

ABSTRACT
Sphingosine kinase 1 (SphK1) is the major source of the bioactive lipid and GPCR agonist sphingosine 1-phosphate (S1P). Although alterations in SphK1 expression and activity have been detected in various human malignancies, its potential molecular mechanisms in the development and sunitinib resistance of clear cell renal cell carcinoma (ccRCC) remain obscure. In this study, we aim to evaluate the clinical significance of SphK1 and to explore the therapeutic implications of combination approach for ccRCC patients. We identify upregulation of SphK1 significantly associated with poor prognosis of large cohort of ccRCC patients, which contributing to cell proliferation, colony formation, migration and survival. Suppression of SphK1 activity either by shRNA or pharmacologic inhibitior FTY720 suppresses cell growth in vitro and in vivo. A comprehensive phosphoprotein antibody array reveals that SphK1 overexpression promoted RCC progression by regulating the Akt/mTOR pathway. Moreover, FTY720 administration enhanced tumor growth inhibition effect of sunitinib treatment on RCC cells in vitro and in vivo. Our results unraveled that increased SphK1 kinase activation defines an important mechanism for sunitinib resistance, therefore contributes to tumour development and represents therapeutic targets for ccRCC.
KEYWORDS: Sphk1, ccRCC, Sunitinib resistance, FTY720, array
Introduction
Renal cell carcinoma (RCC) accounting for 3% of adult malignancies is the most lethal urological malignancy.1 Metastatic RCC (mRCC), characterised by high resistance to radiotherapy and chemotherapy, has a poor prognosis.2 Therapeutic agents targeting VEGF or mTOR signaling have been successfully developed and clinically useful.3–5 Unfortunately, the vast majority of treated patients with mRCC eventually develop progressive disease because of acquired resistance or other reasons.6 Clear cell RCC (ccRCC), the commonest histological type of RCCs, has the worst prognosis with an average 5-year survival at 71%.7 Altered cell metabolism greatly influence tumor development and progression of ccRCC.8,9 Thus, targeting metabolic abnormalities in ccRCC may provide novel opportunities for developing more effective treatments.
Accumulating evidence indicates that sphingolipid metabolite, such as sphingosine-1-phosphate (S1P), belongs to a new class of potent bioactive molecules, which have been shown to be involved in a variety of cancers.10 Sphingosine kinases 1 (SphK1) is homologous isoenzyme that catalyze the phosphorylation of sphingosine to generate S1P.11,12 A number of studies have shown that SphK1 exert a variety of functions at different steps of cancer progression including RCC.13–16 Moreover, recent studies showed that SphK1 may be involved in resistance to both chemotherapeutics and targeted agents, suggesting that inhibitors of SphK1 in combination with chemotherapeutic treatments can synergistically act to induce tumor cell death.17,18 Zhang et al. reported that SphK1 expression and levels of S1P were significantly increased in RCC, and the murine anti-S1P mAb, sphingomab, can slow tumor growth in treatment naïve murine RCC xenograft models.16 However, the molecular mechanisms of SphK1 in the development and sunitinib resistance of ccRCC remain undefined. In this study, we aim to evaluate the clinical significance of SphK1 and to explore the therapeutic implications of combination approach for ccRCC patients.
Materials and methods
Patients and tissues
Paraffin samples (paired normal and cancerous tissues) were obtained from 358 patients with ccRCC in Department of Urology, Ren Ji Hospital, School of Medicine, Shanghai Jiaotong University (Shanghai, China). Fifteen fresh samples were frozen in liquid nitrogen and stored at −80°C for RNA and protein extraction. Informed consent was obtained from patients and the study was approved by the Institutional Review Board of Shanghai Jiaotong University.
Cell lines
Human ccRCC cell lines (Caki-1, ACHN, A498, 786-O, and 769-P), and immortalized normal human proximal tubule epithelial cell line HK-2 were obtained from the American Type Culture Collection (ATCC). Cells were maintained in RPMI-1640 (for Caki-1, 786-O, and 769-P) or minimum essential medium (MEM; for ACHN and A498) media (Gibco) supplemented with 10% FBS (Invitrogen) in a humidified incubator containing 5% CO2 at 37°C. HK-2 cells were cultured in keratinocyte-SFM (GIBCO/Invitrogen).
Immunohistochemistry
Immunohistochemistry was performed on 5 mm-paraffin sections with an indirect immunoperoxidase method using antibodies against SphK1 (Abcam). The IHC staining score was calculated by multiplying the frequency score (0 = no cell positivity, 1 = 1–25%, 2 = 26–50% and 3 = 51–100%) and the intensity score (0 = no staining, 1 = weak, 2 = moderate and 3 = strong). SphK1 IHC score greater than 6 was positive. SphK1-negative was less than or equal to 6.
Western blot and qrt-pcr
Western blot and qRT-PCR assay were carried out as previously described.19
Liquid chromatography (LC) and mass spectrometry (MS)
Sphingolipids, including S1P and sphingosine, were assayed using a liquid chromatography-mass spectrometry (LC/MS/MS)-based lipidomics approach.20 Each plasma sample was assayed in duplicate.
Expression constructs, lentivirus production and infection
The SphK1 expression lentivirus was constructed by inserting the SphK1 open reading frame into the pLenti6.3 vector (Invitrogen). For SphK1 depletion, human SphK1-targeting small interfering RNA sequences (GGCTGAAATCTCCTTCACG) were cloned into pENTR vectors to generate pENTR-SphK1-RNAi lentiviruses (Invitrogen). Lentiviral infection was performed as previously described.
Cell proliferation assay
Cell proliferation was measured by the MTT, colony formation, and EdU incorporation assays. Cells were seeded in 96-well plates and incubated with the indicated concentrations of reagents at 37°C. After incubation, 0.1mg of MTT was added to each well and the absorbance was measured at 490nm by spectrophotometry. For clonogenic assay, cells were plated in each well of a six-well plate. When there was visible colony by naked eye, cells were fixed with methanol and stained with 0.1% crystal violet (Sigma, USA). Colonies were then counted. An EdU assay was conducted using the Cell-Light EdU imaging kit (RiboBio, Guangzhou, China) according to the manufacturer’s instruction.
Cell cycle and apoptosis assays
For the cell cycle analysis, the transfected cells were collected and fixed in 70% ethanol overnight at − 20°C and stained with propidium iodide (Kaiji, Nanjing, China) in a phosphate-buffered saline solution containing RNase. For the apoptosis assay, the cells were stained with 50 μg/ml propidium iodide and Annexin V-fluorescein isothiocyanate (Kaiji) following the manufacturer’s instructions. The data were analyzed using the ModFit 3.3 software (BD Bioscience, Sparks, MD, USA).
Cell invasion assay
The extent of cell invasion was assessed using the BD BioCoatTM MatrigelTM Invasion Chamber (BD Biosciences, Bedford, MA), according to the manufacturer’s protocol.
Animal experiments
Nude mice (5 to 6 weeks old, Shanghai Laboratory Animal Center, Shanghai, China) were injected subcutaneously into the right flank with a total of 1 × 107 786-O cells stably transfected with either SphK1-shRNA or negative control. Tumor volume was calculated using the equation (L × W2)/2, and the tumor weights were measured and recorded in grams. The mice were killed on the 30th day after injection. All animal studies were approved by the Institutional Animal Care and Use Committee of the Shanghai Jiao Tong University.
Phosphoprotein profiling by the cancer signaling phosphor-antibody microarray
To further reveal the mechanisms of how SphK1 may influence RCC progression, we performed the Cancer Signaling Phospho Antibody microarray CSP100, which was designed and manufactured by Full Moon Biosystems, Inc. (Sunnyvale, CA) and contains 248 antibodies. Each of the antibodies has two replicates that are printed on coated glass microscope slides, along with multiple positive and negative controls. The antibody array experiment was performed by Wayen Biotechnology (Shanghai, China), according to their established protocol. The fluorescence signal of each antibody was obtained from the fluorescence intensity of this antibody spot. A ratio computation was used to measure the extent of protein phosphorylation. The phosphorylation ratio was calculated as follows: phosphorylation ratio = phospho value/unphospho value.
Statistical analysis
The statistical significance of differences between the means of two groups was evaluated by unpaired Student’s t test. The χ2 test was used to analyze the relationship between SphK1 expression and the clinicopathologic characteristics. Survival curves were plotted by the Kaplan-Meier method and compared by the log-rank test. All statistical tests were two-sided, and p values less than 0.05 were considered to be statistically significant. SPSS software (version 17.0; SPSS, Chicago, IL) was used for all statistical analyses.
Results
Clinical implications of sphk1 pathway in ccrcc
In order to investigate the role of SphK1 in ccRCC, firstly the SphK1 expression was evaluated by IHC analysis in TMAs among 358 ccRCC patients. The results of IHC showed that SphK1 was found to be overexpressed in ccRCC tissues with positive expression rate of 67.88% (243/358). In contrast, the expression of SphK1 was greatly inhibited in adjacent normal tissues with positive expression in 41.6% (149/358) of cases (P < 0.001). Moreover, expression of SphK1 was observed within the cytoplasm of cancer cells with weak staining in the stage I localized ccRCCs, weak to intermediate staining in II and III ccRCCs, and strong staining in stage IV metastatic ccRCCs (Figure 1A). In the case for which primary and metastasis were available, both the primary and the corresponding lung metastasis were positive for SphK1 expression (Supplementary Figure S1A). The qRT-PCR experiments for SphK1 mRNA expression in 15 pairs of matched tumor and normal tissue confirmed elevated expression in tumor samples (Figure 1B). Increased SphK1 expression in ccRCC samples is confirmed by Western blot (Figure 1C).
Figure 1.
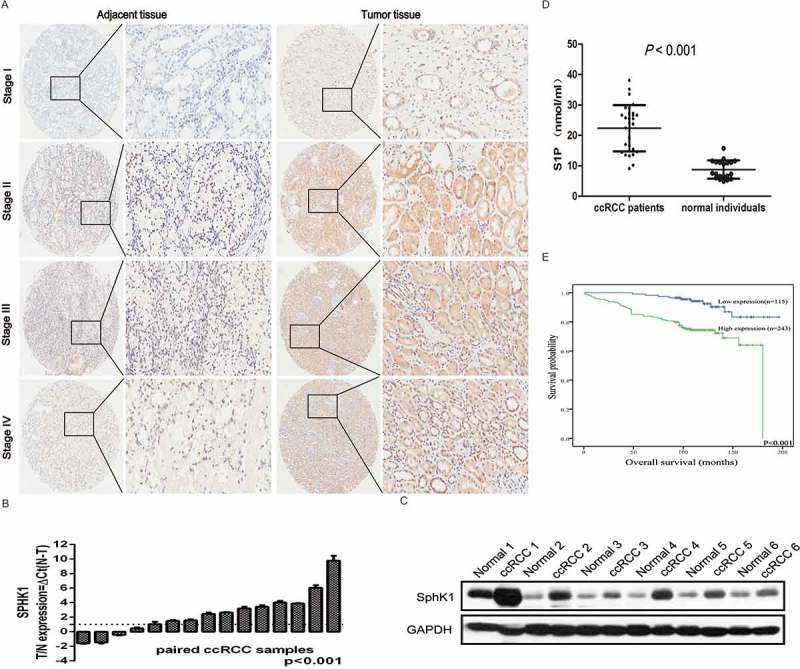
SphK1 expression is upregurated in ccRCC.
Representative immunohistochemistry micrographs of SphK1 expression from the TMAs among 290 ccRCC patients (left 5 × magnification, right 200 × magnification). The expression of SphK1 was observed within the cytoplasm of cancer cells with weak staining in the stage I localized ccRCCs, weak to intermediate staining in II and III ccRCCs, and strong staining in stage IV metastatic ccRCCs. B. Quantitative RT-PCR assay for SphK1 mRNA level in 15 pairs of human clinical ccRCC tumor tissue and adjacent normal tissue. SphK1 mRNA expression levels were normalized to GAPDH mRNA expression. C. Western blot analysis of SphK1 protein in primary ccRCC tumor tissues and adjacent normal tissues. Expression levels were normalized with GAPDH. D. LC-MSMS analysis for S1P levels in plasma from a small group of ccRCC patients (N = 30) and normal individuals (N = 20). Average S1P concentration was 22.36 ± 7.62 nmol/ml for ccRCC patients versus 8.78 ± 3.04 nmol/ml for normal control with a significant difference (P < 0.001). E. Kaplan-Meier survival analysis revealed that patients with high SphK1 expression had significantly lower 5-year overall survival rates, P < 0.001.
Moreover, we measured the S1P levels in plasma from a group of ccRCC patients (N = 30) and normal individuals (N = 20) using LC-MSMS to determine if circulating S1P levels might be a useful biomarker. Average S1P concentration was 22.36 ± 7.62 nmol/ml for ccRCC patients versus 8.78 ± 3.04 nmol/ml for normal control with a significant difference (Figure 1D, P < 0.001). However, there were no significant differences of sphingosine between the two groups (Supplementary Figure S1B), indicating that elevated levels of sphingosine are likely to be localised within the tumours and surrounding tissues.
High expression of sphk1 correlates with disease progression and shortened patient survival in ccrcc
Statistical analyses revealed that SphK1 expression was significantly associated with tumor size, Fuhrman grade, lymph node status and TNM stage of patients with ccRCC (Table 1). Kaplan-Meier survival analysis revealed that patients with high SphK1 expression had significantly lower 5-year overall survival rates (P < 0.001; Figure 1E), indicating that SphK1 could be a valuable prognostic marker for ccRCC patients.
Table 1.
Association between SphK1 and clinicopathologic characteristics
| Characteristics | N | SphK1 expression |
χ2 | P-value | |
|---|---|---|---|---|---|
| (–) |
(+) |
||||
| 115(32.12%) | 243(67.88%) | ||||
| Sex | 0.022 | 0.883 | |||
| Male | 254 | 81(%) | 173(%) | ||
| Female | 104 | 34(%) | 70(%) | ||
| Age, years | 9.789 | 0.002** | |||
| ≤55 | 178 | 71(%) | 107(%) | ||
| >55 | 180 | 44(%) | 136(%) | ||
| Tumor size, cm | 5.393 | 0.020* | |||
| ≤4 cm | 186 | 70(%) | 116(%) | ||
| >4 cm | 172 | 45(%) | 127(%) | ||
| Fuhrman grade | 8.343 | 0.004** | |||
| G1–G2 | 297 | 105(%) | 192(%) | ||
| G3–G4 | 61 | 10(%) | 51(%) | ||
| T classification | 6.895 | 0.006** | |||
| T1–T2 | 344 | 115(%) | 229(%) | ||
| T3–T4 | 14 | 0(%) | 14(%) | ||
| N classification | 1.869 | 0.282 | |||
| N0 | 349 | 114(%) | 235(%) | ||
| N1 | 9 | 1(%) | 8(%) | ||
| Distant metastasis | 2.888 | 0.183 | |||
| No | 352 | 115(%) | 237(%) | ||
| Yes | 6 | 0(%) | 6(%) | ||
| TNM stage | 5.636 | 0.018* | |||
| I–II | 341 | 114 (%) | 227(%) | ||
| III–IV | 17 | 1(%) | 16(%) | ||
*, P < 0.05; **, P < 0.01.
Knockdown of sphk1 inhibits growth and tumorigenesis of ccrcc
We further assessed the expression of SphK1 in a panel of ccRCC-derived cell lines. Both mRNA and protein levels of SphK1 were higher in the ccRCC cell lines, including Caki-1, ACHN, A498, 786-O, and 769-P than in the immortalized human renal tubular epithelial cells line HK-2 which is not tumorigenic (P < 0.01; Supplementary Figure S1C and D). We used 786-O, A-498, and Caki-1 cells for further experiments in our study.
To gain insight into the biological function of SphK1 in ccRCC, we knocked down SphK1 expression in the 786-O and Caki-1 cells. Protein and mRNA levels of SphK1 were markedly decreased after stable transfection of the short hairpin RNA (shRNA)-SphK1 vectors (P < 0.01; Supplementary Figure S1E and F). Cell viability was suppressed significantly by knockdown of SphK1 in 786-O and Caki-1 cells (P < 0.01; Figure 2A and Supplementary Figure S2A). Moreover, 786-O and Caki-1 had a marked lower number of colonies after knockdown SphK1 compared to control shRNA-transfected cells (P < 0.01; Figure 2B). As detected by the EdU incorporation assay, EdU-positive cells were significantly decreased in 786-O and Caki-1 cells transfected with shRNA-SphK1, compared with the control cells (P < 0.01, Figure 2C and Supplementary Figure S2B). More apoptotic cells were found in SphK1 knockdown cells compared to the control shRNA-transfected cells (P < 0.05; Figure 2D and Supplementary Figure S2C). Knockdown of SphK1 led to a significant increase of cells in G0/G1 phase in both 786-O and Caki-1 cells (P < 0.05; Figure 2E and Supplementary Figure S2D). We next investigated the effect of SphK1 on cell migration and invasion using transwell assay. Knockdown of SphK1 markedly slowed the migration and invasion of 786-O and Caki-1 (P < 0.05; Figure 2F and Supplementary Figure S3A).
Figure 2.
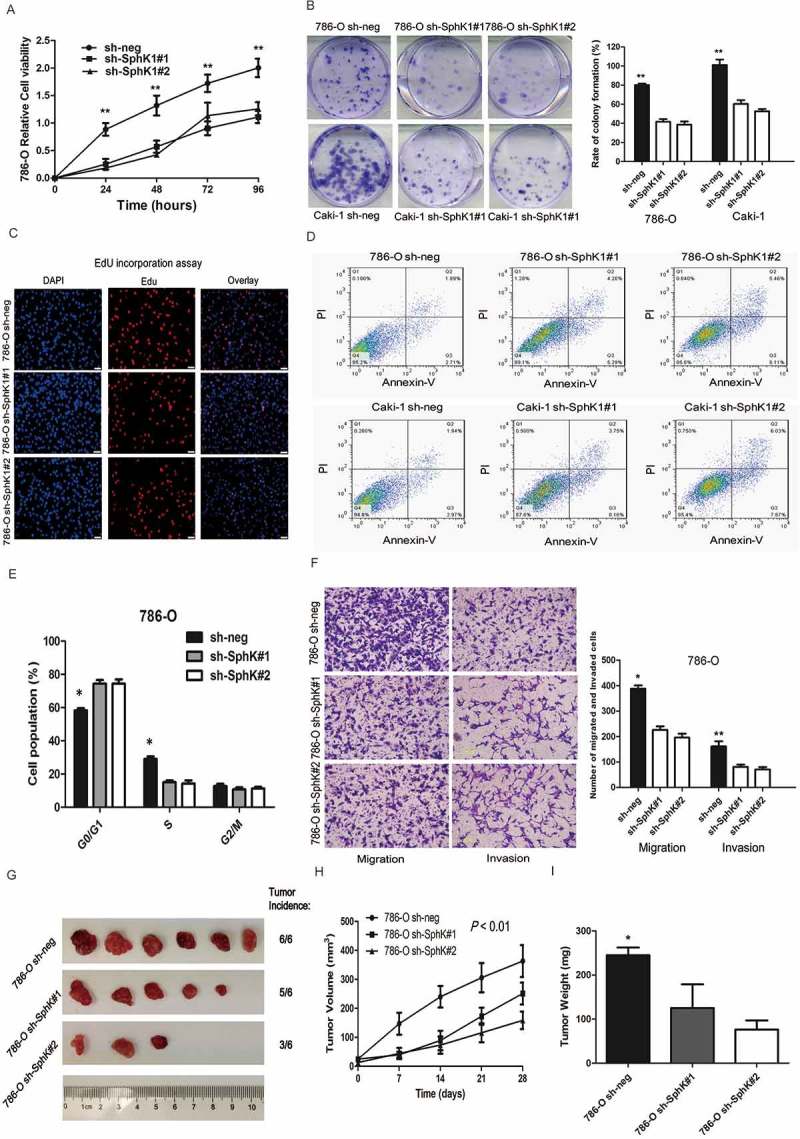
SphK1 knockdown inhibits the growth of ccRCC in vitro and in vivo.
Cell viability was measured by MTT assay at 0, 24, 48, 72 and 96 hours in 786-O cells stably transfected with SphK1 short hairpin RNAs (sh#1 and #2) or control shRNA (sh-neg). The data are presented as the means ± S.D. from three independent experiments. B. Representative images of clonogenic assays of 786-O and Caki-1 cells stably expressing SphK1 shRNAs (sh#1 and #2) or control shRNA. ** P < 0.01. The colonies were counted and captured. C. Representative images of EdU incorporation assay of 786-O and Caki-1 cells stably expressing SphK1 shRNAs (sh#1 and #2) or control shRNA. The Click-it reaction revealed EdU staining (red).The cell nuclei were stained with Hoechst 33342 (blue). D. Representative images of apoptotic 786-O and Caki-1 cells stably expressing SphK1 shRNAs (sh#1 and #2) or control shRNA. E. The bar chart represents the percentage of 786-O cells stably expressing SphK1 shRNAs (sh#1 and #2) or control shRNA in the G0/G1, S, or G2/M phase, as indicated. The data are presented as the means ± SD from three independent experiments. *P < 0.05. F. Representative images of migration and invasion assays in 786-O cells stably expressing SphK1 shRNAs (sh#1 and #2) or control shRNA (left) and quantification of the relative migration and invasion cell number (right). Scale bar = 100 μm. * P < 0.01, ** P < 0.01. G. Representative images of xenografts derived from 786-O cells stably transfected by control or SphK1-shRNA lentiviruses. (n = 6/group). H. Tumor volume of xenografts derived from control or SphK1-knockdowned 786-O cells were evaluated. I. Tumor weights of xenografts derived from control or SphK1-knockdowned 786-O cells were evaluated. Each bar represents Mean ± SD. Two-sided t test. * P < 0.05.
To determine whether the stimulating role of SphK1 in RCC tumor growth in vitro can be extended in vivo, we performed xenograft model assays using 786-O cells stably transfected by control or SphK1-shRNA lentiviruses. We found that knockdown of SphK1 significantly inhibited tumor growth in nude mice (Figure 2G). Tumor volume and tumor weight were significantly decreased in the xenografts derived from SphK1 knockdown cells compared to the control cells (Figure 2H and I). IHC staining of Ki67, a cell proliferation marker, showed fewer proliferative cells in SphK1-shRNA xenograft tumors than the control group (Supplementary Figure S3B). These in vitro and in vivo data collectively indicate that SphK1 acts as a novel tumor-promoting molecule and positively regulates ccRCC growth.
Sphk1 promoted ccrcc cell growth and migration by regulating the akt/mtor pathway
To further confirm that the functional impact of SphK1 in ccRCC, 786-O and Caki-1 cells were transfected with SphK1 expression vector or empty control vector. Overexpression of SphK1 was confirmed by western blotting (Supplementary Figure S4A). Functional studies showed that overexpression of SphK1 significantly increased cell proliferation (P < 0.05; Figure 3A and B, and Supplementary Figure S4B). Moreover, overexpression of SphK1 promoted migration ability of 786-O and Caki-1 cells (Figure 3C and Supplementary Figure S4C).
Figure 3.
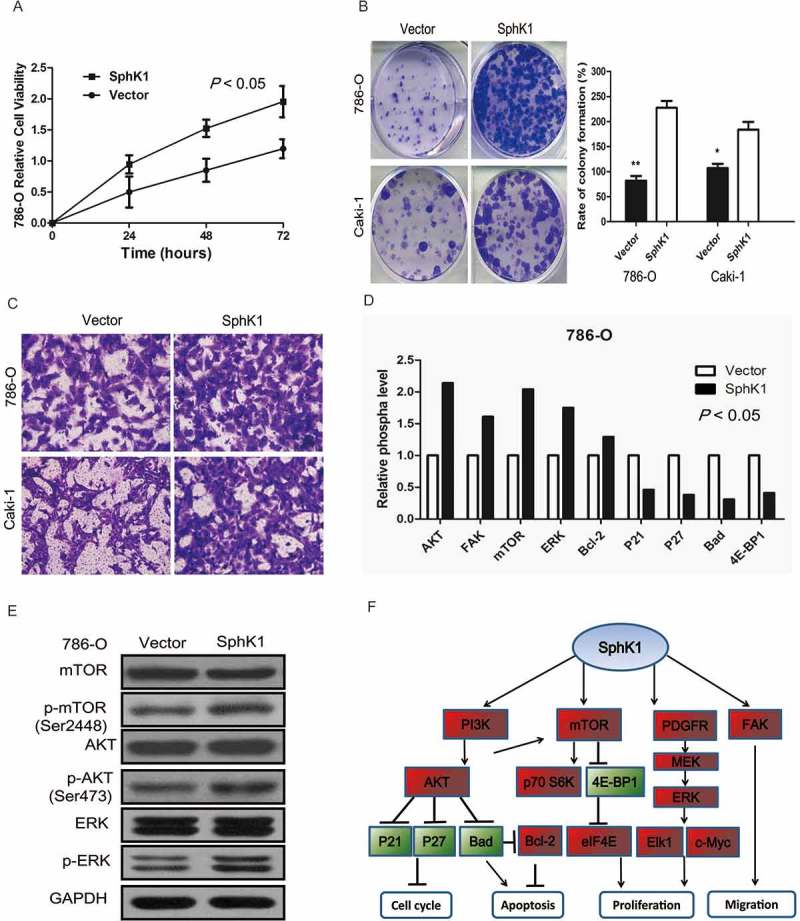
SphK1 promotes ccRCC progression by regulating the Akt/mTOR pathway.
Cell viability was measured by MTT assay at 0, 24, 48 and 72 hours in 786-O cells with overexpression of SphK1 or control vector. The data are presented as the means ± S.D. from three independent experiments. B. Representative images of clonogenic assays of 786-O and Caki-1 cells with overexpression of SphK1 or control vector. * P < 0.05, ** P < 0.01. The colonies were counted and captured. C. Representative images of migration and invasion assays in 786-O cells with overexpression of SphK1 or control vector. Scale bar = 100 μm. D. Downstream effectors of SphK1 were identified by phosphoprotein microarray. The bar chart indicates that phosphorylation levels were downregulated or upregulated by more than 20%. E. Expression of phosphorylation of Akt, mTOR and ERK genes were validated by western blotting. GAPDH was used as a loading control. F. Schematic diagram of the molecular events for SphK1’s function as a oncogene through regulating cell cycle, proliferation, apoptosis and migration effectors.
To comprehensively find mechanistic insight into the role of SphK1 in regulating ccRCC progression, we conducted a phosphoprotein antibody array to identify ectopic phosphorylated proteins induced in 786-O cells with/without SphK1 expression vector. We identified a spectrum of proteins whose phosphorylation levels were altered by more than 20% when SphK1 was stably upregulated, including Akt, mTOR, focal adhesion kinase (FAK), MAPK, c-Jun, and so on. Among these proteins, we confirmed by western blot that targeted upregulation of SphK1 resulted in increased phosphorylation of Akt, mTOR and ERK (Figure 3D-F).
Sunitinib-induced sphk1 activation leads to sunitinib resistance
Our data showed that sunitinib treatment upregulated SphK1 expression that could be reversed by SphK1 knockdown or FTY720 co-treatment in ccRCC cells (Figure 4A). Thus we hypothesize that sunitinib resistance is due, at least in part, to the SphK1 activity after sunitinib administration. Notably, circulating S1P levels were significantly higher at the time of sunitinib resistance in ccRCC patients (N = 20) who received sunitinib as first line targeted therapy (Figure 4B, P < 0.01).
Figure 4.
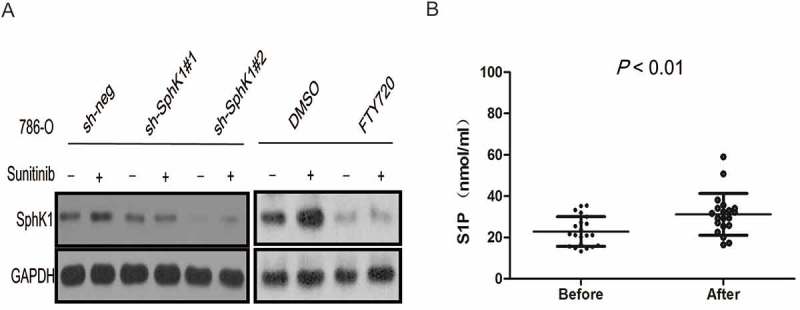
Sunitinib-induced SphK1 activation leads to sunitinib resistance.
Western blot analysis of SphK1 for 786–0 cells stably transfected with SphK1 short hairpin RNAs (sh#1 and #2) or treated with FTY720. GAPDH was used as a loading control. B. LC-MSMS analysis for S1P levels in plasma from a small group of ccRCC patients (N = 20) who received sunitinib as first line targeted therapy. Average S1P concentration was 31.19 ± 10.13 nmol/ml for ccRCC patients at the time of sunitinib resistance versus 22.89 ± 7.17 nmol/ml at the time of sunitinib treatment.
Sphk1 signaling inhibition leads to increased efficacy of sunitinib in vitro and in vivo
Previous studies suggest that FTY720 is an inhibitor of SphK1 and induces its proteasomal degradation.21,22 Concomitantly, FTY720 administration also prevented the increased S1P levels. We previously reported that FTY720 decreases cell proliferation and induces apoptosis in adrenocortical carcinoma cell lines.19 In our study, FTY720 significantly inhibited tumor growth of ccRCC in vitro and in vivo (date not show). Moreover, we wondered whether inhibition of SphK1 activity by FTY720 could enhance sunitinib sensitivity in RCC. Cell proliferation assay showed that 786-O cells with sunitinib exhibited lower growth rate in the presence of FTY720 compared with sunitinib alone (Figure 5A). Furthermore, we sought to determine whether FTY720 could also lead to increased effectiveness of sunitinib treatment in vivo. 786-O cells were injected subcutaneously in nude mice The tumor-bearing mice were treated with sunitinib, FTY720 or sunitinib in combination with FTY720. As expected, sunitinib treatment inhibited tumor formation of 786-O cells in nude mice. More importantly, the efficacy of sunitinib was enhanced significantly when combined with FTY720 (Figure 5B-D). IHC staining analysis of tumors resected from each treatment group was analyzed for proliferation and cell apoptosis. All treatment groups (sunitinib, FTY720, and sunitinib in combination with FTY720) when compared to the placebo control exhibited decreased proliferation as marked by reduction in percent positivity of nuclear Ki67 staining, with the combinatorial group showing the most significant decline (Figure 5E). Cell apoptosis as examined by TUNEL showed significant increases in the combinatorial group when compared to sunitinib or FTY720 (Figure 5F). Taken together, these data show that inhibition of SphK1 could promote the efficacy of sunitinib in RCC in vitro and in vivo.
Figure 5.
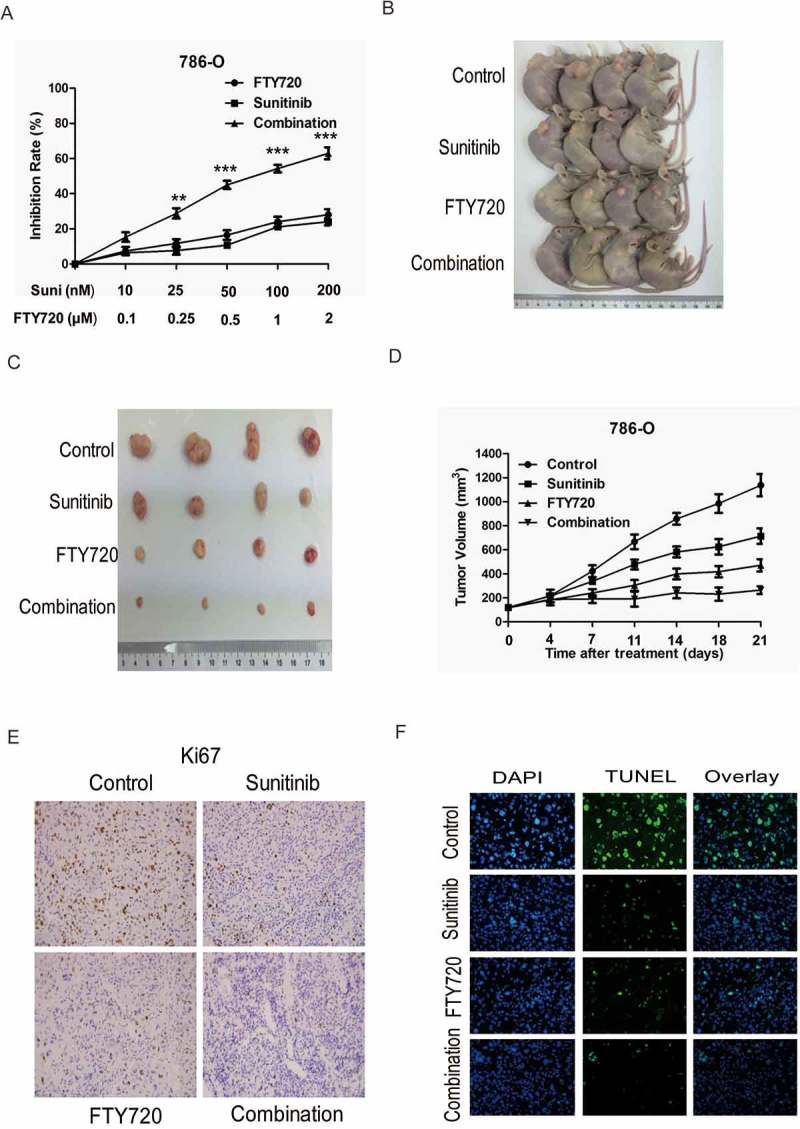
SphK1 signaling inhibition leads to increased efficacy of sunitinib in vitro and in vivo.
Cell proliferation assay showed that 786-O cells with sunitinib exhibited lower growth rate in the presence of FTY720 compared with sunitinib alone. B and C. Representative images of xenografts derived from 786-O cells. (n = 4/group). The tumor-bearing mice were treated with sunitinib, FTY720 or sunitinib in combination with FTY720. D. Tumor volume of xenografts derived from 786-O cells. (n = 4/group). The tumor-bearing mice were treated with sunitinib, FTY720 or sunitinib in combination with FTY720. E. Representative immunohistochemistry micrographs of Ki67 expression from xenograft tumors treated with sunitinib, FTY720 or sunitinib in combination with FTY720. F. Representative immunohistochemistry micrographs of TUNEL expression from xenograft tumors treated with sunitinib, FTY720 or sunitinib in combination with FTY720.
Discussion
In this study, we identify SphK1 overexpression significantly associated with poor prognosis of ccRCC patients, which contributing to cell proliferation, colony formation, migration and survival. Suppression of SphK1 either by shRNA or pharmacologic inhibitior FTY720 suppresses cell growth in vitro and in vivo. A comprehensive phosphoprotein antibody array reveals that SphK1 overexpression promoted RCC progression by regulating the Akt/mTOR pathway. Moreover, FTY720 administration enhanced tumor growth inhibition effect of sunitinib treatment on ccRCC cells in vitro and in vivo. Our results unraveled that increased SphK1 kinase activation defines an important mechanism for sunitinib resistance, therefore contributes to tumour development and represents therapeutic targets for ccRCC.
Recent studies provided clinical and experimental evidence to support the oncogenic role of SphK1 in different types of cancers, including ccRCC.23 Zhang et al. reported that patients with ccRCC show SphK1 overexpression in their tumors and elevated plasma S1P levels.16 Salama et al. analyzed the TCGA RNA seq datasets, and found that SphK1 mRNA expression was higher in patients with ccRCC.24 This study highlighted the clinical significance of SphK1 in our large cohort of ccRCC patients. In our cohort of 358 patients, we found that patients whose tumor exhibited a high level of SphK1 had higher risk of lymph node metastasis, distant metastasis, and poor survival, which is an independent prognostic factor in ccRCC. In addition, ccRCC are likely to produce elevated amounts of S1P resulting from the deregulation of SphK1 expression. Furthermore, both our in vitro and in vivo data confirmed that SphK1 has a critical role in the regulation of tumor cell growth, tumor migration and invasive capacity and survival of cells, suggesting its oncogenic role in ccRCC.
We conducted a phosphokinase array, which encompassed 248 phosphoprotein antibody (AKT, ERK1/2 etc.), to examine the signaling activation upon SphK1 expression vector. Among these signal molecules, we observed a remarkable increase in the level of phosphorylation of Akt (p-Ser473 and p-Ser474), mTOR (p-Ser2448), FAK (p-Tyr397), MAPK (p-Tyr182), c-Jun (p-Ser73 and p-Thr239) following overexpression of SphK1. We finally confirmed by Western blot that targeted upregulation of SphK1 resulted in increased phosphorylation of Akt, mTOR and ERK, which are important for cell proliferation, migration, invasion, and HIF-2α expression when ectopic phosphorylated.25,26
Although the tyrosine kinase inhibitor (TKI) sunitinib has shown single agent activity in patients with metastatic RCC, its clinical use is limited by intrinsic or acquired resistance.27 To date, multiple experimental findings support that SphK1 overactivation has been involved in resistance to anticancer-targeted agents in human solid tumors including ccRCC, prostate cancer, and colorectal cancer.16,28–30 In this study, SphK1 was found overexpressed and overactivated in ccRCC cells with intrinsic or acquired resistance to sunitinib. The circulating S1P levels were significantly upregulated in the plasma from a small group of metastatic ccRCCs at the time of sunitinib resistance, suggesting that S1P may be a predictive biomarker for sunitinib response and resistance in RCC treatment. Furthermore, FTY720 administration significantly enhanced tumor growth inhibition effect of sunitinib treatment on ccRCC cells. The combination of sunitinib and FTY720 caused a potent and long-lasting cooperative antitumor activity, with inhibition of tumor growth, interference with signal transduction, induction of apoptosis, and prolongation of mice survival.
In conclusion, our results indicate that increased SphK1 expression is tumor specific in ccRCC, and upregulation of SphK1 kinase activity contributes to tumor progression and sunitinib resistance in ccRCC. Genetic and molecular targeting of SphK1 activity results in tumor-specific inhibition of cell growth and induction of apoptosis both in vitro and in vivo. Finally, inhibiting SphK1 activation might be a promising strategy to reverse sunitinb resistance or enhance the efficacy of sunitinb for ccRCC patients.
Supplementary Material
Funding Statement
This work was supported by grants from the Advanced technology promotion project of Shanghai Municipal Commission of Health and Family Planning (NO. 27 HYR 2013).
Disclosure statement
The authors report no conflict of interest.
References
- 1.Shuch B, Amin A, Armstrong AJ, Eble JN, Ficarra V, Lopez-Beltran A, Martignoni G, Rini BI, Kutikov A.. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol. 2015;67:85–97. doi: 10.1016/j.eururo.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 2.Kroeger N, Xie W, Lee JL, Bjarnason GA, Knox JJ, Mackenzie MJ, Wood L, Srinivas S, Vaishamayan UN, Rha SY, et al. Metastatic non-clear cell renal cell carcinoma treated with targeted therapy agents: characterization of survival outcome and application of the International mRCC Database Consortium criteria. Cancer. 2013;119:2999–3006. doi: 10.1002/cncr.28151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 4.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 5.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/s0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 6.Buchler T, Bortlicek Z, Poprach A, Pavlik T, Veskrnova V, Honzirkova M, Zemanova M, Fiala O, Kubackova K, Slaby O, et al. Outcomes for Patients with Metastatic Renal Cell Carcinoma Achieving a Complete Response on Targeted Therapy: A Registry-based Analysis. Eur Urol. 2016;70:469–475. doi: 10.1016/j.eururo.2015.12.031. [DOI] [PubMed] [Google Scholar]
- 7.Keegan KA, Schupp CW, Chamie K, Hellenthal NJ, Evans CP, Koppie TM. Histopathology of surgically treated renal cell carcinoma: survival differences by subtype and stage. J Urol. 2012;188:391–397. doi: 10.1016/j.juro.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, Tan WW, Tun HW, Copland JA. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res. 2013;19:2368–2380. doi: 10.1158/1078-0432.ccr-12-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anastasiou D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br J Cancer.2016. doi: 10.1038/bjc.2016.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitatani K, Taniguchi M, Okazaki T. Role of Sphingolipids and Metabolizing Enzymes in Hematological Malignancies. Mol Cells. 2015;38:482–495. doi: 10.14348/molcells.2015.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, Yu CP, Xia JT, Zhang L, Weng GX, Zheng HQ, Kong QL, Hu LJ, Zeng MS, Zeng YX, et al. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2009;15:1393–1399. doi: 10.1158/1078-0432.ccr-08-1158. [DOI] [PubMed] [Google Scholar]
- 12.Long JS, Edwards J, Watson C, Tovey S, Mair KM, Schiff R, Natarajan V, Pyne NJ, Pyne S. Sphingosine kinase 1 induces tolerance to human epidermal growth factor receptor 2 and prevents formation of a migratory phenotype in response to sphingosine 1-phosphate in estrogen receptor-positive breast cancer cells. Mol Cell Biol. 2010;30:3827–3841. doi: 10.1128/mcb.01133-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pyne S, Edwards J, Ohotski J, Pyne NJ. Sphingosine 1-phosphate receptors and sphingosine kinase 1: novel biomarkers for clinical prognosis in breast, prostate, and hematological cancers. Front Oncol. 2012;2:168. doi: 10.3389/fonc.2012.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang YL, Ji C, Cheng L, He L, Lu CC, Wang R, Bi ZG. Sphingosine kinase-1 inhibition sensitizes curcumin-induced growth inhibition and apoptosis in ovarian cancer cells. Cancer Sci. 2012;103:1538–1545. doi: 10.1111/j.1349-7006.2012.02335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitson SM, Powell JA, Bonder CS. Regulation of sphingosine kinase in hematological malignancies and other cancers. Anticancer Agents Med Chem. 2011;11:799–809. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Wang X, Bullock AJ, Callea M, Shah H, Song J, Moreno K, Visentin B, Deutschman D, Alsop DC, et al. Anti-S1P Antibody as a Novel Therapeutic Strategy for VEGFR TKI-Resistant Renal Cancer. Clin Cancer Res. 2015;21:1925–1934. doi: 10.1158/1078-0432.ccr-14-2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V, Mazerolles C, Rischmann P, Teissie J, Malavaud B, et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005;65:11667–11675. doi: 10.1158/0008-5472.can-05-2702. [DOI] [PubMed] [Google Scholar]
- 18.Song L, Xiong H, Li J, Liao W, Wang L, Wu J, Li M. Sphingosine kinase-1 enhances resistance to apoptosis through activation of PI3K/Akt/NF-kappaB pathway in human non-small cell lung cancer. Clin Cancer Res. 2011;17:1839–1849. doi: 10.1158/1078-0432.ccr-10-0720. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Dong B, Huang J, Kong W, Xue W, Zhu Y, Zhang J, Huang Y. Sphingosine kinase 1 is overexpressed and promotes adrenocortical carcinoma progression. Oncotarget. 2016;7:3233–3244. doi: 10.18632/oncotarget.6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Lim KG, Tonelli F, Li Z, Lu X, Bittman R, Pyne S, Pyne NJ. FTY720 analogues as sphingosine kinase 1 inhibitors: enzyme inhibition kinetics, allosterism, proteasomal degradation, and actin rearrangement in MCF-7 breast cancer cells. J Biol Chem. 2011;286:18633–18640. doi: 10.1074/jbc.M111.220756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tonelli F, Lim KG, Loveridge C, Long J, Pitson SM, Tigyi G, Bittman R, Pyne S, Pyne NJ. FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell Signal. 2010;22:1536–1542. doi: 10.1016/j.cellsig.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pyne NJ, Sphingosine PS. 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 24.Salama MF, Carroll B, Adada M, Pulkoski-Gross M, Hannun YA, Obeid LM. A novel role of sphingosine kinase-1 in the invasion and angiogenesis of VHL mutant clear cell renal cell carcinoma. FASEB J. 2015;29:2803–2813. doi: 10.1096/fj.15-270413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, Zipkin RE, Dent P, Kordula T, Milstien S, et al. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009;69:6915–6923. doi: 10.1158/0008-5472.can-09-0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Sphingosine BS. 1-phosphate receptor signaling. Annu Rev Biochem. 2009;78:743–768. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 27.Stone L. Kidney cancer: sunitinib resistance is futile. Nat Rev Urol. 2016;13:566. doi: 10.1038/nrurol.2016.180. [DOI] [PubMed] [Google Scholar]
- 28.Ader I, Gstalder C, Bouquerel P, Golzio M, Andrieu G, Zalvidea S, Richard S, Sabbadini RA, Malavaud B, Neutralizing S1P CO. inhibits intratumoral hypoxia, induces vascular remodelling and sensitizes to chemotherapy in prostate cancer. Oncotarget. 2015;6:13803–13821. doi: 10.18632/oncotarget.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aoyama Y, Sobue S, Mizutani N, Inoue C, Kawamoto Y, Nishizawa Y, Ichihara M, Kyogashima M, Suzuki M, Nozawa Y, et al. Modulation of the sphingolipid rheostat is involved in paclitaxel resistance of the human prostate cancer cell line PC3-PR. Biochem Biophys Res Commun. 2017;486:551–557. doi: 10.1016/j.bbrc.2017.03.084. [DOI] [PubMed] [Google Scholar]
- 30.Rosa R, Marciano R, Malapelle U, Formisano L, Nappi L, D’Amato C, D’Amato V, Damiano V, Marfe G, Del Vecchio S, et al. Sphingosine kinase 1 overexpression contributes to cetuximab resistance in human colorectal cancer models. Clin Cancer Res. 2013;19:138–147. doi: 10.1158/1078-0432.ccr-12-1050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.