a natural setting. The longevity of such mice is revealed only in a protected laboratory environment, and thus these model organisms do not provide examples of how to combine excellent adult health with exceptional resistance to the aging process. It is possible that the loci that slow aging in mice might also encode, in humans, polymorphisms that modulate the rate of aging. The value of antiaging mutations in mice, however, does not rest on this shaky assumption but instead on the realization that they provide invaluable clues to the central unsolved problem in the biology of aging: How do antiaging defenses slow down the signs and symptoms of aging in parallel? Demonstration that a specific mutation extends life span is just the first step in building a case that the life-span extension is caused by slower aging, but such a case is slowly developing for the best studied of the mouse single-gene mutants (9,35–37). Once the evidence that excess longevity is the result of slow aging becomes convincing for a particular mutation, analysis of the pathways by which the mutant alters cell and developmental biology and midlife physiology is currently our best source of insights into how antiaging defenses work in mammals. Extension of these insights to human pathophysiology, and to the search for antiaging pharmaceuticals, does not require evidence that homologous loci modulate life span in people.
CAN STUDYOF MUTATIONS THAT APPEARTO ACCELERATE AGING,IN HUMANSOR RODENTS, GIVE IMPORTANT CLUESTOTHE BIOLOGYOF AGING?
My own answer to this, presented at greater length elsewhere (38), is no, not much. A great deal of effort has gone into the study of mutations of mice and humans that appear, to some observers, to recapitulate some aspects of the normal aged phenotype, that is, to progeria syndromes. A great deal of interesting cell biology has come from studies of these mutations, but in my view, they have major defects as guides to problems in biogerontology. A central concern is that there are very many ways to shorten life span, and thus, little a priori reason to infer that a mutation that shortens life span is doing so through pathways that are in any way related to those that postpone death and preserve health in slow-aging animals. A detailed phenotypic survey will often reveal an assortment of abnormalities in these progeroid mice or people, but resemblances to normal aging often reflect the consequences of severe illness seen both in old individuals and in those who become ill in other ways, or failure of specific organs through pathways demonstrably distinct from those seen in normal aged individuals of the same species (39). In each case, the progeroid syndrome includes many features not seen in normal aging and fails to demonstrate many changes that are indeed characteristic of aged individuals. When we say that aging goes faster in (for example) mice compared with dogs, we base this assertion on evidence that immune failure, loss of hearing, loss of cardiopulmonary reserve, collagen cross-linking, cataracts, loss of muscle strength, neoplastic diseases of multiple cell types, loss of cognitive abilities, and many other problems develop more quickly in mice than in dogs. None of the mutations proposed as examples of accelerated aging in mice or humans comes remotely close, yet, to meeting this standard of evidence.
In a few cases, a more detailed understanding of the basis for the lethal illness has shown that the pathobiology is easily distinguished from normal aging. In one prominent example, the klotho mutant (40) was eventually shown to reflect an abnormality of phosphate clearance by the kidney with associated vitamin D toxicity, whose effects could be mitigated by diets from which vitamin D had been removed (41,42). It seems unlikely that aging in mice or people is attributable to vitamin D toxicity or amenable to prevention by vitamin D-deficient diets. In another case (43), a mutant originally considered to be an excellent example of accelerated aging, the Snell dwarf mouse, was later shown to display extended longevity and many other features of delayed aging when housed in appropriate laboratory conditions (9). The implication here is that the terminal effects of severe illness can be confused with those produced by old age and should not be allowed to substitute for a careful multisystem evaluation of age-sensitive properties and pathogenetic mechanisms in a mouse (or human) thought to be aging with unusual speed.
Proponents of the value of progeroid mutations for elucidation of mechanisms of aging often argue that these syndromes are examples of “segmental” aging, valuable for the evaluation of how aging works on specific tissues or organ systems. This fallback position dodges the challenge of proving that the mechanism by which the mutation leads to functional deficits is indeed also responsible for parallel deficits in nonmutant aged subjects. More importantly, the claim that a specific mutation can lead to aging in some but not other cells and tissues misses the main problem in aging research, that is, elucidating the connections that postpone, in synchrony, the effects of aging in all tissues together. Dissection of the mechanisms by which mutations can lead to premature failure of several organ systems can often lead to important discoveries in cell biology and pathogenesis, which can then in turn be exploited for the study of aging, but in my view, the assumption that these mutations provide valuable guideposts for elucidation of aging is unjustified.
DO STUDIESOF ANIMALS THATARE PRONETO SPECIFIC DISEASES, SUCH AS MOUSE MODELSOF ALZHEIMER’S DISEASE, GIVE ANY INSIGHTS INTOTHE BIOLOGYOF AGINGANDITS RELATIONSHIPTO LATE-LIFE DISEASES?
Analyses of rodent models of human disease always confront the uncertainty about the extent to which the disease resembles the human condition it is intended to represent, but investigation of rodent models of disease has been and will continue to be an exceptionally useful element in medical investigation. Exploitation of these genetic models to learn more about aging is still in its infancy. The biological question is fascinating: For diseases that only become apparent in later life, how do mechanisms that time aging postpone the onset of the disease and do so until ages when so many other diseases of aging first become problematic? A straightforward initial approach is to construct synthetic double mutants that combine genes that promote a specific disease syndrome with alleles that delay aging, to test the prediction that symptoms will be postponed by the antiaging defenses that extend longevity in mice that do not bear the disease-susceptibility alleles. Experiments of this sort in C. elegans have been very productive, leading to important new ideas about how prolonged maintenance of systems that remove damaged proteins and protein aggregates can also delay onset of cellular pathology and whole organism dysfunction (44). If analogous systems in mice do show postponement of analogs of human adult-onset neurodegenerative illness, or diabetes, or bone disease, this will both provide clues to the mechanism that times these illnesses in aging people as well as impetus to pursue pharmacological strategies that reduce disease incidence by manipulation of pathways that time aging and link diseases to biological age.
COMPARATIVE GENETICSOF AGING—A NON-TRADITIONAL GENETIC PROBLEM
The genetic differences that have the most dramatic impact on the rate of aging—those that slow aging more in dogs than in mice and still more in humans and whales—are impervious to the traditional methods of genetics, which focus on analysis of the effects of polymorphic genes segregating within a species. Dogs don’t mate with mice, and introduction of dog genes into mice, while technically feasible, faces all the problems of candidate gene designs compounded by complications of molecular coevolution following the divergence of rodents from carnivores. Yet interspecies comparison is where the action is: Genetic effects on life span within a species have, so far, been limited to effect sizes of not more than 50%, whereas evolution routinely produces 5- to 10-fold variation in life span and aging rate within orders of mammals and approximately 30- to 100-fold within the class Mammalia. The rapidly growing collection of genomic sequences for mammalian species makes it possible to compile intimidatingly long sets of interspecies differences, haystacks of sequence data in which needles of unknown number, size, shape, and color have been strewn. Even pairwise comparisons of closely related species—human/chimp, for example, instead of human/mouse—are likely to be unenlightening without filtration through functional criteria. If we knew, for example, that long-lived primates typically differed from short-lived primates in some specific biochemical or cellular pathway, such as membrane lipid composition, or effectiveness of repair of a specific form of DNA damage, or activation of a set of genes controlled by a specific transcription factor, then interrogation of the sequence-contrast databases seeking systematic variation in control of the corresponding genetic elements would acquire greater focus and rationale. The need to have specific questions in mind when confronting tables of species-specific DNA sequence variation provides strong motivation for a comprehensive program of comparative cell biology and physiology comparing young adults of closely related species that are similar in ecological niche but differ widely in aging rate and maximal life span. A catalog of such contrasts might on its own suggest aging-related pathways for pharmacological manipulation independent of any genetic information but would also provide the needed framework on which to pin and arrange sequence information as it emerges. Large multispecies surveys of genomic structure have documented a number of provocative effects, such as a tendency (45) for mitochondrial genes of longer-lived mammals to avoid the use of methionine codons (particularly AUA codons) and for threonine usage to be higher among long-lived primates (46) and mammals more generally (45). Studies of skin-derived fibroblasts have shown correlations between resistance to lethal stress and species life span in rodents (47), birds (48), and mammals (49), suggesting that resistance to injury may be one element regulating life span across evolutionary lineages (see Figure 1). The observations of codon usage have not yet been linked to physiological consequences, and the data on stress resistance have not yet been traced to genetic variation among species, but studies of this kind may, once extended, guide the interpretation of genomic differences among species that differ in life span.
Figure 1.
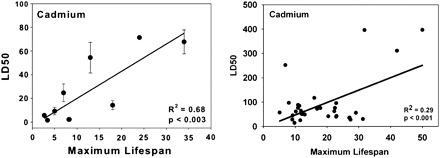
Scatterplots showing maximum recorded life span versus resistance of skin-derived fibroblast cells to toxic levels of cadmium. Y-axis shows LD50 (micromoles per liter), that is, the dose of cadmium needed to kill 50% of the cells. Left panel: eight species of rodents, with one species of bat (life span = 34 years) included for comparison, from (47). Right panel: 35 species of birds from temperate climate zone, from (48). Note changes in y-axis scale: The most resistant of the bird species has LD50 ∼400 μM compared with <80 μM for rodents and the bat species tested.
SUMMARY STATEMENT
Research by geneticists has, then, led to some interesting news about what mechanisms can postpone aging and how these mechanisms postpone so many forms of age-dependent dysfunction roughly in parallel, but these productive paths are entangled in a maze of work that may lead to important insights on tangential questions but is less likely to shed light on aging per se.
Work on single-gene mutants has established beyond a reasonable doubt that remarkably simple alterations in cell biology and endocrine patterns can slow aging in mammals, leading to life-span extensions about 10-fold greater, proportionally, than would be expected by demographic models of a cancer-free population or one free of ischemic heart disease (50,51). These studies, inspired, preceded, and bolstered by faster and more systematic work in yeast, flies, and worms have also aimed a spotlight at specific cellular pathways, some with defined molecular architecture, whose exploration seems very likely to produce new ideas and perhaps even antiaging medicines.
Studies of polymorphism in human populations have not yet given much insight into the genetics, or biology, of aging, in part because loci that regulate aging (if they exist at all) are hard to discern among the many strong loci that modulate risk of common lethal illnesses and in part because such studies have to date focused on a phenotype, extreme age, that is a poor surrogate for slow aging.
Studies of mutations that lead to early death and/or dysfunction, the so-called progerias, suffer from two fundamental misconceptions: First, that poor health and early death are typically caused, and regulated, by the same processes that produce and postpone aging and second, that it is useful to consider the mechanisms that prevent or delay aging as though they might work on each segment of the body separately.
Connections between antiaging mechanisms and disease pathogenesis are likely to be detected in mouse stocks in which disease-susceptibilty alleles are engineered into mice with antiaging mutations.
The strongest evidence for genetic influence on the pace of aging, that is, for genes against aging, comes from comparisons across species, where longevity differences are about an order of magnitude larger than those obtainable within (mammalian) species. Delineating the way in which evolution molds the rate of aging will require development of new methods that exploit in synergy the skills of geneticists, bioinformatics experts, and biologically oriented gerontologists.
Not all lamp posts have keys at their feet, and more thoughtful, obsessive attention to the central problem of biogerontology—figuring out how slow-aging animals delay so many bad things in parallel—could lead to more rapid progress in working out the molecular details connecting aging to pathobiology.
References
- 1.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 2.Charlesworth B. Evolution in Age-Structured Populations. Cambridge, UK: Cambridge University Press; 1980. [Google Scholar]
- 3.Medawar PT. An Unsolved Problem of Biology. London, UK: H. K. Lewis; 1952. [Google Scholar]
- 4.Austad SN. Retarded senescence in an insular population of Virginia opossums (Didelphis virginiana) J Zool. 1993;229:695–708. [Google Scholar]
- 5.Partridge L, Barton NH. Optimality, mutation and the evolution of ageing. Nature. 1993;362:305–311. doi: 10.1038/362305a0. [DOI] [PubMed] [Google Scholar]
- 6.Partridge L, Harvey PH. Methuselah among nematodes. Nature. 1993;366:404–405. doi: 10.1038/366404a0. [DOI] [PubMed] [Google Scholar]
- 7.Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- 8.Coschigano KT, Clemmons D, Bellush LL, Kopchick JJ. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology. 2000;141:2608–2613. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- 9.Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98:6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conover CA, Bale LK. Loss of pregnancy-associated plasma protein A extends lifespan in mice. Aging Cell. 2007;6:727–729. doi: 10.1111/j.1474-9726.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- 11.Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 12.Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 2003;299:1342–1346. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- 13.Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- 14.Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 15.Schachter F, Faure-Delanef L, Guenot F, et al. Genetic associations with human longevity at the APOE and ACE loci. Nat Genet. 1994;6:29–32. doi: 10.1038/ng0194-29. [DOI] [PubMed] [Google Scholar]
- 16.Bloss CS, Pawlikowska L, Schork NJ. Contemporary human genetic strategies in aging research. Ageing Res Rev. 2011;10:191–200. doi: 10.1016/j.arr.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller RA, Austad SN. Growth and aging: why do big dogs die young? In: Masoro EJ, Austad SN, editors. Handbook of the Biology of Aging. 6th ed. New York: Academic Press; 2006. pp. 512–533. [Google Scholar]
- 18.Miller RA. Kleemeier award lecture: are there genes for aging? J Gerontol A Biol Sci Med Sci. 1999;54:B297–B307. doi: 10.1093/gerona/54.7.b297. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Deeb B, Pendergrass W, Wolf N. Cellular proliferative capacity and life span in small and large dogs. J Gerontol A Biol Sci Med Sci. 1996;51:B403–B408. doi: 10.1093/gerona/51a.6.b403. [DOI] [PubMed] [Google Scholar]
- 20.Michell AR. Longevity of British breeds of dog and its relationships with sex, size, cardiovascular variables and disease. Vet Rec. 1999;145:625–629. doi: 10.1136/vr.145.22.625. [DOI] [PubMed] [Google Scholar]
- 21.Patronek GJ, Waters DJ, Glickman LT. Comparative longevity of pet dogs and humans: implications for gerontology research. J Gerontol A Biol Sci Med Sci. 1997;52:B171–B178. doi: 10.1093/gerona/52a.3.b171. [DOI] [PubMed] [Google Scholar]
- 22.Eigenmann JE, Patterson DF, Froesch ER. Body size parallels insulin-like growth factor I levels but not growth hormone secretory capacity. Acta Endocrinol. 1984;106:448–453. doi: 10.1530/acta.0.1060448. [DOI] [PubMed] [Google Scholar]
- 23.Bonnett BN, Egenvall A, Hedhammar A, Olson P. Mortality in over 350,000 insured Swedish dogs from 1995-2000: I. Breed-, gender-, age- and cause-specific rates. Acta Vet Scand. 2005;46:105–120. doi: 10.1186/1751-0147-46-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams DL, Heath MF, Wallis C. Prevalence of canine cataract: preliminary results of a cross-sectional study. Vet Ophthalmol. 2004;7:29–35. doi: 10.1111/j.1463-5224.2004.00317.x. [DOI] [PubMed] [Google Scholar]
- 25.Brosnahan MM, Paradis MR. Demographic and clinical characteristics of geriatric horses: 467 cases (1989–1999) J Am Vet Med Assoc. 2003;223:93–98. doi: 10.2460/javma.2003.223.93. [DOI] [PubMed] [Google Scholar]
- 26.Davey SG, Hart C, Upton M, et al. Height and risk of death among men and women: aetiological implications of associations with cardiorespiratory disease and cancer mortality. J Epidemiol Community Health. 2000;54:97–103. doi: 10.1136/jech.54.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albanes D, Jones DY, Schatzkin A, Micozzi MS, Taylor PR. Adult stature and risk of cancer. Cancer Res. 1988;48:1658–1662. [PubMed] [Google Scholar]
- 28.Hebert PR, Rich-Edwards JW, Manson JE, et al. Height and incidence of cardiovascular disease in male physicians. Circulation. 1993;88:1437–1443. doi: 10.1161/01.cir.88.4.1437. [DOI] [PubMed] [Google Scholar]
- 29.Tretli S. Height and weight in relation to breast cancer morbidity and mortality. A prospective study of 570,000 women in Norway. Int J Cancer. 1989;44:23–30. doi: 10.1002/ijc.2910440105. [DOI] [PubMed] [Google Scholar]
- 30.Petrelli JM, Calle EE, Rodriguez C, Thun MJ. Body mass index, height, and postmenopausal breast cancer mortality in a prospective cohort of US women. Cancer Causes Control. 2002;13:325–332. doi: 10.1023/a:1015288615472. [DOI] [PubMed] [Google Scholar]
- 31.Tretli S, Robsahm TE. Height, weight and cancer of the oesophagus and stomach: a follow-up study in Norway. Eur J Cancer Prev. 1999;8:115–122. doi: 10.1097/00008469-199904000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Barker DJP, Osmond C, Golding J. Height and mortality in the counties of England and Wales. Ann Hum Biol. 1990;17:1–6. doi: 10.1080/03014469000000732. [DOI] [PubMed] [Google Scholar]
- 33.Schaumberg DA, Glynn RJ, Christen WG, Hankinson SE, Hennekens CH. Relations of body fat distribution and height with cataract in men. Am J Clin Nutr. 2000;72:1495–1502. doi: 10.1093/ajcn/72.6.1495. [DOI] [PubMed] [Google Scholar]
- 34.Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3:70ra13. doi: 10.1126/scitranslmed.3001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinney BA, Coschigano KT, Kopchick JJ, Steger RW, Bartke A. Evidence that age-induced decline in memory retention is delayed in growth hormone resistant GH-R-KO (Laron) mice. Physiol Behav. 2001;72:653–660. doi: 10.1016/s0031-9384(01)00423-1. [DOI] [PubMed] [Google Scholar]
- 36.Silberberg R. Articular aging and osteoarthritis in dwarf mice. Pathol Microbiol. 1972;38:417–430. doi: 10.1159/000162458. [DOI] [PubMed] [Google Scholar]
- 37.Vergara M, Smith-Wheelock M, Harper JM, Sigler R, Miller RA. Hormone-treated Snell dwarf mice regain fertility but remain long-lived and disease resistant. J Gerontol A Biol Sci Med Sci. 2004;59:1244–1250. doi: 10.1093/gerona/59.12.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller RA. ‘Accelerated aging’: a primrose path to insight? Aging Cell. 2004;3:47–51. doi: 10.1111/j.1474-9728.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- 39.Miller RA. Evaluating evidence for aging. Science. 2005;310:441–443. doi: 10.1126/science.310.5747.441. [DOI] [PubMed] [Google Scholar]
- 40.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 41.Lanske B, Razzaque MS. Premature aging in klotho mutant mice: cause or consequence? Ageing Res Rev. 2007;6:73–79. doi: 10.1016/j.arr.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Razzaque MS, Lanske B. Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med. 2006;12:298–305. doi: 10.1016/j.molmed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 43.Fabris N, Pierpaoli W, Sorkin E. Lymphocytes, hormones and ageing. Nature. 1972;240:557–559. doi: 10.1038/240557a0. [DOI] [PubMed] [Google Scholar]
- 44.Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol. 2010;694:138–159. doi: 10.1007/978-1-4419-7002-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aledo JC, Li Y, de Magalhaes JP, Ruiz-Camacho M, Perez-Claros JA. Mitochondrially encoded methionine is inversely related to longevity in mammals. Aging Cell. 2011;10:198–207. doi: 10.1111/j.1474-9726.2010.00657.x. [DOI] [PubMed] [Google Scholar]
- 46.Kitazoe Y, Kishino H, Hasegawa M, Nakajima N, Thorne JL, Tanaka M. Adaptive threonine increase in transmembrane regions of mitochondrial proteins in higher primates. PLoS One. 2008;3:e3343. doi: 10.1371/journal.pone.0003343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harper JM, Salmon AB, Leiser SF, Galecki AT, Miller RA. Skin-derived fibroblasts from long-lived species are resistant to some, but not all, lethal stresses and to the mitochondrial inhibitor rotenone. Aging Cell. 2007;6:1–13. doi: 10.1111/j.1474-9726.2006.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harper JM, Wang M, Galecki AT, Ro J, Williams JB, Miller RA. Fibroblasts from long-lived bird species are resistant to multiple forms of stress. J Exp Biol. 2011;214:1902–1910. doi: 10.1242/jeb.054643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kapahi P, Boulton ME, Kirkwood TB. Positive correlation between mammalian life span and cellular resistance to stress. Free Radic Biol Med. 1999;26:495–500. doi: 10.1016/s0891-5849(98)00323-2. [DOI] [PubMed] [Google Scholar]
- 50.Olshansky SJ, Carnes BA, Cassel C. In search of Methuselah: estimating the upper limits to human longevity. Science. 1990;250:634–640. doi: 10.1126/science.2237414. [DOI] [PubMed] [Google Scholar]
- 51.Miller RA. Extending life: scientific prospects and political obstacles. Milbank Q. 2002;80:155–174. doi: 10.1111/1468-0009.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]