

ABSTRACT
Despite their promise, tumor-specific peptide vaccines have limited efficacy. CD27 is a costimulatory molecule expressed on CD4+ and CD8+ T cells that is important in immune activation. Here we determine if a novel CD27 agonist antibody (αhCD27) can enhance the antitumor T cell response and efficacy of peptide vaccines. We evaluated the effects of αhCD27 on the immunogenicity and antitumor efficacy of whole protein, class I-restricted, and class II-restricted peptide vaccines using a transgenic mouse expressing human CD27. We found that αhCD27 preferentially enhances the CD8+ T cell response in the setting of vaccines comprised of linked class I and II ovalbumin epitopes (SIINFEKL and TEWTSSNVMEERKIKV, respectively) compared to a peptide vaccine comprised solely of SIINFEKL, resulting in the antitumor efficacy of adjuvant αhCD27 against intracranial B16.OVA tumors when combined with vaccines containing linked class I/II ovalbumin epitopes. Indeed, we demonstrate that this efficacy is both CD8- and CD4-dependent and αhCD27 activity on ovalbumin-specific CD4+ T cells is necessary for its adjuvant effect. Importantly for clinical translation, a linked universal CD4+ helper epitope (tetanus P30) was sufficient to instill the efficacy of SIINFEKL peptide combined with αhCD27, eliminating the need for a tumor-specific class II-restricted peptide. This approach unveiled the efficacy of a class I-restricted peptide vaccine derived from the tumor-associated Trp2 antigen in mice bearing intracranial B16 tumors. CD27 agonist antibodies combined with peptide vaccines containing linked tumor-specific CD8+ epitopes and tumor-specific or universal CD4+ epitopes enhance the efficacy of active cancer immunotherapy.
Keywords: CD27, tumor immunotherapy, peptide vaccine, adjuvant, monoclonal antibody
Introduction
Tumor immunotherapy has emerged as a promising treatment modality for advanced malignancies. Specifically, peptide vaccines derived from MHC class I-restricted tumor antigens offer the promise of inducing robust tumor-specific CD8+ T cell responses1,2 to promote effective antitumor immunity.3–6 Unfortunately, the efficacy of class I-restricted peptide vaccines has proven to be limited,7,8 with overall clinical response rates as low as 3%.9 Class II CD4+ T cell helper epitopes are also capable of inducing potent antitumor immune responses,10–13 but peptide vaccines consisting of co-administered class I/II epitopes have also not yet experienced widespread clinical success.7 Novel adjuvant strategies are clearly needed to enhance the clinical utility of peptide vaccines in cancer immunotherapy.
Immunomodulatory antibodies targeting T cell checkpoint molecules have emerged as promising therapies that may be capable of promoting robust tumor-specific immunity in the setting of tumor vaccines that are otherwise ineffective.14–16 CD27, a member of the tumor necrosis factor receptor (TNFR) superfamily, is a costimulatory molecule expressed on naïve and activated CD4+ and CD8+ T cells17 and is known to be important in T cell activation,18 maturation,19 cytokine secretion,20 and survival,21 making it a promising target for T cell-based immunomodulation. Recently, a novel, fully human anti-human CD27 monoclonal antibody (αhCD27) was developed which binds with high affinity to induce potent human T cell responses in the context of T cell receptor stimulation.22 Because CD27 stimulation on both CD4+ T cells and CD8+ T cells can lead to their enhanced effector function and concomitant vaccine-induced CD4+ T cell help strengthens CD8+ T cell vaccine responses, we hypothesized that αhCD27 could be leveraged as an adjuvant for peptide vaccines and that it would provide a therapeutic benefit preferentially in the setting of peptide vaccines comprised of class I and II epitopes.
In this study, we evaluated the therapeutic effect of αhCD27 as a vaccine adjuvant in the setting of a highly aggressive intracranial tumor model. We demonstrate that αhCD27 enhances the immune response to class I–restricted tumor antigens, and that its adjuvant effect is potentiated in the setting of linked class I- and class II-restricted peptides. Additionally, we show that a universal CD4+ epitope is sufficient to enhance the tumor-specific CD8+ T cell response and increase the efficacy of tumor-derived class I-restricted peptide vaccines in the setting of adjuvant αhCD27, eliminating the need for class II-restricted tumor antigens. Taken together, our data suggest that αhCD27 coordinates CD8+ and CD4+ T cell responses to enhance the antitumor immune response. These findings highlight the potential for CD27 agonist antibodies to improve the clinical benefit of peptide vaccines for cancer immunotherapy.
Materials and methods
Study design
The goal of this study was to characterize the adjuvant activity of a clinically available immunomodulatory agonist anti-CD27 antibody and its therapeutic potential in a mouse model of advanced stage intracranial malignancy. The experimental design involves studies of vaccine-induced immunogenicity and survival of mice bearing intracranial B16 melanoma tumors. All mice were of the C57BL/6 background, aged 6–12 weeks, and female; naïve or tumor-bearing animals were randomized into treatment groups before the start of each experiment. Immunogenicity experiments were performed in groups of 5 mice each, while survival studies were performed with group sizes in excess of 7 mice each. Sample sizes were calculated using F-power analysis (α = 0.05) to yield at least 80% power to detect interactions, based on pilot data. For survival studies, pre-defined humane endpoints were used, according to the Duke University Institutional Animal Care and Use Committee (IACUC) guidelines. All experimental protocols and procedures were approved by the Duke University IACUC. All experiments were performed at least three times, and all outliers were included in the data analysis.
Mice and tumor cell lines
All mice were bred and maintained under pathogen-free conditions at Duke University Medical Center (DUMC). C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, NC, USA), and transgenic OT-I and OT-II mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Human CD27 transgenic (hCD27) mice, which express both murine and human CD27 molecules under the native murine CD27 promoter,23 were obtained from Celldex Therapeutics (Hampton, NJ, USA) and bred at DUMC. Homozygous hCD27 males were bred with C57BL/6, OT-I, or OT-II females to generate heterozygous hCD27, hCD27xOT-I, or hCD27xOT-II mice, respectively, for use in experiments. All animal experiments were performed according to protocols approved by the Duke University IACUC (Protocol Number: A283–15–11). The tumor cell lines B16.F10 and B16.OVA were a kind gift from Dr. Richard G. Vile at Mayo Clinic.24,25 All cell lines used were submitted for cell line authentication (Cell Check) and pathogen testing including mycoplasma testing (IMPACT) at IDEXX BioResearch prior to use. We confirmed species of origin, cell line specific markers and assessed for possible cross-contamination with other cell lines or pathogens
Evaluation of vaccine-induced t cell responses
Whole ovalbumin (Ova) protein was purchased from Sigma Aldrich (St. Louis, MO, USA), and custom peptides were purchased from JPT (Berlin, Germany) (for peptide sequences see Table 1). For whole Ova protein vaccination, hCD27 mice received intraperitoneal (ip) vaccination on day 0 with 2.5 mg Ova resuspended at 10 mg/mL in water. For peptide vaccination, hCD27 mice received intradermal (id) vaccination on day 0 with peptide emulsified in incomplete Freund’s adjuvant (IFA) (ThermoFisher, Waltham, MA, USA). As indicated by the experiment, the following peptides were used for vaccination: 50 µg of H2-Kb class I-restricted (Ova(I)); 100 µg of H2-IAb class II restricted Ova(IIA); 100 µg of H2-IAb class II restricted Ova(IIB); 150 µg of class II-restricted P30; 150 µg of linked Ova(I-IIA); 200 µg of linked Ova(I)-P30; 50 µg of H2-Kb class I-restricted Trp2 (Trp2(I)); or, 200 µg of linked Trp2-P30. The quantity of Ova-derived peptide used corresponds to the equimolar mass of the peptide in relation to 2.5 mg of whole Ova protein. Administration of αhCD27 was performed ip three days prior to (day −3) and on the day of vaccination (day 0) with 100 µg αhCD27 (Celldex Therapeutics) or 100 ug recombinant human IgG1 Fc isotype control (Bio X Cell, West Lebanon, NH, USA). On day 7 after vaccination, whole blood (50–100 µL) was collected by retro-orbital puncture for flow cytometric analysis of Ova(I)-specific cells, and spleens were harvested for ELISpot assays.
Table 1.
Peptide sequences.
| Peptide | Residue | Sequence |
|---|---|---|
| Ova(I) | Ova(257–264) | SIINFEKL |
| Ova(IIA) | Ova(265–280) | TEWTSSNVMEERKIKV |
| Ova(IIB) | Ova(323–339) | ISQAVHAAHAEINEAGR |
| Ova(I-IIA) | Ova(257–280) | SIINFEKLTEWTSSNVMEERKIKV |
| P30 | TT(948–968) | FNNFTVSFWLRVPKVSASHLE |
| Ova(I)-P30 | Ova(257–264)-Linker-TT(948–968) | SIINFEKLRVKRFNNFTVSFWLRVPKVSASHLE |
| Trp2(I) | Trp2(180–188) | SVYDFFVWL |
| Trp2(I)-P30 | Trp2(180–188)-Linker-TT(948–968) | SVYDFFVWLRVKRFNNFTVSFWLRVPKVSASHLE |
Flow cytometric analysis of ova(i)-specific CD8+ t cells
For the detection of circulating Ova(I)-specific CD8+ T cells, 50 µL whole blood was incubated with rat anti-mouse CD8-FITC (BD Biosciences, San Jose, CA, USA) and H2-Kb(SIINFEKL)-PE murine tetramer (MBL International, Woburn, MA, USA) in 100 µL PBS in the dark for 30 min at room temperature. Red blood cells (RBCs) were lysed, and cells were fixed with 1 mL 1X FACS Lysing Solution (BD Biosciences) in the dark for 15 min at room temperature and washed in PBS. All samples were analyzed on a FACSCalibur flow cytometer (BD Biosciences); absolute numbers per µL blood were calculated using Flowcount® beads (Beckman Coulter, Indianapolis, IN, USA), according to the manufacturer’s instructions.
Ifnγ ELISPOT
Vaccine-specific T cell responses were evaluated ex vivo by IFNγ ELISPOT. MultiScreen® 96-well filter plates (EMD Millipore, Billerica, MA, USA) were coated with 10 µg/mL anti-mouse IFNγ antibody (Mabtech, Cincinnati, OH, USA) overnight at 4°C. A total of 2.5 x 105 splenocytes/well were incubated in duplicate in RPMI media supplemented with 10% FBS (Gemini Bio-Products, West Sacramento, CA, USA), 1X non-essential amino acids (Life Technologies, Carlsbad, CA, USA), 1 mM L-glutamine (Life Technologies), and 100 IU/mL penicillin + 100 µg/mL streptomycin (Life Technologies), in the presence or absence of 1 µg/mL of the indicated peptide overnight at 37°C in a 5% CO2 incubator. Spots were developed using 1 µg/mL biotinylated anti-mouse IFNγ mAb (Mabtech), a VECTASTAIN® Elite ABC horseradish peroxidase kit (Vector Laboratories, Burlingame, CA, USA), and AEC substrate chromogen (Sigma); spots were quantified by ZellNet Consulting (Fort Lee, NJ, USA).
Tumor implantation
B16.F10 and B16.OVA cells were grown in DMEM (Life Technologies), 10% FBS and 2 mM L-glutamine at 37°C in 5% CO2. For intracranial tumor implantation, cells were harvested, resuspended at 3 × 106 cells/mL (B16.OVA) or 2 × 105 cells/mL (B16.F10), mixed 1:1 with 10% methylcellulose in PBS, and loaded into a 250 mL syringe (Hamilton, Reno, NV) with an attached 25-gauge needle. The needle was positioned 2 mm to the right of bregma and 4 mm below the surface of the skull at the coronal suture using a stereotactic frame (Kopf Instruments, Tujunga, CA). A dose of 7,500 cells (B16.OVA) or 500 cells (B16.F10) in a total volume of 5 µL was injected into hCD27 mice. For therapeutic survival studies, tumors were implanted on day 0, followed by 100 µg of αhCD27 or isotype ip on days 3 and 6 after tumor implantation. On day 6, the same day as the second dose of αhCD27, vaccination was administered (either 2.5 mg of ip injected whole Ova protein in water, or the indicated amount of id injected peptide emulsified in IFA). Tumor-bearing mice were monitored daily for morbidity endpoints and survival according to the Duke University IACUC guidelines.
Analysis of tumor-infiltrating lymphocytes
Tumors were harvested at day 14 after implantation and homogenized in a Stomacher® 80 Biomaster (Seward, Islandia, NY) in 6 mL digestion buffer [RPMI 1640 supplemented with 100 IU/mL penicillin + 100 µg/mL streptomycin, 1 mM L-glutamine, 1X non-essential amino acids, 1 mM sodium pyruvate (Life Technologies), 25 µM β-mercaptoethanol (ThermoFisher), 10% FBS, 133 µg/mL DNase I (Roche, Indianapolis, IN, USA), and 133 units/mL Type IV collagenase (Life Technologies)] for 20 min at 37°C. The resultant cell suspension was filtered through a 40 µm strainer and washed twice with PBS. The cells were stained with LIVE/DEAD® (ThermoFisher), H2-Kb(SIINFEKL) tetramer, and antibodies for CD3, CD4, and CD8 cell surface markers (BD Biosciences), according to the manufacturers’ instructions. The cells were resuspended in 150 µL PBS and analyzed on a FACSCalibur flow cytometer.
T cell depletion studies
For immunogenicity studies, mice were depleted of CD4+ or CD8+ cells in the priming phase by once daily intraperitoneal doses of 200 µg αCD4 (GK1.5, Bio X Cell) or αCD8 (2.43, Bio X Cell), respectively, for three consecutive days prior to vaccine/αhCD27 administration (as previously described), and immune responses were assessed at day 7 after vaccination. For survival studies, CD8+ cells were depleted by once daily intraperitoneal administration of 200 µg αCD8 for three consecutive days immediately after intracranial tumor implantation and before Ova/αhCD27 treatment. For CD4 depletion studies in tumor-bearing mice, a tumor challenge model was employed in which mice were implanted with intracranial B16.OVA tumors seven days after vaccination with whole Ova protein and αhCD27; CD4+ cells were depleted by once daily intraperitoneal administration of 200 µg αCD4 for three consecutive days prior to Ova/αhCD27 vaccination (priming phase) or for three consecutive days immediately after intracranial tumor implantation (effector phase).
Adoptive lymphocyte transfers
CD4+ and CD8+ T cells were purified from the spleens of OTII and OTI mice, respectively, by magnetic labeling in an autoMACs® Pro Separator (Miltenyi Biotec, San Diego, CA). Briefly, spleens were disaggregated and filtered into single cell suspensions, and RBCs were removed by incubating for 5 min in 1X lysis buffer (BD Biosciences). The splenocytes were then washed once in RPMI media and once in MACs buffer (Miltenyi Biotec) and resuspended at 2.5 x 108 cells per mL. The cells were then subject to magnetic labeling and T cell isolation using CD4+ and CD8+ T cell isolation kits (Miltenyi Biotec), according to the manufacturer’s instructions. The purified CD4+ and CD8+ T cells were then mixed at a 2:1 ratio in PBS at a total cell concentration of 3 × 107 per mL, and 100 µL of the appropriate cell mixture was injected intravenously in the tail veins of wildtype C57BL/6 mice.
Statistical analysis
Overall survival was computed from the date of tumor implantation to the date of humane endpoint or death. Survival distributions are described using Kaplan-Meier methods, and the Gehan-Breslow-Wilcoxon test was used to compare survival distributions between treatment groups. Student’s unpaired t-test was used to compare IFNγ SFU and tetramer values upon adjuvant αhCD27 administration versus isotype control. One-way ANOVA was used to analyze TIL levels and the effect of T-cell depletion on the ovalbumin vaccine response. Two-way ANOVA was used to assess the magnitude of the effect of αhCD27 on the CD8+ T cell response in animals receiving different vaccine types. Associations of class II responses with class I responses were assessed using the Pearson correlation coefficient.
Results
αhcd27 enhances the immune response to a whole protein vaccine and promotes antitumor efficacy
To explore the use of CD27 costimulation as a vaccine adjuvant, we first evaluated the effect of αhCD27 on the CD8+ T cell response to vaccination with whole protein using ovalbumin as a model antigen. Groups of hCD27 mice received αhCD27 or hIgG1 3 days before and on the day of vaccination, vaccine responses were evaluated by the frequency (as determined by tetramer staining of peripheral blood leukocytes) and effector function (as determined by ex vivo re-stimulation in a IFNγ ELISPOT) of T cells specific for the immunodominant ovalbumin class I epitope (Ova(I)) (Figure 1). We found that the frequency of peripheral blood Ova(I)-specific CD8+ T cells increased from ~0.5% to ~7% in mice that received whole ovalbumin protein combined with αhCD27 compared to human IgG1 (hIgG1) controls (Figure 1A, P = 0.0035). These vaccine-induced responses peaked a week after vaccination and consisted largely of a CD44+CD62L− effector memory phenotype (Supplemental Figure 1). Additionally, we observed an increase in the level of IFNγ-producing splenic lymphocytes upon ex vivo re-stimulation with Ova(I) peptide, from ~110 ± 37 IFNγ+ spot forming units (SFUs) per 106 splenocytes in control mice to 1,656 ± 139 SFUs per 106 splenocytes in αhCD27-treated mice (Figure 1B, P < 0.0001). Both altering the timing of αhCD27 administration or increasing the number of vaccinations did not further enhance vaccine immunogenicity; therefore, our original regimen was continued for all subsequent experiments (Supplemental Figure 2).
Figure 1.

αhCD27 enhances the ovalbumin protein response and promotes antitumor efficacy.(A) hCD27 mice (n = 5 per group) received ip 2.5 mg whole Ova protein alongside 100 µg of αhCD27 or hIgG1 isotype control (3 days prior to and on the day of whole Ova protein vaccination). Ova(I)-specific CD8+ T cell responses were evaluated by H2-Kb(SIINFEKL)-PE tetramer staining of peripheral blood cells seven days after vaccination. Flow cytometric analysis was done by FWD vs SSC identification of the lymphocyte population, followed by gating on CD3+ cells, with subsequent gating on tetramer+ CD8+ cells. (B)Vaccine-induced CD8+ T cell responses were also evaluated by ex vivo re-stimulation of splenocytes with Ova(I) peptide in an IFNγ ELISPOT assay. (C) hCD27 mice bearing intracranial B16.OVA tumors received 100 µg of αhCD27 or hIgG1isotype control at days 3 and 6 after tumor implantation, with or without whole Ova protein vaccination on day 6 after tumor implantation. Tumors were harvested on day 14 after implantation and analyzed for the frequency of tumor-infiltrating Ova(I)-specific CD8+ T cells by H2-Kb(SIINFEKL)-PE tetramer staining. Flow cytometric analysis was done by gating on live CD3+ cells, followed by gating on tetramer+ CD8+ cells. (D) The efficacy of combinatorial whole Ova protein vaccination and αhCD27 administration (n = 7 per group) was evaluated in hCD27 mice bearing 3-day established intracranial B16.OVA tumor. Schema on top of the survival curve indicates the experimental design. B16.OVA tumors were implanted on day 0, followed by 100 µg of αhCD27 or isotype ip on days 3 and 6 after tumor implantation. On day 6, same day after the last dose of αhCD27, vaccination with 2.5 mg of whole Ova protein was given ip and survival was monitored. Statistical analyses were performed using Student’s unpaired t-test (A and B), one-way ANOVA with Tukey post-hoc comparisons (C), or the Gehan-Breslow-Wilcoxon test (D). Statistical significance was determined at a *P value < 0.05.
Figure 2.
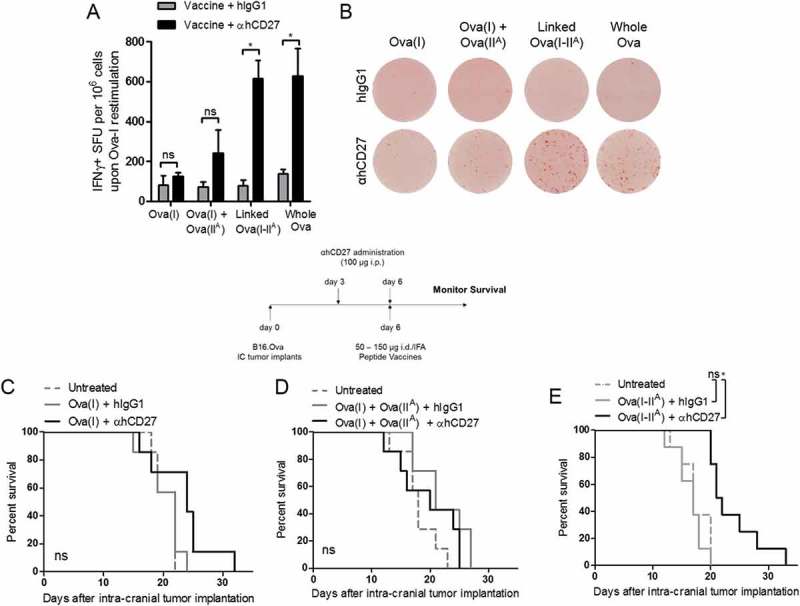
The adjuvant effect of αhCD27 on the class I peptide response is enhanced by a linked class II epitope. (A) hCD27 mice (n = 5 per group) were vaccinated with either: 50 µg of Ova(I) peptide, 50 µg Ova(I) and 100 µg Ova(IIA) peptides, 150 µg of the linked Ova(I-IIA) peptide, or 2.5 mg whole Ova protein. Peptide vaccines were emulsified in IFA and given id and whole Ova protein was administered ip in water. 100 µg αhCD27 or hIgG1 isotype control were given ip three days before (day −3) and on the day of vaccination (day 0). CD8+ T cell responses were evaluated by ex-vivo splenocyte re-stimulation with the Ova(I) peptide in a IFNγ ELISpot (B) representative ELISpot results are shown. (C-E) hCD27 mice (n = 7 per group) were implanted with intracranial B16.OVA tumors on day 0, received 100 µg of ip αhCD27 or hIgG1 isotype control 3 days and 6 days after tumor implantation, and received vaccination 6 days after implantation. Schema on top of the survival curves indicates the experimental design. Vaccination consisted off: 50 µg of Ova(I) peptide (C), co-administration of 50 µg Ova(I) and 100 µg of Ova(IIA) peptides (D), or 150 µg linked Ova(I-IIA) peptide (E). Statistical analyses were performed using two-way ANOVA with Tukey post-hoc comparisons (A) or the Gehan-Breslow-Wilcoxon test (C, D, E). Statistical significance was determined at a *P value < 0.05.
We next asked whether adjuvant αhCD27 combined with whole ovalbumin protein would potentiate the tumor immune response and be efficacious against intracranial B16.OVA, a highly aggressive melanoma model that is consistently resistant to vaccine immunotherapy.26 hCD27 mice bearing 3-day established intracranial B16.OVA tumors and treated with whole ovalbumin protein with adjuvant αhCD27 demonstrated an increased frequency and absolute number of Ova-specific CD8+ T cells both peripherally and intratumorally in comparison to mice treated with isotype control (Supplemental Figure 3). Flow cytometric analysis of the tumor-infiltrating lymphocytes (TILs) revealed a marked increase in the level of Ova(I)-specific CD8+ T cells, from ~20% of the CD8+ TIL population in mice that received ovalbumin alone to ~50% in mice treated with ovalbumin + αhCD27 (Figure 1C, Ova vs Ova + αhCD27: P = 0.0162). This enhanced level of tumor-infiltrating CD8+ T cells corresponded to an 18-day increase in median survival in mice that received ovalbumin + αhCD27 compared to control animals (Figure 1D, Ova + αhCD27 vs. Ova + hIgG1: P = 0.0024).
Figure 3.
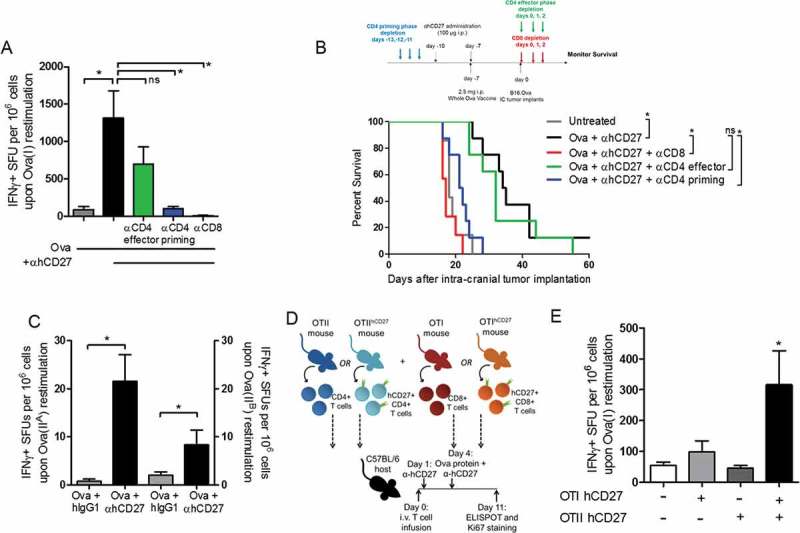
CD8+ and CD4+ T cells are required for the adjuvant effect and survival benefit of αhCD27. (A) hCD27 mice (n = 5 per group) received 2.5 mg whole Ova protein vaccine on day 0 with or without 100 µg of αhCD27 on days −3 and 0. To assess the impact of T cell depletion, CD8+ T cells were depleted by 200 µg of αCD8 antibody (clone 2.43) administered ip on days −6, −5, and −4 immediately prior to the first αhCD27 administration on day −3. CD4+ T cells were depleted by 200 µg αCD4 antibody (clone GK1.5) administered ip on days −6, −5, and −4 to deplete during the priming phase, or on days 5, 6 and 7 to deplete during the T cell effector phase. CD8+ T cell responses were determined 8 days after vaccination by ex vivo re-stimulation of splenocytes with Ova(I) peptide in an IFNγ ELISpot. (B) The effect of T cell depletion on antitumor efficacy was evaluated in a tumor challenge (see schema on top of the survival curve). For all experiments, hCD27 mice (n = 7 per group) received 2.5 mg of whole Ova protein vaccine on day −7, 100 µg of αhCD27 on days −10 and −7, and tumor implantation on day 0, survival was monitored. For CD8+ T cell depletion, 200 µg of αCD8 was administered on days 0, 1 and 2 immediately after tumor implantation (red). For CD4+ T cell depletion during priming, 200 µg of αCD4 was administered on days −13, −12, and −11 immediately prior to αhCD27 treatment on day −10 (blue). For CD4+ T cell depletion during the effector phase, αCD4 was administered on days 0, 1, and 2 immediately after tumor implantation (green). (C) The immune responses to two class II Ova epitopes, Ova(IIA) and Ova(IIB), were evaluated in hCD27 mice (n = 5 per group) vaccinated with 2.5 mg whole Ova protein ip on day 0 and treated with 100 µg of αhCD27 or hIgG1 isotype control on days −3 and 0. Seven days after vaccination, splenocytes were harvested and T cell responses were evaluated by ex vivo re-stimulation of splenocytes with Ova(IIA) or Ova(IIB) peptide in an IFNγ ELISpot. (D-E) hCD27 transgenic mice were crossed with transgenic mice that possess Ova-specific CD8+ T cells (OTI) or Ova-specific CD4+ T cells (OTII) to generate Ova-specific T cells with hCD27 expression. As described in the methods, C57BL/6 hosts (n=5 per group) received adoptive transfer of one of four combinations of T cells on day 0: OTI and OTII cells (no hCD27), OTIxhCD27 and OTII cells, (hCD27 on CD8) OTI and OTIIxhCD27 cells (hCD27 on CD4), or OTIxhCD27 and OTIIxhCD27 (hCD27 on CD4 and CD8). αhCD27 was administered ip on days 1 and 4 and whole Ova protein was given on day 4 (D). CD8+ T cell responses were measured by IFNγ ELISPOT upon ex vivo re-stimulation with Ova(I) peptide (E). Statistical analyses were performed using a one-way ANOVA with Tukey post-hoc comparisons (A), the Gehan-Breslow-Wilcoxon test (B), Student’s unpaired t-test (C), and two-way ANOVA with Tukey post-hoc comparisons (E). Statistical significance was determined at a *P value < 0.05.
The adjuvant effect of αhCD27 on the class I peptide response is enhanced by a linked class II epitope
Given the potent therapeutic effect of αhCD27 combined with whole ovalbumin protein and to inform the use of αhCD27 in combination with peptide vaccines, we next examined which peptide components of whole ovalbumin protein were contributing to the enhanced ovalbumin-specific CD8+ T cell response in the setting of adjuvant αhCD27. We compared the adjuvant effect of αhCD27 when combined with four different ovalbumin-derived vaccines (Table 1): 1) the single immunodominant class I-restricted ovalbumin peptide epitope, SIINFEKL (Ova(I)); 2) Ova(I) co-administered with an immunodominant class II-restricted peptide epitope, TEWTSSNVMEERKIKV (Ova(IIA), located immediately downstream of Ova(I); 3) a long peptide comprised of the continuous Ova(I) and Ova(IIA) sequences (Ova(I-IIA)); and 4) the whole ovalbumin protein. We administered each of these four vaccines with combined αhCD27 or hIgG1 isotype control and evaluated the CD8+ T cell response seven days after vaccination by ex vivo restimulation with the Ova(I) peptide in a IFNγ ELISPOT assay. We observed only slight (but statistically insignificant) increases in the Ova(I)-specific CD8+ T cell response in the settings of αhCD27 alongside the single Ova(I) peptide vaccine or Ova(I) co-administered with Ova(IIA) (Figures 2A and 2B). In contrast, αhCD27 in the setting of Ova(I-IIA) and whole ovalbumin protein resulted in a 5-fold increase in the level of Ova(I)-specific CD8+ T cells compared to controls (Figure 2A, linked Ova(I-IIA) + hIgG1 vs linked Ova(I-IIA) + αhCD27: P = 0.00045; whole Ova + hIgG1 vs whole Ova + αhCD27: P = 0.0016).
These results were mirrored in our tumor efficacy experiments with the three peptide vaccines. hCD27 mice bearing 3-day established intracranial tumors were treated with Ova(I), Ova(I)+Ova(IIA), or Ova(I-IIA), with or without αhCD27. None of these vaccines were therapeutic in the absence of adjuvant αhCD27, and αhCD27 combined with Ova(I) (Figure 2A) or Ova(I)+Ova(IIA) (Figure 2B) had no effect on survival. In contrast, we found that adjuvant αhCD27 in the setting of linked Ova(I-IIA) resulted in a significant therapeutic benefit (Figure 2C, Ova(I-IIA) + hIgG1 vs Ova(I-IIA) + αhCD27: P = 0.0004), reminiscent of our previous findings with whole ovalbumin protein. Taken together, these results suggest that linked class I and II epitopes drive the adjuvant activity of αhCD27 on the CD8+ T cell response such that αhCD27 prolongs survival preferentially in the setting of vaccines comprised of linked class I and II epitopes.
CD8+ T cells and CD4+ T cells during the priming phase of the vaccine response are required for the adjuvant effect of αhCD27
The failure of a class I-restricted peptide vaccine to induce effective antitumor immune responses is consistent with prior clinical experience with peptide vaccines,9 but we found that αhCD27 could enhance the tumor-specific CD8+ T cell response and resultant antitumor efficacy when combined with vaccines containing linked class I and II epitopes. We thus hypothesized that effector CD8+ T cells and CD4+ T cells, in either the priming or effector phase, were necessary for the antitumor effect of adjuvant αhCD27. To test this hypothesis, we first examined the effect of CD4+ T cell depletion on the CD8+ T cell response to whole ovalbumin protein vaccination, during the priming phase (depletion prior to vaccination) and effector phase (depletion 6 days after vaccination) of the vaccine response. We found that a lack of CD4+ T cells in the priming phase significantly reduced the adjuvant effect of αhCD27 on the CD8+ T cell response to whole ovalbumin protein vaccination, as determined by ex vivo re-stimulation with Ova(I) peptide in a IFNγ ELISPOT (Figure 3A, Ova + αhCD27 vs Ova + αhCD27 + αCD4 priming: P = 0.00316), while the depletion of CD4+ T cells later in the vaccine response had only a slight but statistically non-significant effect (Figure 3A). Additionally, the depletion of CD8+ T cells at the effector phase completely abrogated the ex vivo response to Ova(I) peptide re-stimulation (Figure 3A, Ova + αhCD27 vs Ova + αhCD27 + αCD8: P = 0.00272).
We next examined the effect of CD8+ or CD4+ T cell depletion on the antitumor effect of adjuvant αhCD27 in a challenge setting of intracranial B16.OVA, in which mice were pretreated with αhCD27 + ovalbumin seven days prior to tumor implantation. To distinguish between the priming and effector phases of the vaccine response, CD8+ T cells were depleted seven days after vaccination, at the time of tumor implantation (effector phase), and CD4+ T cells were depleted immediately prior to vaccination (priming phase) or at the time of tumor implantation (effector phase). We found that the depletion of CD8+ T cells seven days after vaccination completely abrogated the efficacy of combined αhCD27 + whole ovalbumin protein (Figure 3B, red). In contrast, the depletion of CD4+ T cells seven days after vaccination had no significant effect on the efficacy of αhCD27 + ovalbumin (Figure 3B, green); however, CD4+ T cell depletion prior to vaccination completely abrogated antitumor efficacy (Figure 3B, blue, Ova + αhCD27 vs Ova + αhCD27 + αCD4 priming: P = 0.0004). These data demonstrate that both CD8+ and CD4+ T cells are necessary for the therapeutic effect of adjuvant αhCD27 but that CD4+ T cells contribute to this effect only during the priming phase of the vaccine response. Consistent with our own data and previous reports showing that class II epitopes are integral for robust class I-restricted vaccine responses,11 these results suggest that CD4+ T cell help is critical for the adjuvant effect of αhCD27.
Direct CD27 stimulation on vaccine-specific CD4+ and CD8+ T cells is necessary for the adjuvant effect of αhCD27
Our previous data reveals the necessity of CD4+ T cells in the priming phase of the vaccine response for αhCD27 to enhance the immunogenicity and antitumor efficacy of a whole ovalbumin protein vaccine, leading us to further investigate the effect of αhCD27 on the ovalbumin-specific CD4+ T cell response. We hypothesized that αhCD27 enhances the CD4+ T cell response to ovalbumin class II epitopes and that direct stimulation of CD27 expressed on ovalbumin-specific CD8+ and CD4+ T cells was necessary for the observed adjuvant effect of αhCD27. To test this hypothesis, we first asked if αhCD27 enhances the CD4+ T cell response to class II epitopes in the whole ovalbumin protein vaccine. We measured changes in the immune response to Ova(IIA) and Ova(IIB), a class II ovalbumin epitope downstream from Ova(IIA) (Table 1), elicited by whole ovalbumin protein vaccination, with or without adjuvant αhCD27. Indeed, we found that the addition of αhCD27 enhanced the T cell response to both of these class II–restricted epitopes, as determined by IFNγ ELISPOT (Figure 3C, Ova(IIA): P = 0.0056; Ova(IIB): P = 0.071,).
Next, to determine if direct engagement of CD27 on antigen-specific CD4+ and CD8+ T cells was necessary for the enhanced CD8+ T cell response to whole ovalbumin protein vaccination, we employed an adoptive transfer wherein wildtype C57BL/6 mice received intravenous infusions of CD8+ T cells purified from OT-I or OT-I x hCD27 transgenic mice, whose CD8+ T cells are specific for Ova(I), and CD4+ T cells purified from OT-II or OT-II x hCD27 transgenic mice, whose CD4+ T cells are specific for Ova(IIB). CD8+ and CD4+ T cells purified from OT-I or OT-II mice express only murine CD27 and so would not be responsive to αhCD27 treatment, while those purified from OT-I x hCD27 or OT-II x hCD27 mice, which express both murine and human CD27, could be responsive to treatment with αhCD27 (Figure 3D). Of note, the cohort of mice receiving OT-I + OT-II T cells, which do not express hCD27, serve as an experimental control for determining the baseline immune response to whole ovalbumin protein vaccination (i.e., in the absence of exogenous CD27 stimulation) in this adoptive transfer setting. We found that vaccine responses were highest in mice that received OT-I x hCD27 CD8+ T cells and OT-II x hCD27 CD4+ T cells, as measured by ex vivo re-stimulation with Ova(I) in a IFNγ ELISPOT (Figure 3E, OTI + OTII vs OTI x hCD27 + OTII x hCD27: P = 0.028), indicating that direct CD27 stimulation on both ovalbumin-specific CD8+ and CD4+ T cells contributes to the immunogenicity of whole ovalbumin protein in the setting of αhCD27. Taken together, our data suggest that αhCD27 enhances both vaccine-induced CD8+ and CD4+ T cell responses, resulting in increased CD4+ T cell helper function to allow for a more pronounced vaccine-induced CD8+ effector response.
A universal CD4+ T cell helper epitope is sufficient to enhance CD8+ tumor-specific vaccine responses in the setting of adjuvant αhCD27
In light of our finding that the therapeutic effect of adjuvant αhCD27 requires vaccine-specific CD4+ T cells in the priming phase of the immune response, we wondered if non-specific CD4+ T cell help would be sufficient to instill antitumor efficacy in the setting of adjuvant αhCD27. To test this hypothesis, we evaluated the adjuvant activity of αhCD27 when combined with a known CD4+ T cell universal class II helper epitope, tetanus toxin (TT) P30 (TT(948–968)).27 We developed a peptide vaccine consisting of Ova(I) covalently linked with the P30 epitope (Ova(I)-P30) (Table 1) and administered it with or without adjuvant αhCD27. A linker sequence consisting of a furin cleavage site28 was included between the Ova(I) and P30 epitopes to ensure that this synthetic long peptide would be processed into two distinct epitopes. Indeed, we found that αhCD27 enhances the vaccine response to Ova(I)-P30 (Figures 4A and 4B, Ova(I)-P30 + hIgG1 vs Ova(I)-P30 + αhCD27: P = 0.0003) and that the CD8+ T cell response to Ova(I) was greater upon vaccination with Ova(I)-P30 compared to Ova(I) in the setting of adjuvant αhCD27 (Figure 4B, Ova(I) + αhCD27 vs Ova(I)-P30 + αhCD27: P = 0.0327). Consistent with our previous findings, this enhanced Ova(I) response was reduced when CD4+ T cells were depleted prior to vaccination (Figure 4B, Ova(I)-P30 + αhCD27 vs Ova(I)-P30 + αhCD27 + αCD4: P = 0.0001). Moreover, αhCD27 combined with Ova(I)-P30 prolonged survival in hCD27 mice bearing intracranial B16.OVA tumors (Figure 4C, Ova(I)-P30 + hIgG1 vs Ova(I)-P30 + αhCD27: P = 0.0005). These data indicate that a universal class II helper epitope can be the source of vaccine-induced helper CD4+ T cells, thereby broadening the utility of this strategy by eliminating the need for a tumor-derived class II epitope in the vaccine. Importantly, however, the addition of the class II P30 epitope alone was not sufficient to enhance the vaccine-induced immune response and promote antitumor efficacy, while combined αhCD27 unveiled the efficacy of this synthetic long peptide vaccine, thus highlighting the synergy between CD4+ T cell help and adjuvant αhCD27.
Figure 4.
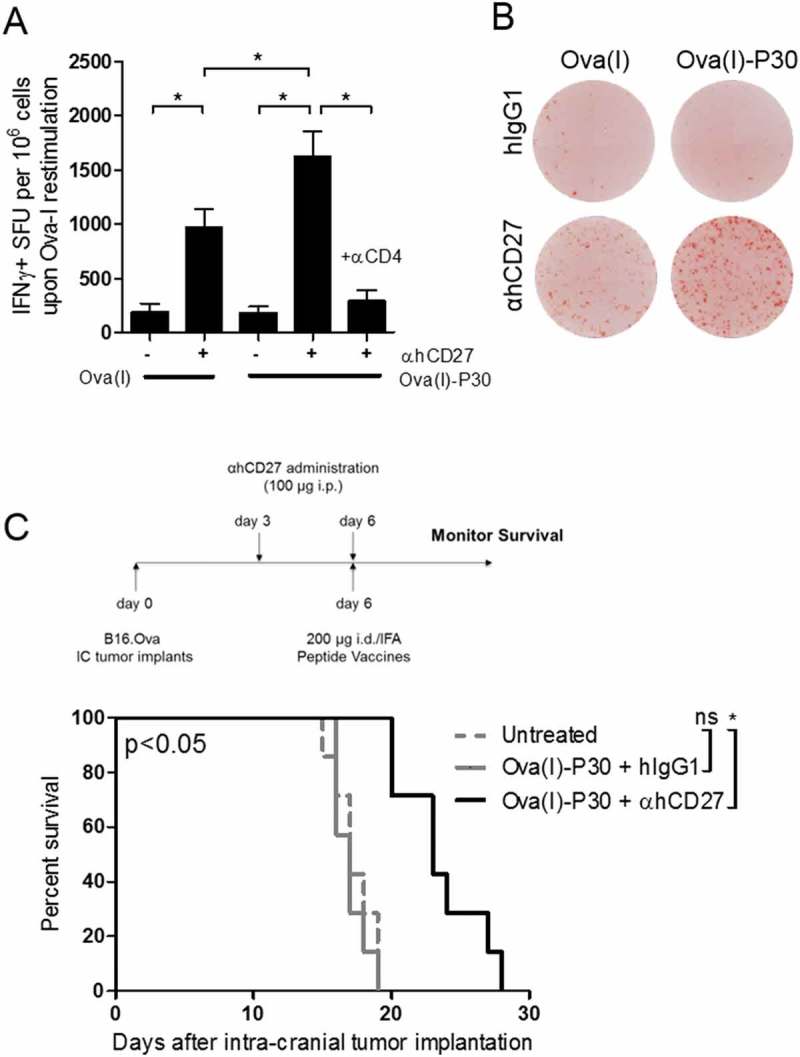
A universal CD4+ T cell helper epitope is sufficient to enhance CD8+ tumor-specific vaccine responses in the setting of adjuvant αhCD27. (A) CD8+ T cell responses were evaluated in mice vaccinated id with 50 µg of Ova(I) or 200 µg of Ova(I)-P30 peptides. 100 µg of αhCD27 or hIgG1 was administered on days −3 and 0 with vaccination on day 0. Seven days after vaccination, splenocytes were harvested and ex vivo re-stimulated with Ova(I) peptide in an IFNγ ELISpot. CD4+ cells were depleted during vaccine priming by αCD4 antibody (GK1.5) administered ip on days −6, −5, and −4. (B) Representative ELISpot images from mice vaccinated with Ova(I) or Ova(I)-P30 that also received αhCD27 or hIgG1 are shown. (C) For survival analysis, hCD27 mice bearing 3-day established intracranial B16.OVA tumors were vaccinated with Ova(I)-P30 and received αhCD27 or hIgG1 isotype control (n = 7 per group). Schema on top of the survival curves indicates the experimental design. B16.OVA tumors were implanted on day 0, 100 µg of αhCD27 or isotype was administered ip on days 3 and 6, and 200 µg of linked Ova(I)-P30 peptide emulsified IFA was given id on day 6 and survival was monitored. The quantity of peptide used correspond to the equimolar mass of the peptides in relation to 2.5 mg of whole Ova protein. Statistical analyses were performed using two-way ANOVA with Tukey post-hoc comparisons (A) or the Gehan-Breslow-Wilcoxon test (C). Statistical significance was determined at a *P value < 0.05.
CD27 stimulation coordinates CD4+ t cell help and vaccine-induced CD8+ t cell responses
We show above that vaccination with linked class I and II epitopes increases the capacity for αhCD27 to enhance the vaccine-induced CD8+ T cell response. We hypothesized that the adjuvant activity of αhCD27 occurs because the enhanced CD4+ T cell response potentiates the CD8+ T cell response in the setting of CD27 stimulation. If correct, this hypothesis would predict that the magnitude of the CD4+ T cell response dictates the magnitude of the CD8+ T cell response. We thus analyzed the relationship between vaccine-induced CD4+ and CD8+ T cell responses in the settings of αhCD27 and hIgG1 isotype control. We plotted the class I and class II T cell responses (as determined by IFNγ ELISPOT) of individual mice vaccinated with Ova(I-IIA) or Ova(I)-P30 peptide upon ex vivo re-stimulation with Ova(IIA) versus Ova(I) (Figures 5A and 5B) or P30 versus Ova(I) (Figures 5C and 5D), respectively, normalized across cumulative experiments. Indeed, for both of these cases, we found that the class II response positively correlates with the class I response in the presence of adjuvant αhCD27 (Ova(IIA) vs Ova(I) with Ova(I-IIA) vaccination, P = 0.0156 and R = 0.631; P30 vs Ova(I) with Ova(I)-P30 vaccination, P = 0.0299 and R = 0.6817) (Figure 5), while no statistically significant relationship between the CD4+ and CD8+ T cell responses was found in the setting of hIgG1 isotype control. This analysis suggests that a coordinated CD4/CD8 response is orchestrated upon CD27 stimulation, further highlighting the potential of αhCD27 as a vaccine adjuvant for peptide vaccines comprised of linked class I and class II epitopes.
Figure 5.
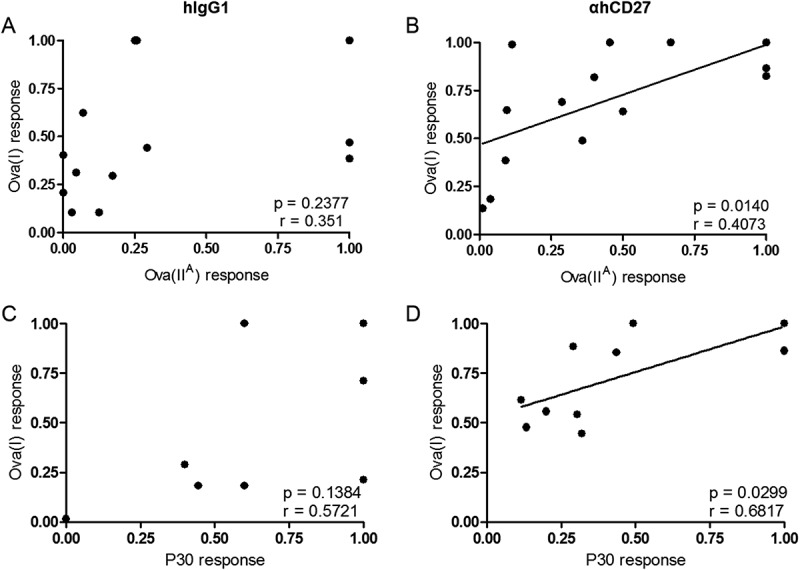
CD27 stimulation coordinates CD4+ T cell help and vaccine-induced CD8+ T cell responses. hCD27 mice (n = 5 per group) were immunized id with 150 µg of Ova(I-IIA) or 200 µg of Ova(I)-P30 on day 0, and 100 µg of αhCD27 or hIgG1 isotype control was given ip on days −3 and 0. The quantity of peptide utilized correspond to equimolar mass of the peptides in relation to 2.5 mg of whole Ova protein. T cell responses to class I and II peptide epitopes were evaluated seven days after vaccination by ex vivo re-stimulation of splenocytes in an IFNγ ELISpot using Ova(I), Ova(IIA), or P30 as indicated. Correlation analyses were performed comparing ex vivo responses to peptide re-stimulation of T cells derived from mice that received Ova(I-IIA) vaccination and hIgG1 (A) or αhCD27 (B), and mice that received Ova(I)-P30 vaccination and hIgG1 (C) or αhCD27 (D). Statistical analyses were performed using Pearson correlation analysis, and statistical significance was determined at a *P value < 0.05.
αhcd27 enhances the immune response to a peptide vaccine derived from a tumor-associated antigen
To demonstrate the clinical feasibility of αhCD27 as a peptide vaccine adjuvant, we evaluated the ability of αhCD27 to enhance the vaccine response to a known tumor-associated antigen, tyrosinase-related protein 2 (Trp2).29,30 Vaccination with the Trp2 immunodominant class I epitope (Trp2(I)) (Table 1) has been shown to promote robust antigen-specific immune responses in C57BL/6 mice.31,32 We found that αhCD27 enhances the vaccine response to both Trp2(I) and a linked Trp2(I)-P30 peptide vaccine (Figures 6A and 6B, Trp2(I) + hIgG1 vs Trp2(I) + αhCD27: P = 0.0220; Trp2(I)-P30 + hIgG1 vs Trp2(I)-P30 + αhCD27: P = 0.0009). Additionally, similar to our previous findings, αhCD27 enhances the vaccine-induced CD8+ T cell response to a greater degree in the setting of Trp2(I)-P30 compared to Trp2(I) alone (P = 0.00147), leading to an overall higher vaccine response in the setting of Trp2(I)-P30 (Figures 6A and 6B, Trp2(I) + αhCD27 vs Trp2(I)-P30 + αhCD27: P = 0.0004). Furthermore, the magnitude of the P30-specific CD4+ T cell response correlates with the magnitude of the Trp2(I)-specific CD8+ T cell response in the setting of αhCD27 (Figure 6D, P30 vs Trp2(I), P = 0.0012 and R = 0.867), but not in the setting of hIgG1 (Figure 6C), indicating the ability for αhCD27 to coordinate CD4+ and CD8+ T cell responses in this vaccine setting as well. Our immunogenicity results were mirrored in a tumor efficacy experiment, in which αhCD27 combined with Trp2(I)-P30 prolonged survival in mice bearing 3-day established intracranial B16.F10 tumors (Figure 6E, Trp2(I)-P30 + hIgG1 vs Trp2(I)-P30 + αhCD27: P = 0.0095), while αhCD27 in combination with Trp2(I) had no effect relative to control groups. Our findings in the setting of αhCD27 combined with a clinically-relevant tumor antigen serve as a proof of principle for the promising translational potential of αhCD27 as a peptide vaccine adjuvant for tumor immunotherapy.
Figure 6.:
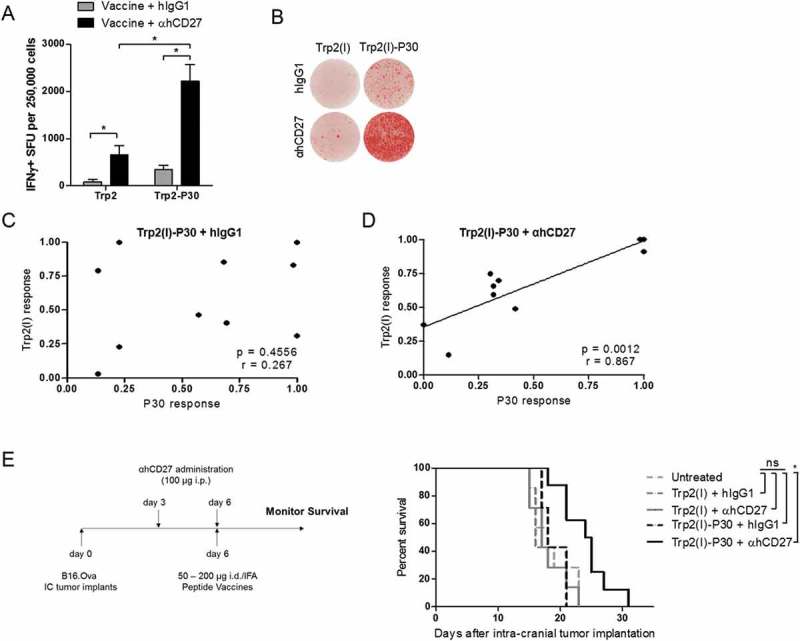
αhCD27 enhances the T cell response to a linked class I/II peptide vaccine derived from Trp2. (A) Mice were vaccinated id with 50 µg of Trp2(I) or 200 µg of Trp2(I)-P30 peptides emulsified in IFA on day 0 with 100 µg of αhCD27 or hIgG1 administered on day −3 or day 0. CD8+ T responses were evaluated seven days after vaccination by ex vivo re-stimulation of splenocytes with Trp2(I) peptide in an IFNγ ELISpot. (B) Representative ELISpot images are shown. (C-D) Mice were vaccinated with Trp2(I)-P30 peptide (day 0) and received αhCD27 or hIgG1 administration (days −3 and 0). Correlation analyses were performed comparing ex vivo responses of T cells derived from vaccinated mice that had received IgG (C) or αhCD27 (D) and were then re-stimulated with Trp2(I) and P30 peptides. (E) For survival analysis, hCD27 mice (n = 7 per group) were implanted with intracranial B16.F10 tumors on day 0, were vaccinated with 50 µg of Trp2(I) or 200 µg of Trp2(I)-P30 on day 6, and received 100 µg of αhCD27 or hIgG1 isotype control on days 3 and 6. Survival was monitored and the results of two separate experiments for each vaccine are shown. Schema on the left of the survival curves indicates the experimental design. Statistical analyses were performed using two-way ANOVA with Tukey post-hoc comparisons (A), Pearson correlation analysis (C, D), or the Gehan-Breslow-Wilcoxon test (E). Statistical significance was determined at a *P value < 0.05.
Discussion
In this study, we show that αhCD27 preferentially enhances the T cell response to peptide vaccines containing linked class I/II epitopes. Similar to clinical experiences with single-epitope class I-restricted peptide vaccines,9 we found that αhCD27 was not efficacious as a vaccine adjuvant in the setting of single class I-restricted peptides. In contrast, when administered alongside linked class I/II peptide vaccines designed to target both CD8+ and CD4+ T cells, we show that adjuvant αhCD27 leads to robust vaccine-induced CD8+ T cell responses in a CD4-dependent manner. Indeed, we found that CD27 stimulation on both antigen-specific CD4+ and CD8+ T cells, rather than either T cell compartment alone, is required for the adjuvant activity of αhCD27. As such, we concluded that αhCD27 preferentially enhances vaccine responses in the setting of linked class I/II-epitopes, which distinctively allows for increased tumor-specific CD8+ T cell responses and prolonged survival in mice bearing aggressive and poorly immunogenic intracranial tumors. Most importantly for clinical translation, we show that the linked class II epitope can be a universal helper epitope and does not need to be derived from a tumor-specific antigen to give rise to antitumor efficacy. We demonstrate the clinical potential of this approach using a synthetic peptide vaccine targeting both CD4+ and CD8+ T cells against a clinically-relevant tumor antigen, Trp2. Our findings represent a significant improvement to class I-restricted tumor-derived peptide vaccines and highlight an increased potential for their clinical translation.
We believe that αhCD27 enhances vaccine immunogenicity by coordinating CD4+ and CD8+ T cell responses, thereby increasing the helper function of vaccine-specific CD4+ T cells and the resultant antitumor effector function of tumor-specific CD8+ T cells. Indeed, while neither adjuvant αhCD27 nor a linked class II epitope alone is sufficient to promote antitumor efficacy, their combination has a substantial therapeutic effect. These data indicate that a synergistic relationship exists between CD27 stimulation and CD4+ T cell help. CD27 signaling has been previously shown to drive TH1 polarization in mouse33 and human34 CD4+ T cells by upregulating IL-12Rβ2 and T-bet expression and was also found to enhance their helper activity towards CD8+ T cells.35 Additionally, we surmise that CD4+ T cell help may render CD8+ T cells more sensitive to CD27 signaling, thus amplifying the effect of αhCD27 on the CD8+ T cell response compared to the effect that αhCD27 would have in the absence of CD4+ T cell help. We believe that these characteristics of CD27 biology make αhCD27 uniquely poised as a promising vaccine adjuvant for enhancing the response to peptide vaccines containing linked class I/II epitopes.
It is also worth noting that CD27 is expressed by other cell types, including natural killer (NK) cells, regulatory T cells (Tregs), medullary thymocytes, and a subset of peripheral blood B cells.36 It is possible that potential off target effects of αhCD27 could result in the activation of these cell subsets and engender bystander functionality. The activation of NK or B cells could contribute to inflammatory immunopathology, while the stimulation of CD27 on Tregs could increase immunosuppression. Additionally, while αhCD27 only stimulates T cells with engaged T cell receptors, αhCD27 could result in the activation of CD4+ and CD8+ T cells specific for available self-antigens. Stimulation of T cells specific for self-antigen could engender autoimmune reactivity and pathology. However, the αhCD27 monoclonal antibody employed in our studies is currently being evaluated in clinical studies as a single agent (Varlilumab®) and in combination therapy with checkpoint inhibitors in patients with solid tumors (NCT02335918). These studies have shown that Varlilumab® is safe and well tolerated, with no severe adverse events, systemic immunosuppression, or autoimmune reactivity reported in over 50 patients that have received this experimental therapy.37 Therefore, while off-target effects are always a possibility for immunomodulatory antibodies, the αhCD27 monoclonal antibody Varlilumab® has shown an extremely safe profile, making it a suitable agent to use as a vaccine adjuvant.
There are a variety of clinically-available peptide vaccines comprised of a tumor-derived class I epitope linked to a universal helper epitope or tumor-derived class II epitope,13,38,39 which would make for promising vaccine candidates for combination with adjuvant αhCD27. In addition to peptide vaccines targeting high-frequency tumor mutations, we believe that adjuvant αhCD27 may enhance the clinical benefit of peptide vaccines derived from class I-restricted tumor neo-antigens, especially when linked to immunogenic class II epitopes. The therapeutic promise of such neo-epitope peptide vaccines has recently been demonstrated in the setting of advanced melanoma, where the administration of long peptides derived from melanoma neo-antigens has resulted in effective antitumor T cell responses.40,41 To broaden the scope of this approach, we envision that putatively any class I tumor neo-antigen could be linked to a helper epitope (e.g., P30) and administered alongside adjuvant αhCD27 to enhance its immunogenicity and efficacy, giving rise to a customizable vaccine/adjuvant treatment modality.
αhCD27 was previously shown to promote effective tumor immune responses as a single agent in mice bearing immunogenic tumors.23 Herein, we demonstrate that this antibody can also be employed as a vaccine adjuvant with a preferential effect on linked class I/II peptide vaccines to engender antitumor efficacy against poorly immunogenic intracranial tumor. However, it is worth noting that while αhCD27 dramatically elevated vaccine immunogenicity, that there was variability in these responses despite age- and gender-matched genetically identical recipients. While vaccine variability in humans is often attributed to differing genetic backgrounds, we attribute vaccine variability in mice to the technical aspects of vaccine emulsification and intradermal administration. These results within genetically identical murine models underscore the importance of establishing standard operating procedures to achieve the most consistent results possible in human clinical trials as well. We believe our treatment strategy of combinatorial αhCD27 and peptide vaccination has high translational relevance, as both a monotherapy for potentiating weak endogenous tumor immune responses and as a vaccine adjuvant for potentiating the immunogenicity of class I-restricted tumor-derived antigens. Importantly, the addition of a class II epitope alone was not sufficient to promote efficacy of a class I–restricted peptide vaccine in the absence of adjuvant αhCD27, indicating the requirement for both CD27 stimulation and CD4+ T cell help in promoting the efficacy of class I-restricted peptides.
However, despite the improved survival observed in vaccinated mice treated with adjuvant αhCD27, our therapy was not curative. Several mechanisms could be responsible for limiting the therapeutic efficacy of our treatment platform, such as the differences in tumor infiltration of CD4+ and CD8+ T cells, tumor antigen loss, decreased or lost CD27 expression on T cells, expression of pro-apoptotic molecules, the expression of checkpoint inhibition molecules, and the activity of immune-suppressive regulatory T cells (Tregs). Recent studies have demonstrated that CD27 engagement can lead to Fas expression and T cell apoptosis, while other studies have seen an increased in the expression of the immune checkpoint PD-1.42,43 In support of these observations, our supplemental Figure 4 demonstrates that administration of αhCD27 with vaccination increases the expression of PD-1 on CD8+ T cells. Importantly, a recent study showed that PD-1 blockade can augment the efficacy of αhCD27 administration in the absence of vaccination through increasing T cell cytotoxicity.44,45 Therefore, despite the expansion we observed in antigen-specific T cells after vaccination and αhCD27, the antitumor efficacy of these T cells could potentially be improved by the incorporation of a checkpoint inhibitor. In addition to effector T cell dysfunction from PD-1 expression, an increase in suppressive regulatory T cell (Treg) levels and/or activity could also limit the therapeutic efficacy we observed. CD27 has been shown to be expressed at high levels on Tregs and to increase their suppressive function.46,47 However, recent studies in both the pre-clinical and clinical settings have demonstrated that αhCD27 administration does not potentiate Treg function but instead results in Treg depletion and a lack of suppressive function.37,42 Therefore, we believe an increase in Treg suppression is unlikely to be impairing efficacy in our model. Finally, tumors can still overcome potent immune responses through tumor editing, a scenario in which antigen-loss variants arise as a result of antigen-specific immune pressure. Overcoming tumor-editing may well depend upon increasing tumor antigen presentation; this could be achieved by tumoricidal chemotherapy to promote the shedding of novel tumor antigens and their subsequent presentation to T cells by professional antigen-presenting cells. Importantly, αhCD27 could function synergistically in this context through the expansion of T cells recognizing linked class I and class II tumor epitopes from newly presented tumor antigen.48 Furthermore, our therapeutic platform lends itself to the use of multivalent vaccination containing multiple known tumor-specific or tumor-associated class I epitopes linked to universal class II epitopes in the context of αhCD27 administration. Targeting multiple tumor antigens could potentially impair the generation of antigen loss variants and further increase the efficacy of our vaccine approach.
We believe that our study serves as a proof-of-principle demonstration of the adjuvant potential of exogenous CD27 stimulation. As such, our findings warrant further clinical investigation into αhCD27 as an adjuvant for peptide vaccines consisting of a class I-restricted tumor antigen linked with a class II helper epitope for instilling effective tumor-specific T cell immunity.
Funding Statement
This work was supported by grants from the National Institutes of Health (NIH) National Cancer Institute [F31-CA210535–01 (KAR), R01-CA177476–04 (JHS), and P50-CA190991–02 (JHS)] and the NIH National Institute of Neurological Disorders and Stroke [R01-NS099463–01 (JHS), R01-NS085412–04 (JHS), R01-NS086943–03 (JHS), and U01-NS090284–02) (JHS)]. Additional support was provided by a National Science Foundation Graduate Research Fellowship (KAR).
Acknowledgments
The authors thank Celldex Therapeutics for kindly providing us with the necessary animals and reagents for this study. We also thank David Snyder for his assistance with the hCD27 breeding colony.
Author contributions
JHS, LSP, and KAR jointly conceived and implemented this study; KAR, PKN, and AS performed ex vivo immune monitoring; KAR and CMS performed animal vaccine and tumor studies; KAR, LSP, EAR, LH, TK, KLC and JHS performed data analysis and interpretation; KAR, LSP, KLC and JHS wrote the manuscript. All authors have provided final approval of this manuscript.
Disclosure of Potential Conflicts of Interests
TK and LH are both shareholders and employees of Celldex Therapeutics, Inc.
Supplementary material
Supplementary data for this article can be accessed here.
References
- 1.Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F, Krieg AM, Cerottini JC, Romero P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest. 2005. March; 115(3): 739–746. doi: 10.1172/JCI23373 PubMed PMID: 15696196; PubMed Central PMCID: PMC546459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, Mrugala MM, Jensen R, Baehring JM, Sloan A, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-Oncology. 2015. June; 17(6): 854–861. doi: 10.1093/neuonc/nou348 PubMed PMID: 25586468; PubMed Central PMCID: PMC4483122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slingluff CL Jr., Lee S, Zhao F, Chianese-Bullock KA, Olson WC, Butterfield LH, Whiteside TL, Leming PD, Kirkwood JM. A randomized phase II trial of multiepitope vaccination with melanoma peptides for cytotoxic T cells and helper T cells for patients with metastatic melanoma (E1602). Clin Cancer Res: an Official J Am Assoc Cancer Res. 2013. August 1; 19(15): 4228–4238. doi: 10.1158/1078-0432.CCR-13-0002 PubMed PMID: 23653149; PubMed Central PMCID: PMC3813832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP, Essahsah F, Fathers LM, Offringa R, Drijfhout JW, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009. November 5; 361(19): 1838–1847. doi: 10.1056/NEJMoa0810097 PubMed PMID: 19890126 [DOI] [PubMed] [Google Scholar]
- 5.Slingluff CL., Jr. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer Journal. 2011. Sep-Oct; 17(5): 343–350. doi: 10.1097/PPO.0b013e318233e5b2 PubMed PMID: 21952285; PubMed Central PMCID: PMC3204371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg SA. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005; 175(9): 6169–6176. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004. September; 10(9): 909–915. doi: 10.1038/nm1100 PubMed PMID: 15340416; PubMed Central PMCID: PMCPMC1435696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, Tadmor AD, Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015; 520: 692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zwaveling S, Ferreira Mota SC, Nouta J, Johnson M, Lipford GB, Offringa R, van der Burg SH, Melief CJ. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol. 2002. July 01; 169(1): 350–358. PubMed PMID: 12077264. [DOI] [PubMed] [Google Scholar]
- 10.Welters MJ, Kenter GG, Piersma SJ, Vloon AP, Löwik MJ, Berends-van der Meer DM, Drijfhout JW, Valentijn AR, Wafelman AR, Oostendorp J, et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res: an Official J Am Assoc Cancer Res. 2008. January 01; 14(1): 178–187. doi: 10.1158/1078-0432.CCR-07-1880 PubMed PMID: 18172269. [DOI] [PubMed] [Google Scholar]
- 11.Knutson KL, Schiffman K, Disis ML. Immunization with a HER-2/neu helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer patients. J Clin Invest. 2001. February; 107(4): 477–484. doi: 10.1172/JCI11752 PubMed PMID: 11181647; PubMed Central PMCID: PMC199268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madan RA, Mohebtash M, Arlen PM, Vergati M, Rauckhorst M, Steinberg SM, Tsang KY, Poole DJ, Parnes HL, Wright JJ, et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012. May; 13(5): 501–508. doi: 10.1016/S1470-2045(12)70006-2 PubMed PMID: 22326924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibney GT, Kudchadkar RR, DeConti RC, Thebeau MS, Czupryn MP, Tetteh L, Eysmans C, Richards A, Schell MJ, Fisher KJ, et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res: an Official J Am Assoc Cancer Res. 2015. February 15; 21(4): 712–720. 10.1158/1078–0432.CCR-14–2468. PubMed PMID: 25524312; PubMed Central PMCID: PMCPMC4620684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clinical Oncology: Official Journal Am Soc Clin Oncol. 2015. June 10; 33(17): 1974–1982. doi: 10.1200/JCO.2014.59.4358 PubMed PMID: 25605845; PubMed Central PMCID: PMCPMC4980573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hintzen RQ, Lens SM, Beckmann MP, Goodwin RG, Lynch D, van Lier RA. Characterization of the human CD27 ligand, a novel member of the TNF gene family. J Immunol. 1994. February 15; 152(4): 1762–1773. PubMed PMID: 8120385. [PubMed] [Google Scholar]
- 16.Hintzen RQ, Lens SM, Lammers K, Kuiper H, Beckmann MP, van Lier RA. Engagement of CD27 with its ligand CD70 provides a second signal for T cell activation. J Immunol. 1995. March 15; 154(6): 2612–2623. PubMed PMID: 7876536 [PubMed] [Google Scholar]
- 17.Taraban VY, Rowley TF, Tough DF, Al-Shamkhani A. Requirement for CD70 in CD4+ Th cell-dependent and innate receptor-mediated CD8+ T cell priming. J Immunol. 2006. September 01; 177(5): 2969–2975. PubMed PMID: 16920932. [DOI] [PubMed] [Google Scholar]
- 18.Soares H, Waechter H, Glaichenhaus N, Mougneau E, Yagita H, Mizenina O, Dudziak D, Nussenzweig MC, Steinman RM. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J Exp Med. 2007. May 14; 204(5): 1095–1106. doi: 10.1084/jem.20070176 PubMed PMID: 17438065; PubMed Central PMCID: PMC2118574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med. 2003. November 3; 198(9): 1369–1380. doi: 10.1084/jem.20030916 PubMed PMID: 14581610; PubMed Central PMCID: PMC2194245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vitale LA, He LZ, Thomas LJ, Widger J, Weidlick J, Crocker A, O'Neill T, Storey J, Glennie MJ, Grote DM, et al. Development of a human monoclonal antibody for potential therapy of CD27-expressing lymphoma and leukemia. Clin Cancer Res: an Official J Am Assoc Cancer Res. 2012. July 15; 18(14): 3812–3821. 10.1158/1078–0432.CCR-11–3308. PubMed PMID: 22589397. [DOI] [PubMed] [Google Scholar]
- 21.He LZ, Prostak N, Thomas LJ, Vitale L, Weidlick J, Crocker A, Pilsmaker CD, Round SM, Tutt A, Glennie MJ, et al. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J Immunol. 2013. October 15; 191(8): 4174–4183. doi: 10.4049/jimmunol.1300409 PubMed PMID: 24026078. [DOI] [PubMed] [Google Scholar]
- 22.Sanchez-Perez L, Kottke T, Diaz RM, Ahmed A, Thompson J, Chong H, Melcher A, Holmen S, Daniels G, Vile RG. Potent selection of antigen loss variants of B16 melanoma following inflammatory killing of melanocytes in vivo. Cancer Res. 2005. March 1; 65(5): 2009–2017. doi: 10.1158/0008-5472.CAN-04-3216 PubMed PMID: 15753401. [DOI] [PubMed] [Google Scholar]
- 23.Daniels GA, Sanchez-Perez L, Diaz RM, Kottke T, Thompson J, Lai M, Gough M, Karim M, Bushell A, Chong H, Melcher A, et al. A simple method to cure established tumors by inflammatory killing of normal cells. Nat Biotechnol. 2004. September; 22(9): 1125–1132. doi: 10.1038/nbt1007 PubMed PMID: 15300260. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez-Perez LA, Choi BD, Archer GE, Cui X, Flores C, Johnson LA, Schmittling RJ, Snyder D, Herndon JE 2nd, Bigner DD, et al. Myeloablative temozolomide enhances CD8(+) T-cell responses to vaccine and is required for efficacy against brain tumors in mice. PloS One. 2013; 8(3): e59082. doi: 10.1371/journal.pone.0059082 PubMed PMID: 23527092; PubMed Central PMCID: PMC3601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demotz S, Lanzavecchia A, Eisel U, Niemann H, Widmann C, Corradin G. Delineation of several Dr-restricted tetanus toxin T-cell epitopes. J Immunol. 1989. January 15; 142(2): 394–402. PubMed PMID: WOS:A1989R647800005; English. [PubMed] [Google Scholar]
- 26.Tian S, Huang QS, Fang Y, Wu J. FurinDB: A database of 20-residue furin cleavage site motifs, substrates and their associated drugs. Int J Mol Sci. 2011. February; 12(2): 1060–1065. doi: 10.3390/ijms12021060 PubMed PMID: WOS:000287732000012; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bloom MB, Perry-Lalley D, Robbins PF, Li Y, el-Gamil M, Rosenberg SA, Yang JC. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997; 185(3): 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynolds SR, Celis E, Sette A, Oratz R, Shapiro RL, Johnston D, Fotino M, Bystryn JC. HLA-independent heterogeneity of CD8+ T cell responses to MAGE-3, Melan-A/MART-1, gp100, tyrosinase, MC1R, and TRP-2 in vaccine-treated melanoma patients. J Immunol. 1998. December 15; 161(12): 6970–6976. PubMed PMID: 9862732. [PubMed] [Google Scholar]
- 29.Cho HI, Celis E. Optimized peptide vaccines eliciting extensive CD8 T-cell responses with therapeutic antitumor effects. Cancer Res. 2009. December 1; 69(23): 9012–9019. doi: 10.1158/0008-5472.CAN-09-2019 PubMed PMID: 19903852; PubMed Central PMCID: PMCPMC2789207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mansour M, Pohajdak B, Kast WM, Fuentes-Ortega A, Korets-Smith E, Weir GM, Brown RG, Daftarian P. Therapy of established B16-F10 melanoma tumors by a single vaccination of CTL/T helper peptides in VacciMax. J Transl Med. 2007. April 23; 5: 20. doi: 10.1186/1479-5876-5-20 PubMed PMID: 17451606; PubMed Central PMCID: PMCPMC1867806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Libregts S, van Olffen RW, van der Sluijs KF, van Lier RA, Nolte MA. Function of CD27 in helper T cell differentiation. Immunol Lett. 2011. May; 136(2): 177–186. doi: 10.1016/j.imlet.2011.01.008 PubMed PMID: 21277898. [DOI] [PubMed] [Google Scholar]
- 32.van Oosterwijk MF, Juwana H, Arens R, Tesselaar K, van Oers MH, Eldering E, van Lier RA. CD27-CD70 interactions sensitise naive CD4+ T cells for IL-12-induced Th1 cell development. Int Immunol. 2007. June; 19(6): 713–718. doi: 10.1093/intimm/dxm033 PubMed PMID: 17548342. [DOI] [PubMed] [Google Scholar]
- 33.Xiao Y, Peperzak V, Keller AM, Borst J. CD27 instructs CD4+ T cells to provide help for the memory CD8+ T cell response after protein immunization. J Immunol. 2008. July 15; 181(2): 1071–1082. PubMed PMID: 18606659. [DOI] [PubMed] [Google Scholar]
- 34.van de Ven K, Borst J. Targeting the T-cell co-stimulatory CD27/CD70 pathway in cancer immunotherapy: rationale and potential. Immunotherapy. 2015; 7(6): 655–667. doi: 10.2217/imt.15.32 PubMed PMID: 26098609. [DOI] [PubMed] [Google Scholar]
- 35.Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, Sikic BI, Taylor MH, Northfelt DW, Carson WE 3rd, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, in patients with advanced solid tumors. J Clinical Oncology: Official Journal Am Soc Clin Oncol. 2017. June 20; 35(18): 2028–2036. doi: 10.1200/JCO.2016.70.1508 PubMed PMID: 28463630. [DOI] [PubMed] [Google Scholar]
- 36.Slingluff CL Jr., Yamshchikov G, Neese P, Galavotti H, Eastham S, Engelhard VH, Kittlesen D, Deacon D, Hibbitts S, Grosh WW, Petroni G, et al. Phase I trial of a melanoma vaccine with gp100(280–288) peptide and tetanus helper peptide in adjuvant: immunologic and clinical outcomes. Clin Cancer Res: an Official J Am Assoc Cancer Res. 2001. October; 7(10): 3012–3024. PubMed PMID: 11595689. [PubMed] [Google Scholar]
- 37.Helling F, Zhang S, Shang A, Adluri S, Calves M, Koganty R, Longenecker BM, Yao TJ, Oettgen HF, Livingston PO. GM2-KLH conjugate vaccine: increased immunogenicity in melanoma patients after administration with immunological adjuvant QS-21. Cancer Res. 1995. July 1; 55(13): 2783–2788. PubMed PMID: 7796403. [PubMed] [Google Scholar]
- 38.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017. July 13; 547(7662): 222–226. doi: 10.1038/nature23003 PubMed PMID: 28678784. [DOI] [PubMed] [Google Scholar]
- 39.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017. July 13; 547(7662): 217–221. doi: 10.1038/nature22991 PubMed PMID: 28678778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wasiuk A, Testa J, Weidlick J, Sisson C, Vitale L, Widger J, Crocker A, Thomas LJ, Goldstein J, Marsh HC, et al. CD27-mediated regulatory T cell depletion and effector T cell costimulation both contribute to antitumor efficacy. J Immunol. 2017. December 15; 199(12): 4110–4123. doi: 10.4049/jimmunol.1700606 PubMed PMID: 29109120; PubMed Central PMCID: PMCPMC5713498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee EM, Hurh S, Cho B, Oh KH, Kim SU, Surh CD, Sprent J, Yang J, Kim JY, Ahn C. CD70-CD27 ligation between neural stem cells and CD4+ T cells induces Fas-FasL-mediated T-cell death. Stem Cell Res Ther. 2013. May 21; 4(3): 56. doi: 10.1186/scrt206 PubMed PMID: 23692980; PubMed Central PMCID: PMCPMC3706991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchan SL, Fallatah M, Thirdborough SM, Taraban VY, Rogel A, Thomas LJ, Penfold CA, He LZ, Curran MA, Keler T, Al-Shamkhani A. PD-1 blockade and CD27 stimulation activate distinct transcriptional programs that synergize for CD8(+) T-cell-driven antitumor immunity. Clin Cancer Res: an Official J Am Assoc Cancer Res. 2018. May 15; 24(10): 2383–2394. doi: 10.1158/1078-0432.CCR-17-3057 PubMed PMID: 29514845; PubMed Central PMCID: PMCPMC5959006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahrends T, Babala N, Xiao Y, Yagita H, van Eenennaam H, Borst J. CD27 agonism plus PD-1 blockade recapitulates CD4+ T-cell help in therapeutic anticancer vaccination. Cancer Res. 2016. May 15; 76(10): 2921–2931. doi: 10.1158/0008-5472.CAN-15-3130 PubMed PMID: 27020860. [DOI] [PubMed] [Google Scholar]
- 44.Coquet JM, Ribot JC, Babala N, Middendorp S, van der Horst G, Xiao Y, Neves JF, Fonseca-Pereira D, Jacobs H, Pennington DJ, et al. Epithelial and dendritic cells in the thymic medulla promote CD4+Foxp3+ regulatory T cell development via the CD27-CD70 pathway. J Exp Med. 2013. April 8; 210(4): 715–728. doi: 10.1084/jem.20112061 PubMed PMID: 23547099; PubMed Central PMCID: PMCPMC3620350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Claus C, Riether C, Schurch C, Matter MS, Hilmenyuk T, Ochsenbein AF. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res. 2012. July 15; 72(14): 3664–3676. doi: 10.1158/0008-5472.CAN-11-2791 PubMed PMID: 22628427. [DOI] [PubMed] [Google Scholar]
- 46.Locher C Conforti R, Aymeric L, Ma Y, Yamazaki T, Antoine Tesnière SR, Ghiringhelli F, Apetoh L, Morel Y, et al. Desirable cell death during anticancer chemotherapy. Ann N Y Acad Sci. 2010. October; 1209: 99–108. doi: 10.1111/j.1749-6632.2010.05763.x PubMed PMID: 20958322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.