

Abstract
Aims:
Heart failure (HF) is a prevalent disease that is considered the foremost reason for hospitalization in the United States. Most protein kinases (PK) are activated in heart disease and their inhibition has been shown to improve cardiac function in both animal and human studies. However, little is known about the direct impact of PKA and PKC inhibitors on human cardiac contractile function.
Material and Methods:
We investigated the ex vivo effect of such inhibitors on force as well as on kinetics of left ventricular (LV) trabeculae dissected from non-failing and failing human hearts. In these experiments, we applied 0.5 μM of H-89 and GF109203X, which are PKA and PKC inhibitors, respectively, in comparison to their vehicle DMSO (0.05%).
Key findings and Conclusion:
Statistical analyses revealed no significant effect for H-89 and GF109203X on either contractile force or kinetics parameters of both non-failing and failing muscles even though they were used at a concentration higher than the reported IC50s and Kis. Therefore, several factors such as selectivity, concentration, and treatment time, which are related to these PK inhibitors according to previous studies require further exploration.
Introduction
HF is an increasing public health problem that represents the foremost reason of hospitalization in the U.S, where it affects over 5.7 million [1, 2]. The most common etiologies are ischemic heart disease, hypertension, and diabetes [3, 4]. Starting from this primary etiology, during its transition to end stage failure many molecular cardiac changes are observed, including excitation-contraction coupling, calcium homeostasis, signal transduction pathways, and cardiomyocyte death pathways [5]. In this regard, PK activity variations have been linked to development as well as to exacerbation of HF in both animal [6, 7] and human studies [8]. Considering the fact that most, but not all, of PKs are activated in end-stage heart failure, such as Ca2+/calmodulin-dependent protein kinase II (CaMKII), protein kinase (PK) D, protein kinase A (PKA) and protein kinase C (PKC) [9, 10] and their inhibition has been shown to improve cardiac function in some studies [11, 12], they clearly appear as an attractive target for HF drug discovery/development.
So far, disputing results about the significance of PKA modulators on cardiac function were found in numerous studies. For example, the PKA activation has been shown to synergistically support the PKC-induced cardiac hypertrophy [13], while its inhibition in different animal studies has revealed remarkable I/R injury protection [14], attenuating cardiomyocyte hypertrophy [15], and improving cardiac contractility [16]. In contrast, decreased PKA-dependent cTnI phosphorylation and its regulatory subunits during human dilated cardiomyopathy has been reported [17]. Also, Dvornikov et al. examined the restrictive cardiomyopathy linked cTnI-R145W mutation regarding in-vitro function of human myofilament and the interplay with PKA/PKC-induced cTnI phosphorylation [18]. On the other side, previous studies have shown that PKC inhibition could attenuate cardiac hypertrophy and improve cardiomyocyte function in animals [19, 20]. Recent clinical trials suggest that systemic delivery of inhibitors and activators of PKC isoenzymes is well tolerated [21, 22].
We have previously shown that the broad-spectrum serine-threonine kinase inhibitor, staurosporine, inhibited the frequency-dependent induction of TnI phosphorylation, which is, at least partially, responsible for frequency-dependent myofilament desensitization in rabbit trabeculae muscle [23], and its inhibition may contribute to cardiac diastolic dysfunction [24]. Since cardiac studies in animal models do not always unambiguously translate to humans, a more robust understanding of the underlying mechanism for the disease development in human is critically needed for strategizing therapeutic progress in this field [25, 26]. Thus, the goal of this study was to investigate the ex vivo effect of PKA and PKC inhibitors on force as well as on kinetics of LV trabeculae dissected from non-failing and failing human myocardium since the activity of both PKA and PKC is still preserved ex vivo as shown in many studies have been done before [23, 25, 27-29]. We tested the efficacy of these inhibitors on the force-frequency relationship (FFR), which is the primary intrinsic modulator of cardiac contractility and relaxation, where changes are strong phenotypical markers of cardiac pathology [30].
Materials and methods
Human Tissue Procurement
Explanted hearts were obtained directly in the operating room and immediately flushed with cardioplegic solution after removal from donors/patients, as described previously [25, 26]. The hearts were transferred to the laboratory (within 10-15 minutes) in cold cardioplegic solution containing (in mM): 110 NaCl, 16 KCL, 16 MgCl2, 10 NaHCO3, and 0.5 CaCl2. All hearts were procured and treated with identical protocols, solutions and timing regardless of their source. All human tissues were experimented on with approval from the Institutional Review Board (IRB) of The Ohio State University and conform to the declaration of Helsinki. Non-transplantable donor hearts were attained in the operating room in collaboration with Lifeline of Ohio Organ Procurement.
Informed consents were acquired from cardiac transplant patients. All end-stage failing hearts were acquired from patients in the operating room undergoing cardiac transplantation at The Ohio State University Wexner Medical Center. The biometric characteristics of these non-failing and failing hearts are provided in Tables 1 and 2, respectively.
Table 1:
Characteristics of Non-failing Hearts.
| Heart # |
Age , yr |
Se x |
Race | Cause of Death |
LVE F (%) |
BP, mmHg |
HR, BP M |
H W (g) |
LVA D |
|---|---|---|---|---|---|---|---|---|---|
| 219852 | 30 | F | Caucasian | Unknown COD, sudden cerebral edema and respiratory failure | 55% | 102/75 | 135 | 299 | No |
| 266541 | 57 | M | Caucasian | ICB/ICH | 53% | 130/68 | 61 | 635 | No |
| 331253 | 42 | F | Hispanic | CVA/ICH | 55% | 118/65 | 85 | 390 | No |
| 394176 | 57 | M | Caucasian | Blunt injury Non-MVA | NA | NA | NA | 748 | No |
| 402879 | 54 | M | Caucasian | ICB/ICH | NA | NA | NA | 474 | No |
| 435578 | 20 | M | Caucasian | DO/ Anoxia | 35% | 157/10 3 | 112 | 324 | No |
| 514489 | 42 | F | Caucasian | Cardiovascula r arrest/Anoxia | 55% | 119/71 | 114 | 327 | No |
| 632941 | 68 | F | Caucasian | Natural causes CVA/Stroke | NA | NA | NA | 402 | No |
| 694855 | 46 | F | Caucasian | CVA/ICH & SAH | 60% | 124/65 | 94 | 356 | No |
| 712301 | 67 | M | Caucasian | Blunt injury/IPH | NA | NA | NA | 527 | No |
| 785258 | 51 | F | Caucasian | CVA/ICH | 60% | 128/58 | 82 | 335 | No |
BP, blood pressure; COD, cause of death; CVA, cerebral vascular accident; DO, drug overdose; F, female; HR, heart rate; HW, heart weight; ICB, intracerebral bleeding; ICH, intracerebral; IPH, intraparenchymal hemorrhage; LVAD, left ventricular assist device; LVEF, left ventricular ejection fraction; M, male; MVA, motor vehicle accident; hemorrhage; NA, not available; SAH, subarachnoid hemorrhage; Yr, year.
Table 2:
Characteristics of Failing Hearts.
| Heart # | Age, yr | Sex | Race | Etiology | HW (g) | LVAD |
|---|---|---|---|---|---|---|
| 250585 | 65 | M | Caucasian | ICM | 621 | No |
| 369452 | 61 | M | African American | NICM | 540 | No |
| 390112 | 60 | F | Caucasian | NICM | 608 | Yes |
| 599014 | 50 | M | Caucasian | NICM | 667 | No |
| 611422 | 68 | M | Caucasian | ICM | 576 | No |
| 631231 | 49 | F | Asian | CCM | 306 | No |
| 645444 | 47 | M | African American | ICM | 411 | Yes |
| 679533 | 47 | M | Caucasian | NICM | 930 | Yes |
| 774694 | 50 | F | Caucasian | ICM | 486 | No |
| 777902 | 59 | M | Caucasian | CAD | 530 | Yes |
| 9116141 | 65 | M | Caucasian | NICM | 716 | Yes |
| 963542 | 60 | F | Caucasian | NICM | 329 | No |
| 971258 | 57 | M | Caucasian | ICM | 619 | Yes |
CAD, coronary artery disease; CCM, chemo-induced cardiomyopathy; CM, cardiomyopathy; F, female; HW, heart weight; LVAD, left ventricular assist device; NICM, non-ischemic cardiomyopathy; M, male; Yr, year.
Multicellular Muscle Preparation and Mounting
The left ventricle of the heart was transferred from the cardioplegic solution to a cold modified Krebs-Henseleit solution (K-H) bubbled with 95% O2-5% CO2 containing (in mM): 137 NaCl, 5 KCl, 0.25 CaCl2, 20 NaHCO3, 1.2 NaH2PO4, 1.2 MgSO4, 10 dextrose, and 20 BDM (2,3-butanedione monoxime) and pH of 7.4. Linear, small, and free-running trabeculae were isolated with the aid of a stereo dissection microscope, and kept in this solution at 0-4 °C until the time of the experiment [25, 26]. Muscles were transferred into custom-made setups as previously described for animal models [31] and the perfusion solution was changed to another modified K-H without the BDM. This solution was maintained at 37 °C and continuously bubbled with 95% O2-5% CO2 resulting in a pH of 7.4. Stimulation was started at baseline frequency of 0.5 Hz while CaCl2 concentration of the solution was slowly raised to 2 mM over ~15 minutes. Muscles were gradually stretched (over a few minutes) until an increase in the developed force was not matched by an increase in the resting tension. This length, designated as L100 or Lopt (optimal length), roughly corresponds to sarcomere length of ~2.2 μm, which is near or at the in vivo sarcomere length at end-diastole then allowed to stabilize for at least 30 minutes before the experimental protocol was initiated [26].
Once muscles had stabilized in the set-up, at 0.5 Hz, baseline contractile force was assessed. Under the stereo microscope, dimensions of the trabeculae were assessed (resolution of 10 μm), and all recorded forces were normalized to the cross-section area of the trabecula. Average dimensions (width × thickness × length) of LV trabeculae from non-failing and failing hearts were: (0.37 ± 0.02 × 0.25 ± 0.02 × 1.58 ± 0.23 mm) and (0.45 ± 0.02 × 0.29 ± 0.02 × 2.00 ± 0.18 mm), respectively, and there were no significant differences in muscle dimensions between the studied groups.
Force- Frequency Experimental Design
In these experiments, a total of 21 trabeculae from 11 different non-failing hearts were used for H-89 (n = 6), GF109203X (n = 8) and DMSO (n = 7). Similarly, a total of 27 trabeculae from 13 different failing hearts were used for H-89 (n = 9), GF109203X (n = 9) and DMSO (n = 9). Once stabilized (at 0.5 Hz), trabeculae from non-failing and failing hearts underwent basal FFR experiments via a custom designed LabVIEW program, as previously published [23]. Steady-state (basal) twitch amplitudes as well as kinetics were recorded at 0.5, 1.0, 1.5, 2.0, 2.5 and 3 Hz, then either PKA inhibitor (H-89; 0.5 μM), PKC inhibitor, (GF109203X; 0.5 μM) or their vehicle DMSO (0.05%) were added to the K-H superfusion solution and allowed to equilibrate for 5 minutes followed by FFR again in continuous presence of the inhibitor as stated before [23]. The 0.5 μM concentration of both PKA and PKC inhibitors was selected because it is around 33 and 100 times the in vitro Ki for the PKA; 15 nM and PKC; 5 nM, respectively [32] however, still lower than the concentration where some of the nonselective PKA inhibitor, H-89, effects have been found to occur [33].
Peak isometric developed force (Fdev) was determined and normalized to the cross-sectional area of the muscle. Additionally, as a force-independent parameter of force decay kinetics, time to peak force (TTP), time from peak force to 50% relaxation (RT50), maximum rate of force increase normalized to developed force (dF/dtmax/F) and maximum rate of force decrease normalized to developed force (dF/dtmin/F) were calculated [26, 34].
Protein Analysis
At the end of contractile experiments trabeculae were immediately frozen (within 1 second of the last contraction) by dousing them with liquid nitrogen while contracting, then are quickly removed from the set-up (and remain frozen) and stored at -80 °C.
Trabeculae were solubilized by heating at 80 °C for 6 minutes with vortexing for 10 seconds every 2 minutes in 20 μl urea sample buffer (8 M urea, 2 M thiourea, 75 mM DTT, 3% SDS, 0.05% bromophenol blue and 50 mM Tris-HCl, pH 6.8) and supernatant stored at -80 °C until use. Total protein phosphorylation was determined by Pro-Q Diamond phosphoprotein staining as previously described [25, 35]. Due to the variable size of the trabeculae recovered, the amount of total protein in each sample varied greatly across all of the samples. To determine optimal loading for each independent sample, 8 μl of its supernatant was fractionated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) on a 12% Laemmli gel with an acrylamide-to-bisacrylamide ratio of 29:1. Total protein phosphorylation was determined in the resultant gel by staining with Pro-Q Diamond Phosphoprotein Gel Stain (Molecular Probes) according to the manufacturer’s instructions and scanning on a Typhoon 9410 Imager (GE Healthcare) using an excitation of 532 nm and a 580 nm BP 30 emission filter. Total protein amount was then determined in the same gel by staining with SYPRO Ruby Protein Gel Stain (Molecular Probes) according to the manufacturer’s instructions and imaged on the Typhoon with an excitation of 457 nm and a 610 nm BP 30 emission filter. The actin band of the SYPRO-stained gel was quantified with ImageQuant TL (GE Healthcare) to generate actin-normalized loadings at an optimal ProQ Diamond staining and the above procedure was repeated using a subset of the above trabecula samples, loaded at similar actin amounts. Since for some muscles no protein was left, or not enough protein, only a sub-set of samples could be run on the normalized try. Resultant gel images were quantified by ImageQuant TL v7.0 (GE Healthcare). Whole-lane total protein phosphorylation was determined as the phospho-protein signal in the entire land divided by the total protein signal in the entire land. Specific protein phosphorylation was determined as the phosphoprotein band / total protein band for a given protein. Site-specific phosphorylation of TnI was determined in a subset of the trabecula samples by Multiplex Western blot similar to that previously described [36, 37]. Trabecular proteins were separated by SDS-PAGE as above and transferred to low-fluorescence PVDF. The membrane was probed with a rabbit anti-pTnI23/24 primary antibody (Phospho-Troponin I (cardiac) (Ser23/24), Cell Signaling) and anti-rabbit Cy3-conjugated secondary antibody and scanned on a Typhoon 9410 Imager using an excitation of 532 nm and a 580 nm BP 30 emission filter. The same blot was then re-probed for total TnI using a mouse anti-TnI primary antibody (C5, Fitzgerald) and anti-mouse Cy5-conjugated secondary antibody and scanned as above using an excitation of 633 nm and a 670 nm BP 30 emission filter. Quantification of pTnI23/24 and total TnI using different host species of the primary antibodies and different excitation wavelengths is critical to ensure no carryover of the pTnI signal into the total TnI detection. Resultant images were quantified with ImageQuant TL. The resultant pTnI23/24 signal was normalized to the total TnI signal for each trabecula and represented as the pTnI23/24 signal / total TnI signal. We anticipated reduction in the phosphorylation level of TnI due to inhibition of PKA and PKC via their inhibitors but our result here did not show any significant change in the phosphorylation level which is in accordance with our negative results on the contractility parameters but due to lower number of samples we were not able to comprehensively evaluate that result.
Statistical analysis
Data are presented as mean ± S.E.M. and were analyzed by ordinary 1-way analysis of variance (ANOVA) followed by Tukey-Kramer’s post hoc multiple comparison test, comparing all pairs of column. A 2-tailed value of P ≤ 0.05 was considered statistically significant.
Results
Effect of PKA and PKC Inhibitors on Contractile Performance of Non-failing and Failing Human Hearts
At basal condition, muscles from non-failing hearts showed the expected positive FFR (Figure 1A, B). Neither H-89 nor GF109203X and their vehicle (DMSO) significantly changed the Fdev (Figure 1A), relative tension (Figure 1B) or Fdia (Figure 1C) at any of the tested frequencies (0.5-3 Hz) compared to the initial basal condition. On the other hand, the failing heart muscles displayed a negative FFR (Figure 1D, E), which is a classical hallmark that has been extensively observed in failing human myocardium [38]. Likewise, H-89, GF109203X and DMSO did not significantly change the Fdev (Figure 1D), relative tension (Figure 1E) and Fdia (Figure 1F) of the failing muscles in the same frequency range compared to the corresponding values at basal condition, with the only exception that the PKC inhibitor, GF109203X, remarkably decreased the Fdia at 0.5 Hz (6.302 ± 0.65) compared to the Basal (8.38 ± 0.39) (P < 0.05) (Figure 1F). Notably, we could not detect a significant difference between the DMSO and any of the PK inhibitors in this study.
Figure 1:
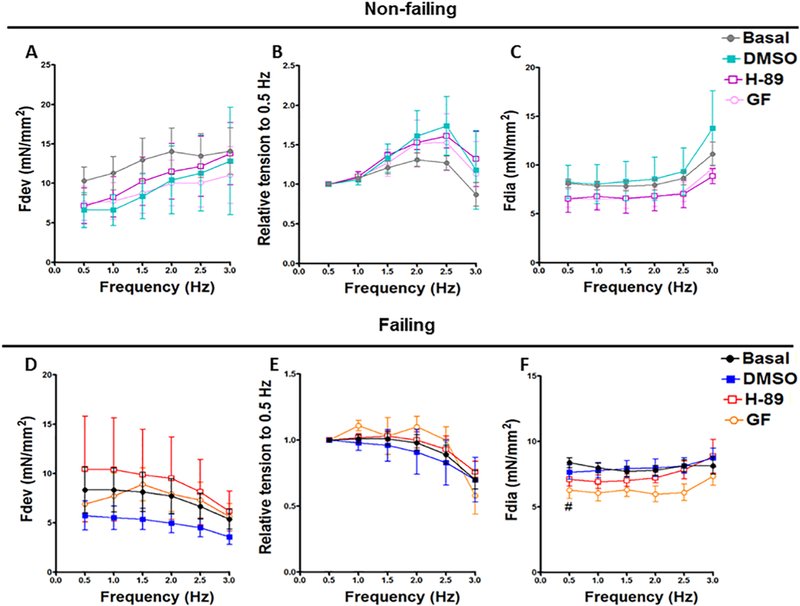
Effect of H-89 and GF109203X on Contractile Performance of Non-failing and Failing Human Hearts. Representative line graphs show average isometric developed force (Fdev) data from non-failing trabeculae muscles treated with DMSO, GF109203X and H-89 (A); their corresponding peak isometric developed force (Fdev) values expressed as a fraction of its corresponding Fdev at the basal frequency of 0.5 Hz (B); diastolic tension (Fdia) (C). Average developed force (Fdev) data from failing trabeculae muscles treated with DMSO, GF109203X and H-89 (D); their corresponding peak isometric developed force (Fdev) values expressed as a fraction of its corresponding Fdev at the basal frequency of 0.5 Hz (E); diastolic tension (Fdia) (F). All data presented as mean ± SEM. DMSO: dimethyl sulfoxide. Non-failing: (n= 7); failing: (n=9). GF: GF109203X. Non-failing: (n= 8); failing: (n=9). H89: Non-failing: (n= 6); failing: (n=9). #: Indicates a significant change in GF group comparing to basal failing group at 0.5 Hz.
Effect of PKA and PKC Inhibitors on Twitch Kinetics of Non-failing and Failing Human Hearts
In this study, twitch kinetic analyses showed that H-89, GF109203X and DMSO could not significantly change the TTP (Figure 2A) or RT50 (Figure 2B) of non-failing muscles at any frequency compared to the Basal. Equally, there was no significant effect for these drugs or their vehicle on the TTP (Figure 2D) or RT50 (Figure 2E) of muscles from failing hearts at any frequency compared to the Basal. Besides, the PK inhibitors and DMSO had no significant effect on dF/dtmax/F or dF/dtmin/F in both non-failing (Figure 3A, B) and failing (Figure 3C, D), respectively. These twitch parameters have the asset to provide purely kinetic process information in s−1 [34]. Again, there is no significant difference between the DMSO and any of the PK inhibitors in all twitch parameters in this study.
Figure 2:
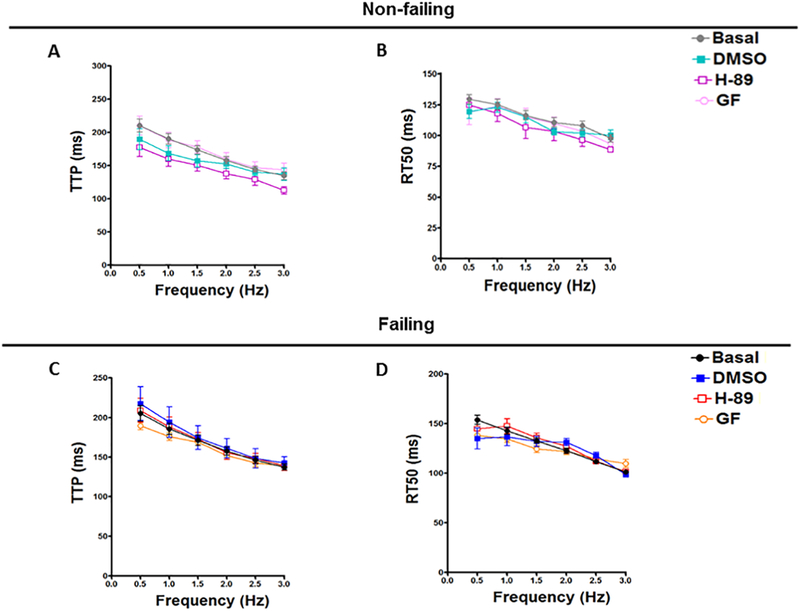
Effect of H-89 and GF109203X on TTP and RT50 of Non-failing and Failing Human Hearts. Representative line graphs show average isometric time to peak (TTP) data from non-failing trabeculae muscles treated with DMSO, H-89 and GF109203X (A); their corresponding 50% relaxation time (RT50) (B). Average time to peak (TTP) data from failing trabeculae muscles treated with DMSO, H-89 and GF109203X (C); their corresponding 50% relaxation time (RT50) values (D). All data presented as mean ± SEM. DMSO: dimethyl sulfoxide. Non-failing: (n= 7); failing: (n=9). H89: Non-failing: (n= 6); failing: (n=9). GF: GF109203X. Non-failing: (n= 8); failing: (n=9).
Figure 3:
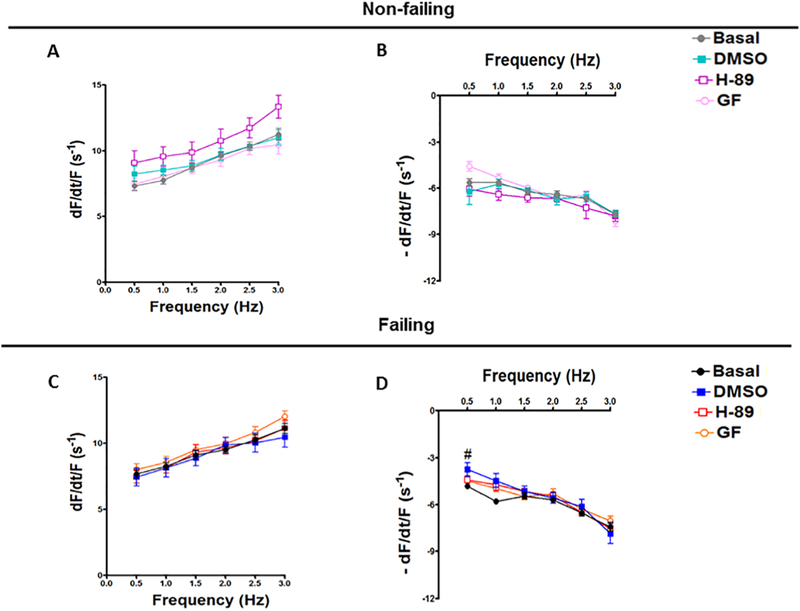
Effect of H-89 and GF109203X on dF/dtmax/F and dF/dtmin/F of Non-failing and Failing Human Hearts. Representative line graphs show the maximum rate of force increase normalized to developed force (dF/dtmax/F) data from non-failing trabeculae muscles treated with DMSO, H-89 and GF109203X (A); and the minimum rate of force decrease normalized to developed force (dF/dtmin/F) (B). The maximum rate of force increase normalized to developed force (dF/dtmax/F) data from failing trabeculae muscles treated with DMSO, H-89 and GF109203X (C); and the minimum rate of force decrease normalized to developed force (dF/dtmin/F) (D). All data presented as mean ± SEM. DMSO: dimethyl sulfoxide. Non-failing: (n= 7); failing: (n=9). H89: Non-failing: (n= 6); failing: (n=9). GF: GF109203X. Non-failing: (n= 8); failing: (n=9). #: Indicates a significant change in DMSO comparing to basal failing group at 0.5 Hz.
Myofilament proteins are phosphorylated by PKA and PKC
From 8 different non-failing and 7 failing hearts, we had (for each heart) a muscle frozen in the presence of DMSO, PKA inhibitor or PKC inhibitor and used for Pro-Q Diamond analysis (Figure 4). Trabecular proteins were separated by SDS-PAGE and the resultant gel stained with Pro-Q Diamond to identify phosphorylated proteins followed by Sypro Ruby staining for total protein detection. Only 9 of the 15 muscles analyzed contained sufficient recovered material to quantify protein phosphorylation. The loading of these 9 samples was normalized as best as possible given the wide range of initial loading that makes exact quantification difficult due to lane bulging, etc. and re-run and the total sample phosphorylation level was determined (Figure 5). Of the initial samples, 4 did not have enough material in the first run, and 2 did not have enough to re-run. Furthermore, we measured the phosphorylation of TnI at Ser22/23 in these same samples with sufficient material by Western blot with the TnI Ser-23/24 phosphorylation specific antibody (Figure 6). As we work with extremely limited amounts of tissue (typically well below 0.1 mg) we can run one gel (lane) per muscle only and thus we decided to focus on phosphorylation of TnI Ser-23/24 because it is the primary PKA site of phosphorylation.
Figure 4:
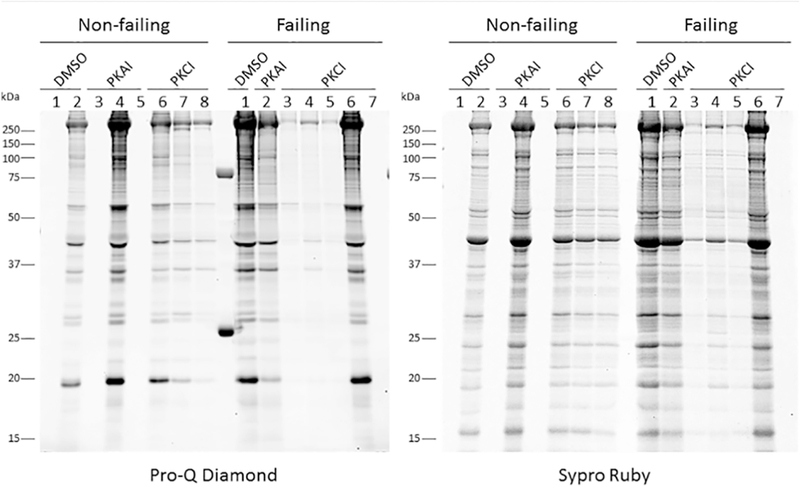
Trabeculae from non-failing and failing hearts were treated with DMSO, PKA inhibitor (PKAI) or PKC inhibitor (PKCI). Trabecular proteins were separated by SDS-PAGE and the resultant gel stained with Pro-Q Diamond to identify phosphorylated proteins followed by Sypro Ruby staining for total protein detection.
Figure 5:
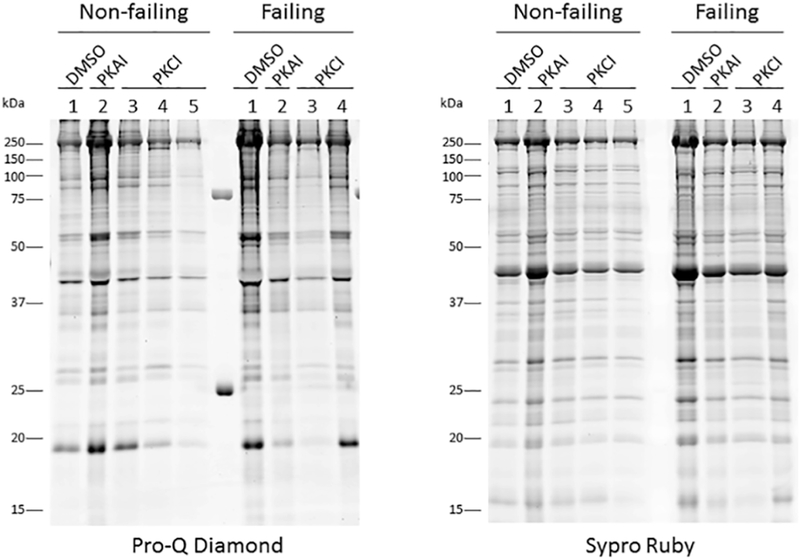
For a subset of the samples from Fig. 4, trabecular protein loading was optimized for phospho-protein detection, the loading normalized and separated by SDS-PAGE. The resultant gel was stained with Pro-Q Diamond to identify phosphorylated proteins followed by Sypro Ruby staining for total protein detection.
Figure 6:
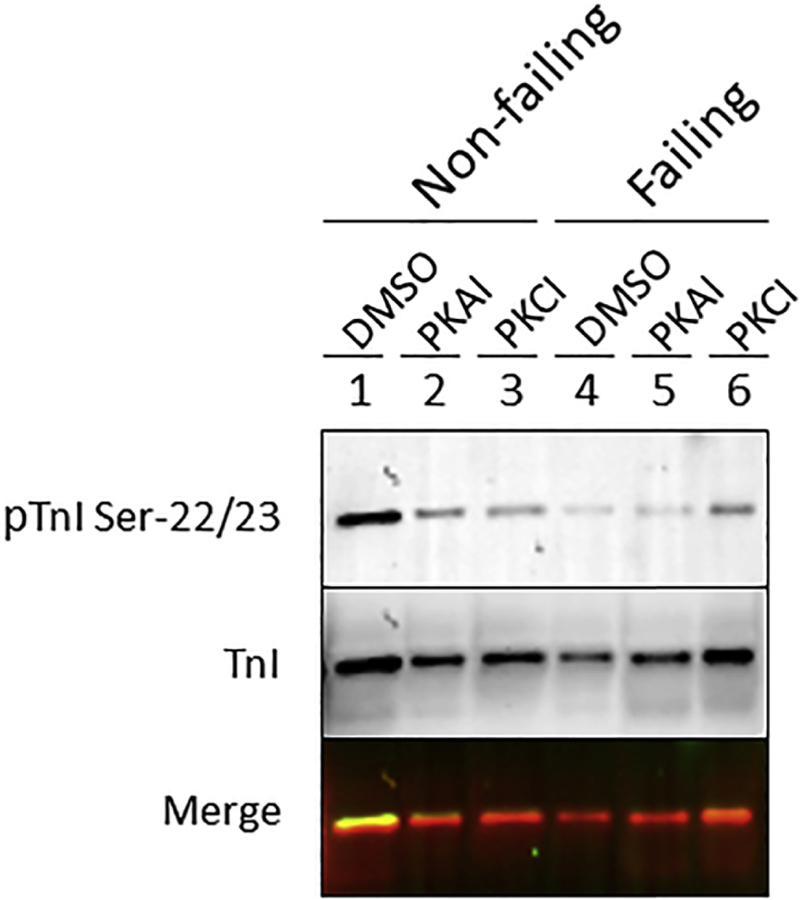
Quantification of TnI Ser-22/23 phosphorylation in non-failing and failing trabeculae treated with DMSO, PKA inhibitor (PKAI) or PKC inhibitor (PKCI) was determined by Western blot with the anti-pTnI Ser-22/23 followed by Total TnI protein detection.
Although we only had an n = 1 available, this proof-of-principle clearly illustrates that for future studies it is technically possible to study total and specific myofilament protein phosphorylation in these small samples, and that the kinase inhibitors may depress or block the increase of protein phosphorylation in failing and non-failing treated muscles.
Discussion
In this study we show that inhibition of PKA and PKC in non-failing and failing LV trabeculae from human myocardium with H-89 and GF 109203X, respectively, did not significantly impact force nor twitch kinetic parameters of the muscles.
There are three main physiological modifiers that alter myocardial contraction in vivo: length-dependent activation [39], frequency-dependent activation [40], and β-adrenergic stimulation [41]. Previously we found that PKA and PKC-βII pathways are essential in length-induced phosphorylation of TnI and MLC-2 in rabbit myocardium [34]. Based on that, we set out to determine the effect of rapid length step changes and the effect of PKA and PKCβII inhibitors on the rate of tension redevelopment (ktr) ktr in ultra-thin non-failing and failing human right ventricular trabeculae [25]. Treatment of human cardiac trabeculae with H-89 and PKCβII peptide inhibitor I in this study did not significantly affect ktr at either L90 (90% of optimal length) or at Lopt. Furthermore, we found that serine threonine kinase pathways activated with changes in frequency mainly, if not exclusively, modulate myofilament function, but not calcium transient regulation in rabbit myocardium [23]. A nonspecific serine-threonine kinase inhibitor, staurosporine, was used in this previous study to determine a potential role of kinases involved with the FFR. Since it is significantly important to elucidate specific kinases and specific targets, we decided in this study to focus on the effect of PKA and PKC in the FFR since their roles have been investigated to some extent [42, 43], but a conclusive mechanism is still lacking. Treatment of human cardiac trabeculae with these inhibitors in this study did not significantly affect the FFR of the muscles. Several factors can contribute to the difference between our results and the expectation that inhibition of PKA and PKC, based on our previous study [23], could alter force and kinetic parameters of treated trabeculae muscles. In our previous study we used rabbit myocardium [23] while in the current study we used human myocardium and there are significant differences between human and rabbit myocardium which can affect expected outcomes as previously reviewed [44]. Although, the use of human tissues in medical research has been critical to the development of current medical care since they are ideal for identifying cell markers, testing drug efficacy, providing insight into the physiological mechanisms of disease, and ultimately creating breakthroughs in drug discovery, the capacity to perform multiple types of experiments in human hearts is limited by the number of available human hearts, time constraint to perform experiments within several hours after explantation, and inherent variability between human myocardial tissues due to differences in factors such as genetics, medications, diets, diseases, exercise, and social habits that cannot be regulated in the human population as one could in a laboratory animal study which at the end could affect the outcome results. Therefore, we did not represent a concentration-dependent curves due to difficulty of repeating the FFR with different concentrations and only focused on one concentration for each inhibitor; 0.5 μM for H-89 and GF109203X, which is 10 times higher than the IC50 of H-89; 0.05 μM [45] and 50 times higher than the IC50 of GF109203X; 0.01 μM [46]. Reaching the most significant concentration(s) should be determined in the future for more confirmation. Moreover, unlike different studies H-89 or GF109203X were added to the KH superfusion solution and allowed to circulate for only 5 minutes in our study before a second force-frequency was measured as we did previously [23]. But in a different study, it has been shown that H-89 was able to achieve maximal inhibition of the bioluminescence signal produced by Renilla luciferase (RLuc) variants after incubation period of 20 min, still the effects of inhibition was seen immediately after H-89 addition regardless of incubation time [47]. While in a different study, H-89 was found to decreases the phosphatidylcholine biosynthesis by 50% when incubated for 1 h with Hela cells [48]. Likely, incubation of different PKA inhibitors for 1 h with skinned rat cardiac trabeculae muscle was able to inhibit the PKA effect on contraction [49]. In addition, GF109203X has been found to significantly reduce the carbachol-stimulated ERK1/2 activation and cell proliferation after 48 h incubation with SNU-407 colon cancer cells [50]. Even though the GF109203X is a well-known selective PKC inhibitor the H-89 is a non-selective PKA antagonist and looking for another selective one should be considered in further studies.
Lowering diastolic tension will efficiently improve cardiac performance since insufficient relaxation of the heart result in inadequate filling of the ventricles before the next excitation stimulus and compromised cardiac output. A multitude of factors are involved in myocardial relaxation [24]. These factors include cytosolic Ca2+ handling, myofilament Ca2+ sensitivity, cross-bridge cycling kinetics, the energetic state of the heart, and membrane potential as well as passive and active elastic properties and the length of the sarcomeres [51]. It has been recently reported that PKC may alter the phosphorylation status of proteins involved in Ca2+-handling, specifically phospholamban (PLN), indirectly via phosphorylation of protein phosphatase inhibitor-1 (I-1) resulting in enhanced protein phosphatase-1 (PP-1) activity [52]. In failing human myocardium increased PKC could, via phosphorylation of I-1 [53], result in increased PP-1 activity [52, 53]. This activation of PP-1 leads to the dephosphorylation of PLN, thus reducing Ca2+ uptake by SERCA-2a [54]. Pathak et al. show that targeted inhibition of PP-1 by increased activity of its inhibitor, inhibitor-1 in the heart, results in enhanced contractility and is protective against the development of cardiac hypertrophy and HF stimulated by hemodynamic load [55]. So it is predicted that inhibition of PKC will decrease PP-1 activity and consequently increase PLN phosphorylation and reduce diastolic Ca2+ levels, which may explain the reduction in the diastolic tension after treating our failing trabeculae muscles with GF109203X specially after our limited study to the phosphorylation of myofilament proteins did not show any significant difference that may be related to that effect. In line with these observations, it has been shown that ablation of PKC-α, a calcium-dependent PKC isoform, which phosphorylates the inhibitor-1 at a different site, Ser67, and reduces its activity [52] is associated with decreased phosphatase activity and enhanced function [52], which further support our findings. Probably because of the dose we used, (0.5 μM), the change on Fdia was observed only at low frequency (0.5 Hz) and not at higher frequencies.
Again, the quantity of available human hearts and the number of free running, thin, cardiac trabeculae that can be excised from each heart is limited. At least 3 muscles from each heart would have to undergo the protocol (treated with DMSO, PKA inhibitor or PKC inhibitor); this was logistically not possible for the majority of hearts studied. We were not able to comprehensively evaluate the phosphorylation of myofilament proteins in response to our inhibitors in the current study. Only 9 of the 15 muscles analyzed contained sufficient recovered material to quantify protein phosphorylation. Although we only had an n = 1 available, which is not appropriate for statistically relevant quantification, our purpose here was to show qualitative effects and the ability to conduct these experiments in the future with more directed pro-Q site or western. N=1 suffices for demonstration of technical possibility to carry out the experiment (i.e. if it works even once, it proves it can technically be done). Furthermore, it was not possible to test our inhibitors on different bigger sized tissues of the heart, as muscles that are bigger than used in this study will unavoidable suffer from core-hypoxia, making the contractile data obtained unreliable. The remaining LV free wall tissue of these human hearts is not frozen while still contracting. As such, the phosphorylation status of this tissue is not the one that is present in a contractile homeostasis. Thus, the available quiescently frozen tissue (unfortunately the vast majority of biochemistry on human tissue in the past has been done on quiescent tissue due to lack of tissue that is frozen in contractile homeostasis) cannot be unambiguously used to show a meaningful phosphorylation profile.
Conclusion
No significant effect for both H-89 and GF109203X on either contractile force or kinetics parameters of both non-failing and failing muscles was observed. This despite the fact that these inhibitors were used at a concentration higher than the reported IC50s and Kis. Therefore, several factors such as selectivity, concentration, and treatment time, which are related to these PK inhibitors according to previous studies may require further exploration.
Acknowledgements
The authors would like to thank Susan Montgomery, Erin Bumgardner, and Emily Jarvis for their help in obtaining consent from the patients for this study. The authors would like to thank Abraham Zawodni (Lifeline of Ohio) for help with obtaining clinical correlates for the donor hearts. Funding for this project was supported by NIH RC1HL099538 (to PMLJ), NIH R01HL113084 (to PMLJ), NIH R01HL114940 (To BJB), AHA 16POST27760155 (to MTE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mozaffarian D, et al. Heart disease and stroke statistics-2015 update: a report from the american heart association. Circulation, 2015, 131(4): e29. [DOI] [PubMed] [Google Scholar]
- [2].Organization WH. Cardiovascular diseases (CVDs). Fact sheet, 2011, 317 [Google Scholar]
- [3].Roger VL, et al. Heart disease and stroke statistics—2011 update a report from the American Heart Association. Circulation, 2011, 123(4): e18–e209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lloyd-Jones DM, et al. Lifetime risk for developing congestive heart failure the framingham heart study. Circulation, 2002, 106(24): 3068–72 [DOI] [PubMed] [Google Scholar]
- [5].Cho GW, et al. Chronic heart failure: Ca2+, catabolism, and catastrophic cell death. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 2016, 1862(4): 763–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Backs J, et al. The δ isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proceedings of the National Academy of Sciences, 2009, 106(7): 2342–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fielitz J, et al. Requirement of protein kinase D1 for pathological cardiac remodeling. Proceedings of the National Academy of Sciences, 2008, 105(8): 3059–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Leineweber K, et al. G-protein-coupled receptor kinase activity in human heart failure: effects of β-adrenoceptor blockade. Cardiovascular research, 2005, 66(3): 512–9 [DOI] [PubMed] [Google Scholar]
- [9].Bossuyt J, et al. Ca2+/calmodulin-dependent protein kinase IIδ and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circulation research, 2008, 102(6): 695–702 [DOI] [PubMed] [Google Scholar]
- [10].Wang J, et al. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. The Canadian journal of cardiology, 1999, 15(6): 683–90 [PubMed] [Google Scholar]
- [11].Boyle AJ, et al. Inhibition of protein kinase C reduces left ventricular fibrosis and dysfunction following myocardial infarction. Journal of molecular and cellular cardiology, 2005, 39(2): 213–21 [DOI] [PubMed] [Google Scholar]
- [12].Komander D, et al. Interactions of LY333531 and other bisindolyl maleimide inhibitors with PDK1. Structure, 2004, 12(2): 215–26 [DOI] [PubMed] [Google Scholar]
- [13].Yamazaki T, et al. Protein Kinase A and Protein Kinase C Synergistically Activate theRaf-1 Kinase/Mitogen-activated Protein Kinase Cascade in Neonatal Rat Cardiomyocytes. Journal of molecular and cellular cardiology, 1997, 29(9): 2491–501 [DOI] [PubMed] [Google Scholar]
- [14].Gesmundo I, et al. Growth hormone-releasing hormone attenuates cardiac hypertrophy and improves heart function in pressure overload-induced heart failure. Proceedings of the National Academy of Sciences, 2017: 201712612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang L, et al. UCR1C is a novel activator of phosphodiesterase 4 (PDE4) long isoforms and attenuates cardiomyocyte hypertrophy. Cellular signalling, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fontes-Sousa AP, et al. Effects of adrenomedullin on systolic and diastolic myocardial function. Peptides, 2009, 30(4): 796–802 [DOI] [PubMed] [Google Scholar]
- [17].Zakhary DR, et al. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation, 1999, 99(4): 505–10 [DOI] [PubMed] [Google Scholar]
- [18].Dvornikov AV, et al. Restrictive cardiomyopathy Troponin-I R145W mutation does not perturb myofilament length dependent activation in human cardiac sarcomeres. Journal of Biological Chemistry, 2016: jbc. M116. 746172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lim J-Y, et al. TGF-β1 induces cardiac hypertrophic responses via PKC-dependent ATF-2 activation. Journal of molecular and cellular cardiology, 2005, 39(4): 627–36 [DOI] [PubMed] [Google Scholar]
- [20].Stebbins EG and Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of (βII protein kinase C. Journal of Biological Chemistry, 2001, 276(32): 29644–50 [DOI] [PubMed] [Google Scholar]
- [21].Aiello LP, et al. The Effect of Ruboxistaurin on Visual Loss in Patients With Moderately Severe to Very Severe Nonproliferative Diabetic Retinopathy: Initial Results of the Protein Kinase C [beta] Inhibitor Diabetic Retinopathy Study (PKC-DRS) Multicenter Randomized Clinical Trial. Diabetes, 2005, 54(7): 2188. [DOI] [PubMed] [Google Scholar]
- [22].Baumal C Effect of Ruboxistaurin in Patients With Diabetic Macular Edema: Thirty-Month Results of the Randomized PKC-DMEs Clinical Trial. Evidence-Based Ophthalmology, 2007, 8(4): 208–9 [DOI] [PubMed] [Google Scholar]
- [23].Varian KD, et al. Staurosporine inhibits frequency-dependent myofilament desensitization in intact rabbit cardiac trabeculae. Biochemistry research international, 2012, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Periasamy M and Janssen PM. Molecular basis of diastolic dysfunction. Heart failure clinics, 2008, 4(1): 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Milani-Nejad N, et al. Insights into length-dependent regulation of cardiac cross-bridge cycling kinetics in human myocardium. Archives of biochemistry and biophysics, 2016, 601: 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Elnakish MT, et al. Effects of zacopride, a moderate IK1 channel agonist, on triggered arrhythmia and contractility in human ventricular myocardium. Pharmacological research, 2017, 115: 309–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Patel HH, et al. Disruption of protein kinase A localization using a transactivator of transcription (TAT)-conjugated A-kinase anchoring peptide reduces cardiac function. Journal of Biological Chemistry, 2010: jbc. M110. 146589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Haddad GE, et al. Modulation of atrial contraction by PKA and PKC during the compensated phase of eccentric cardiac hypertrophy. Basic research in cardiology, 2004, 99(5): 317–27 [DOI] [PubMed] [Google Scholar]
- [29].Ferrero P, et al. Ca2+/calmodulin kinase II increases ryanodine binding and Ca2+-induced sarcoplasmic reticulum Ca2+ release kinetics during β-adrenergic stimulation. Journal of molecular and cellular cardiology, 2007, 43(3): 281–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Endoh M Force-frequency relationship in intact mammalian ventricular myocardium: physiological and pathophysiological relevance. European journal of pharmacology, 2004, 500(1): 73–86 [DOI] [PubMed] [Google Scholar]
- [31].Janssen PML, et al. Myofilament properties comprise the rate-limiting step for cardiac relaxation at body temperature in the rat. Am J Physiol Heart Circ Physiol, 2002, 282: H499–H507 [DOI] [PubMed] [Google Scholar]
- [32].Meggio F, et al. Different susceptibility of protein kinases to staurosporine inhibition. The FEBS Journal, 1995, 234(1): 317–22 [DOI] [PubMed] [Google Scholar]
- [33].Lochner A and Moolman J. The many faces of H89: a review. Cardiovascular therapeutics, 2006, 24(3 - 4): 261–74 [DOI] [PubMed] [Google Scholar]
- [34].Monasky MM, et al. Length-Dependent Prolongation of Force Relaxation Is Unaltered by Delay of Intracellular Calcium Decline in Early-Stage Rabbit Right Ventricular Hypertrophy. Frontiers in physiology, 2017, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Biesiadecki BJ, et al. Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice. Journal of Biological Chemistry, 2010, 285(25): 19688–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Biesiadecki BJ, et al. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circulation research, 2007, 100(10): 1486–93 [DOI] [PubMed] [Google Scholar]
- [37].Nixon BR, et al. Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca 2+ sensitivity and accelerates deactivation in an acidic environment. Journal of molecular and cellular cardiology, 2014, 72: 177–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rossman EI, et al. Abnormal frequency-dependent responses represent the pathophysiologic signature of contractile failure in human myocardium. Journal of molecular and cellular cardiology, 2004, 36(1): 33–42 [DOI] [PubMed] [Google Scholar]
- [39].Starling EH. The Linacre lecture on the law of the heart: Longmans, Green, & Company, 1918 [Google Scholar]
- [40].Bowditch HP. Uber die Eigenthumlichkeiten der Reizbarkeit, welche die Muskelfasern des Herzens zeigen. Arb Physiol Inst Leipzig, 1871, 6: 139–76 [Google Scholar]
- [41].Bristow MR, et al. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. New England Journal of Medicine, 1982, 307(4): 205–11 [DOI] [PubMed] [Google Scholar]
- [42].Takimoto E, et al. Frequency-and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circulation research, 2004, 94(4): 496–504 [DOI] [PubMed] [Google Scholar]
- [43].Lamberts RR, et al. Frequency - dependent myofilament Ca2+ desensitization in failing rat myocardium. The Journal of physiology, 2007, 582(2): 695–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Milani-Nejad N and Janssen PM. Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharmacology & therapeutics, 2014, 141(3): 235–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chijiwa T, et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino) ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. Journal of Biological Chemistry, 1990, 265(9): 5267–72 [PubMed] [Google Scholar]
- [46].Toullec D, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. Journal of Biological Chemistry, 1991, 266(24): 15771–81 [PubMed] [Google Scholar]
- [47].Herbst KJ, et al. The cAMP-dependent protein kinase inhibitor H-89 attenuates the bioluminescence signal produced by Renilla Luciferase. PLoS One, 2009, 4(5): e5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Geilen CC, et al. A selective inhibitor of cyclic AMP - dependent protein kinase, N - [2 - bromocinnamyl (amino) ethyl] - 5 - isoquinolinesulfonamide (H - 89), inhibits phosphatidylcholine biosynthesis in HeLa cells. Febs letters, 1992, 309(3): 381–4 [DOI] [PubMed] [Google Scholar]
- [49].Janssen PM and De Tombe PP. Protein kinase A does not alter unloaded velocity of sarcomere shortening in skinned rat cardiac trabeculae. American Journal of Physiology-Heart and Circulatory Physiology, 1997, 273(5): H2415–H22 [DOI] [PubMed] [Google Scholar]
- [50].Park Y-S and Cho NJ. EGFR and PKC are involved in the activation of ERK1/2 and p90 RSK and the subsequent proliferation of SNU-407 colon cancer cells by muscarinic acetylcholine receptors. Molecular and cellular biochemistry, 2012, 370(1-2): 191–8 [DOI] [PubMed] [Google Scholar]
- [51].Biesiadecki BJ, et al. Tri-modal regulation of cardiac muscle relaxation; intracellular calcium decline, thin filament deactivation, and cross-bridge cycling kinetics. Biophysical reviews, 2014, 6(3-4): 273–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Braz JC, et al. PKC-α regulates cardiac contractility and propensity toward heart failure. Nature medicine, 2004, 10(3): 248–54 [DOI] [PubMed] [Google Scholar]
- [53].Champion HC and Kass DA. Calcium handler mishandles heart. Nature medicine, 2004, 10(3): 239–40 [DOI] [PubMed] [Google Scholar]
- [54].Schwinger RH, et al. cAMP - Dependent Protein Kinase A - Stimulated Sarcoplasmic Reticulum Function in Heart Failurea. Annals of the New York Academy of Sciences, 1998, 853(1): 240–50 [DOI] [PubMed] [Google Scholar]
- [55].Pathak A, et al. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circulation research, 2005, 96(7): 756–66 [DOI] [PubMed] [Google Scholar]