

ABSTRACT
Objective:Ovarian cancer (OC) is often diagnosed at an advanced stage with two thirds of patients experiencing recurrent disease with a poor prognosis. Adoptive cell therapy (ACT) with tumor-infiltrating lymphocytes (TIL) has shown curative potential in malignant melanoma, but has only been investigated scarcely in other cancers. In this pilot study, we tested TIL based ACT in patients with metastatic OC.
Methods:Six patients with progressive platinum-resistant metastatic OC were treated with an infusion of TIL preceded by standard lymphodepleting chemotherapy and followed by decrescendo intravenous interleukin-2 (IL-2). Primarily, the feasibility and tolerability of the treatment was assessed. Secondarily, disease control rate was described and immune responses against tumor cells were monitored.
Results:Treatment was well tolerated with manageable toxicities. Four patients had stable disease for three months and two patients for five months with five patients having a decrease in target lesions. Progression was primarily due to new lesions while target lesions in general remained stable or in regression. Antitumor reactivity was observed in TIL infusion products from five patients but no antitumor reactivity was detectable in peripheral blood lymphocytes collected after treatment. High numbers of infused TIL expressed exhaustion markers including LAG3 and PD-1, and immunostaining of tumor tissue demonstrated substantial MHCII and PD-L1 expression.
Conclusions:ACT with TIL in combination with decrescendo IL-2 is feasible in patients with metastatic OC. Early indications of clinical activity were found. However, TIL ACT efficacy was incomplete with possible involvement of the inhibitory immune checkpoint pathways LAG3/MHCII and PD1/PD-L1.
KEYWORDS: Adoptive cell therapy, T-cell therapy, TIL therapy, tumor-infiltrating lymphocytes, immune therapy, immunotherapy, metastatic ovarian cancer
Introduction
In 2012, approximately 250,000 women were diagnosed with ovarian cancer (OC) with 150,000 estimated deaths worldwide1. OC is often diagnosed at an advanced stage2 with platinum-based chemotherapy being the standard of care 3, but more than two thirds of these patients will develop recurrent disease4 with a poor prognosis3,4. Therefore, new and improved treatment options are highly wanted.
The presence of intratumoral T cells in patients with OC has been associated with improved overall survival (OS) and progression-free survival (PFS)5. Thus, strengthening natural T-cell responses might represent a successful therapeutic option in OC. Adoptive cell therapy (ACT) with tumor infiltrating lymphocytes (TIL) is based on an infusion of autologous T cells obtained from the tumor microenvironment of the individual patient. These T cells are expanded and activated in vitro before reinfusion6. This treatment has shown remarkable results in patients with malignant melanoma with objective response rates ranging from 40–70% and up to 20% durable complete responses7-11. Promising results are reported in epithelial cancers12,13. In OC, TIL based ACT has previously been tested with limited success14,15, but the protocols used for expansion of TIL have been extensively changed and optimized over the last decades. At our center we have recently successfully expanded TIL with antitumor reactivity from OC16.
Here we report the clinical and immunological results of a pilot study investigating TIL based ACT in metastatic OC.
Results
Patient characteristics
Eleven patients were enrolled in the study. Five patients did not receive treatment due to: benign surgical biopsy for TIL production (n = 1); unsuccessful TIL expansion (n = 1); clinical deterioration (n = 3). Table 1 summarizes characteristics of treated patient. Untreated patients are described in the supplementary data. Six patients were treated between October 2015 and November 2016. They were 50–65 years old (median 59 years) with Cancer antigen (CA-125) levels ranging from 10–4320 kU/L (median 214) and FIGO stage IIIc (n = 4) and IV (n = 2) disease, and had received two to six prior treatment regimens (median three).
Table 1.
Patient characteristics and objective response.
| Patient ID | At debut | At screening | Treatment | Objective response | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary diagnose | FIGO |
CA-125 (kU/L) |
Age | PS | Comorbidity | Disease sites |
Prior treatmentsa No Regimens |
Time from diagnosis to ACT (months) | Best overall response RECIST v 1.1 | PFS (months) | OS (months) | ||
| GY1508.01 | 2014 | IV | 4320 | 58 | 0 | Factor V Leiden mutation, DVT x 3 | Lower abdomen, carcinomatosis (abdomen) | 2 | Carbo/tax (N), carbo/tax/beva (A), beva (M) | 12 | SD | 3 | 11 |
| GY1508.04 | 2007 | IIIc | 66 | 50 | 0 | None | LN (right hilus, retroperitoneum, at iliac vessels) | 6 | Carbo/tax (A), carbo/tax/vori (P), caelyx (P), topo (P), beva (P), veli (P) | 97 | SD | 3 | 5 |
| GY1508.06 | 2015 | IV | 107 | 65 | 0 | Hypertension | LN (retroperitoneum, mesenteria), carcinomatosis (at liver, at spleen, paracolic spaces) | 3 | Carbo/tax (N), carbo/caelyx/beva (A), carbo/caelyx (P) |
12 | SD | 3 | 4 |
| GY1508.08 | 2010 | IIIc | 10 | 59 | 1 | Ileus x 1 | Liver, mesenteria, carcinomatosis (above liver) | 6 | Carbo/tax (A), carbo/tax (P), carbo/caelyx (P), topo (P), gemci (P), tax (P) | 71 | SD | 5 | 13+ |
| GY1508.10 | 2013 | IIIc | 320 | 65 | 1 | Hypertension, myxedema | Pleura, liver, spleen, LN (groin) | 2 | Carbo/tax/beva (A), carbo/caelyx (P) | 39 | SD | 3 | 11+ |
| GY1508.11 | 2014 | IIIc | 400 | 55 | 1 | Hypertension, psoriasis arthritis, splenectomy | LN (paraaortal, axil), carcinomatosis (upper abdomen) | 2 | Carbo/tax (A), tax/caelyx/beva (P), beva (M) | 33 | SD | 5 | 8 |
|
Median (Range) |
2014 (2007–2015) |
NA (IIIc-IV) |
214 (10–4320) |
59 (50–65) |
1 (0–1) |
NA | NA |
3 (2–6) |
NA |
36 (12–97) |
NA |
3 (3–5) |
10 (4–13+) |
a Neoadjuvant therapy was assessed as a separate treatment regimen and maintenance therapy with Bevacizumab following Carboplatin/paclitaxel was assessed as a continuation of the same treatment regimen.
Abbreviations: LN = Lymph node, DVT = Deep venous thrombosis, NA = Not applicable, No = number, Carbo = Carboplatin, Tax = Paclitaxel, Beva = Bevacizumab, Vori = Vorinostat, Topo = Topotecan, Veli = Veliparib, Gemci = Gemcitabine, N = Neoadjuvant, A = Adjuvant, P = Palliative, M = maintenance,/ = given in combination, + = ongoing.
TIL characteristics
Young TIL (YT) expansion was successful in nine of ten patients. Table 2 summarizes treated patients’ TIL characteristics. Data from untreated patients are described in the supplementary data. YT culture generation took a median of 25 days (range 10–33). The rapid expansion protocol (REP) led to a median expansion fold of 2810 (range 890–4560) with median percentage of CD3+ cells increasing from 89.9% (range 56.7–97) in YT to 98.3% (range 95.5–99.4) in the infusion product (INF PROD). A small increase in CD4+ cells (from median 55.5% to 58.3%) and CD8+ cells (from median 35.5% to 37.4%) was seen, but with considerable interpatient variation (largest median CD4+ cell variation 17.8% and largest median CD8+ cell variation 24.4%). The total number of cells in the final INF PROD was median 56.2x10^9 cells (range 17.8–91.2x10^9 cells).
Table 2.
TIL characteristics.
| Patient ID |
Tumor |
YT |
INF PROD |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Site | Cryo before REP | Days in culture | % of singlets | % of CD3+ | REP expansion fold | % of singlets | % of CD3+ | Total cells (x10^9) | |||||||
| CD3+ | CD4+ | CD8+ | CD4-CD8- |
CD4+ CD8+ |
CD3+ | CD4+ | CD8+ | CD4-CD8- |
CD4+ CD8+ |
||||||
| GY1508.01 | IA | No | 33 | 97 | 41.2 | 51.1 | 7.5 | 0.1 | 3000 | 99.4 | 23.4 | 75.5 | 0.5 | 0.6 | 60 |
| GY1508.04 | IA | Yes | 10 | 56.7 | 23.9 | 51.8 | 23.9 | 0.8 | 2420 | 97.9 | 31.2 | 55.5 | 11 | 2.8 | 48.4 |
| GY1508.06 | IA | Yes | 21 | 94 | 48 | 46.6 | 4.2 | 0.4 | 4560 | 98.6 | 38.7 | 60.3 | 0.5 | 0.4 | 91.2 |
| GY1508.08 | IA | No | 33 | 92.8 | 95.6 | 2.5 | 1.8 | 0.2 | 2820 | 96.9 | 82.1 | 17.1 | 0.4 | 0.5 | 56.4 |
| GY1508.10 | LN | Yes | 19 | 87 | 81.6 | 15.6 | 2.6 | 0.2 | 2800 | 95.5 | 80.3 | 16 | 2.6 | 0.4 | 56 |
| GY1508.11 | LN | No | 28 | 75.7 | 63 | 24.4 | 12.6 | 0.2 | 890 | 98.8 | 77.9 | 19.3 | 2.4 | 0.5 | 17.8a |
| Median | - | NA | 25 | 89.9 | 55.5 | 35.5 | 5.9 | 0.2 | 2810 | 98.3 | 58.3 | 37.4 | 1.5 | 0.5 | 56.2 |
| Range | - | NA | 10–33 | 56.7–97 | 23.9–95.6 | 2.5–51.8 | 1.8–23.9 | 0.2–0.8 | 890–4560 | 95.5–99.4 | 23.4–82.1 | 16–75.5 | 0.4–11 | 0.4–2.8 | 17.8–91.2 |
a Technical problem with the wave bioreactor.
Abbreviations: Cryo = Cryopreserved, IA = Intraabdominal, LN = Lymph node, NA = Not applicable.
Treatment and toxicity
Timeline
It lasted a median of 64 days (range 54–76) from inclusion until infusion of TIL; median 21 days (range 7–29) from inclusion until surgery and a median TIL expansion period of 39 days (range 24–47).
Surgery
Eleven patients underwent surgery with the purpose of removing tumor tissue for TIL expansion. Of the 6 patients treated, four had an intraabdominal lesion removed either by explorative laparotomy (n = 3) or laparoscopic procedure (n = 1), while two had an accessible lymph node (LN) removed from the left axillary region (in local anesthesia) and right groin, respectively. One patient was initially discharged, but then hospitalized for 3 days due to a fever. No signs infection was found and no treatment was administered. Another patient had a wound infection after surgery that was treated with 10 days of oral antibiotics. Treatment characteristics, treatment related grade 3–4 adverse events (AEs) and serious adverse events (SAEs) are listed in Table 3.
Table 3.
Treatment characteristics and adverse events.
| GY1508.01 | GY1508.04 | GY1508.06 | GY1508.08 | GY1508.10 | GY1508.11 | All patients | |
|---|---|---|---|---|---|---|---|
| Treatment characteristics | Median (range) | ||||||
| Days in hospital | 28 | 20 | 20 | 21 | 18 | 20 | 20 (18–28) |
| Units RBC transfusiona | 4 | 8 | 6 | 3 | 4 | 3 | 4 (3–8) |
| Units PLT transfusionb | 7 | 10 | 4 | 7 | 2 | 4 | 6 (2–10) |
| Days with neutrophils < 0,5x10^9/L | 10 | 10 | 8 | 8 | 8 | 9 | 9 (8–10) |
| Dose IL-2 administered, MIU | 78 | 126 | 109.8 | 113 | 121.5 | 135 | 117 (78–135) |
| % IL-2 administered | 61 | 100 | 100 | 94 | 90 | 100 | 97 (61–100) |
| Treatment related AEs (grade 3–4)c | Total | ||||||
| Surgery | |||||||
| Fever without infection | 3 | 1 | |||||
| Leukapheresis | |||||||
| Fewer with infection | 3* | 1 | |||||
| TIL therapyd | |||||||
| Performance status | 3 | 3 | 3 | 3 | 3 | 3 | 6 |
| Dyspnea | 4* | 1 | |||||
| Fever without infection | 3 | 1 | |||||
| Febrile neutropenia | 3 | 3 | 3 | 3 | 3 | 3 | 6 |
| Confusion | 3 | 1 | |||||
| Fatigue | 3 | 1 | |||||
| Maculopapular rash | 3 | 1 | |||||
| Hallucinations | 3 | 1 | |||||
| Hyponatremia | 3 | 3 | 3 | 3 | 3 | 3 | 6 |
| Hypophosphatemia | 3 | 3 | 3 | 3 | 3 | 5 | |
| Hypokalemia | 3 | 3 | 3 | 3 | 3 | 5 | |
| Anemia | 3 | 3 | 3 | 3 | 3 | 3 | 6 |
| Lymphocytopenia | 4 | 4 | 4 | 4 | 4 | 4 | 6 |
| Thrombocytopenia | 4 | 4 | 4 | 4 | 4 | 4 | 6 |
| All | 10 | 8 | 11 | 8 | 9 | 8 | 54 |
| SAEs | Total | ||||||
| Deep venous thrombosis | Xe | 1 | |||||
| Dyspnea | X | 1 | |||||
| Fever with infection | X | 1 | |||||
| All | 2 | 1 | 3 |
a Transfusion when hemoglobin < 6.0 mmol/L, b Transfusion when platelets < 20x10^9/L or < 30x10^9/L if the patient was in risk of bleeding, c According to the CTCAE v. 4.0, d Includes lymphodepleting chemotherapy, TIL infusion and IL-2 stimulation, e Found on baseline scan after leukapheresis and before TIL therapy, * SAE.
Leukapheresis
For the treated patients, duration of hospitalization for leukapheresis ranged from two to nine days with the procedure performed for up to two consecutive days. Two patients received increased granulocyte colony stimulating factor (G-CSF) dosages to obtain a sufficient number of stem cells, which was possible in five of six patients. Common adverse reactions observed in relation to G-CSF were headache (n = 3), myalgia (n = 2) and arthralgia (n = 2). One patient had a prolongation after hospitalization following the procedure due to a fever and urinary infection that was treated with antibiotics for a total of 7 days. Another patient had a deep venous thrombosis found on the baseline scan before TIL therapy. She was known with a factor V Leiden mutation, had no symptoms and was treated with an increase in her usual anticoagulation treatment for 4 weeks.
T-cell therapy
The median duration of hospitalization was 20 days (range 18–28). Lymphodepleting chemotherapy was administered without the occurrence of unexpected toxicities. All patients experienced expected grade 3–4 electrolyte derangements and expected hematologic toxicities requiring transfusions. All patients recovered without the need of stem cell support. Expected leukopenia was observed in all patients within a few days prior to TIL administration. Four of six patients were treated with antibiotics from before or during lymphodepleting chemotherapy. Two patients experienced known and expected low-grade reversible adverse events in relation to TIL infusion. These were chills (n = 2) and a minor rise in temperature to 38.7 °C (n = 1).
IL-2 infusion was generally well tolerated. Three of six patients received 100% of the planned IL-2 dose (median 97%, range 61–100). All patients experienced a rise in temperature during IL-2 and were treated with antibiotics according to local guidelines for neutropenic fever. One patient experienced a rise in temperature close to 41 °C and later dyspnea and confusion, which lead to early termination of IL-2 treatment after 61% of the total IL-2 dosage. The patient recovered quickly after IL-2 withdrawal. Other reasons for early termination were mental strain and hallucinations after the administration of 94% and 90% of the total IL-2 dose, respectively.
The hospitalization of one patient for 28 days was due to a prolonged recovery period with respiratory distress and rising body temperature without an infectious focus, alleviated by pleurocentesis and treated with antibiotics, respectively. The remaining five patients had uncomplicated recovery periods. Patients had expected need of correction of electrolyte derangements (n = 6), diuretics due to excessive fluid (n = 5) and increased doses of fungal treatment due to oral candidiasis (n = 2).
Clinical efficacy
All patients had stable disease (SD) six weeks after TIL therapy (Table 1) with five of six patients having a decrease in target lesions (largest decrease 23%) (Figure 1A). Four patients had SD for three months and two patients maintained SD for five months. Decreasing (n = 3) or stable (n = 3) CA-125 values were seen at first evaluation time point (Figure 1B). Five of six patients developed progressive disease (PD) due to the occurrence of new lesions only, while target lesions were generally stable.
Figure 1.
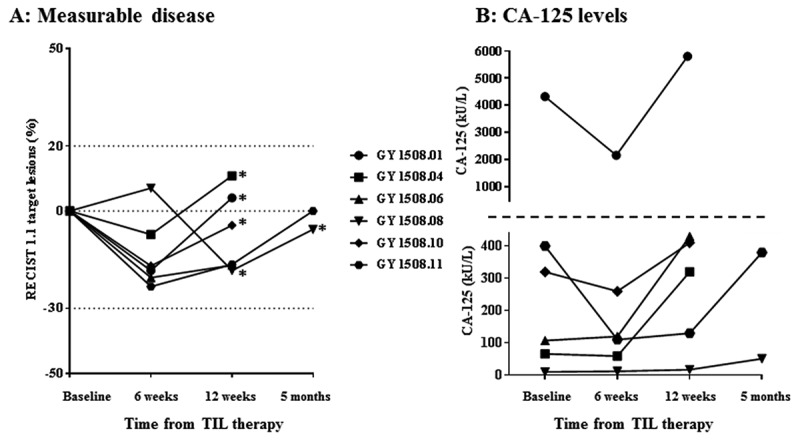
Characteristics of disease control rates over time in patients receiving TIL therapy. A, spider-plot of changes in target lesions. Horizontal dotted lines at 20 and −30 indicate the threshold for defining PD and PR from baseline according to RECIST 1.1., respectively. * = new lesion(s). B, changes in CA-125 levels in kU/L. The horizontal dotted line marks a change in Y axis values and intervals.
There was a difference between the RECIST and the PERCIST assessment in one or more of the evaluation scans in five of six patients. Thus, at the six weeks scan, three patients had a partial metabolic response (PMR) and one had progressive metabolic disease (PMD) in accordance with the PERCIST criteria, with all patients having SD in accordance with the RECIST criteria. At the five months evaluation, one patient had stable metabolic disease (SMD), but PD in accordance with RECIST due to new lesions that was not seen by PET. Vice versa, in the other patient evaluated after five months, new lesions were seen only by PET.
Antitumor reactivity of expanded t cells
Autologous tumor cell lines (TCL) were established from the transport media in five of ten included patients and four of six treated patients. In the treated patient YT, specific antitumor reactivity in CD4+ TIL was seen in two of six patients against fresh tumor digest (FTD) (4.4% and 9.3% reactive cells) (Figure 2). Specific antitumor reactivity in CD8+ TIL was seen in two of six patients, in one patient against TCL alone (1.9% reactive cells) and in another patient against TCL alone (0.5% reactive cells) with increased reactivity after pretreatment of tumor cells with interferon-gamma (IFN-γ) (1.1% reactive cells).
Figure 2.
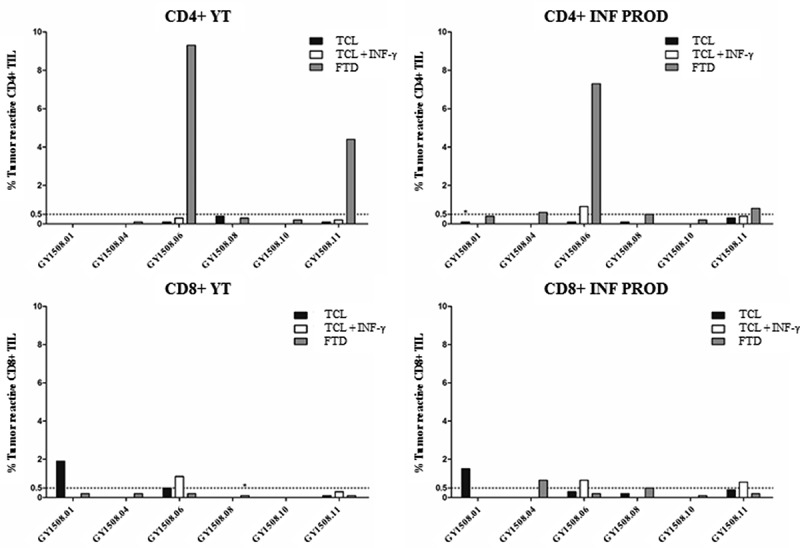
Antitumor reactivity of in vitro expanded TIL. The figure shows the frequency of T cells expressing at least one of the T-cell functions: TNF, IFN-γ or CD107a, upon stimulation with autologous TCL (with or without low-dose IFN-γ (100 IU/ml) stimulation) or FTD with FACS analysis. A specific anti-tumor response was defined at minimum 0.5% responding cells (horizontal line), with a minimum of 50 positive events (*). The frequency of tumor-reactive cells in stimulated samples was subtracted from un-stimulated samples.
In the INF PROD, specific antitumor reactive CD4+ TIL were seen in four of six patients, with all responding against FTD (0.5–7.3% reactive cells) and one also against TCL + IFN-γ (0.9% reactive cells) (Figure 2). Specific antitumor reactivity in CD8+ TIL were seen in five of six patients, with one responding against TCL (1.5% reactive cells), two responding against TCL + IFN-γ (0.8% and 0.9% reactive cells, respectively) and two responding against FTD (0.5% and 0.9% reactive cells).
To further evaluate the activation stage of the infused T-cells, INF PROD were analyzed using a panel of T-cell differentiation and exhaustion markers (Table 4). Looking into the T cell differentiation stages, defined by Sallusto17, the majority of both CD4+ TIL and CD8+ TIL were effector memory T cells, median 99.6% (range 99.3–99.9) and median 91.1% (range 87–96.9, respectively (Figure 5A). Regarding exhaustion markers, LAG-3 was highly expressed with a median of 85.2% in CD4+ TIL (range 65.9–93.6), and a median of 98.2% in CD8+ TIL (range 95.6–99.8), respectively (Figure 5B). PD-1 expression was lower and more variable with a median of 8.7% in CD4+ TIL (range 1.5–17.7), and a median of 16.4% in CD8+ TIL (range 6–51.1) (Figure 5B).
Table 4.
T-cell exhaustion markers in infusion products.
| INF PROD |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Live TIL |
CD3+ |
CD4+ |
CD8+ |
||||||
| Patient ID | (%) | CD4+ (%) | CD8+ (%) | LAG-3+ (%) | PD-1+ (%) |
PD-1+ LAG-3+ (%) |
LAG-3+ (%) | PD-1+ (%) | PD-1+ LAG-3+ (%) |
| GY1508.01 | 99.5 | 24.6 | 72.7 | 80.9 | 17.7 | 14.9 | 97.5 | 51.5 | 49.9 |
| GY1508.04 | 97.9 | 39.2 | 40.7 | 93.6 | 7.1 | 7.1 | 98.8 | 11.3 | 11.3 |
| GY1508.06 | 99.5 | 35.9 | 60.4 | 87.4 | 10.3 | 9.4 | 98.9 | 32.5 | 32.1 |
| GY1508.08 | 96.6 | 85.8 | 9 | 83 | 18.7 | 2.3 | 95.6 | 6 | 5.8 |
| GY1508.10 | 91.8 | 91.7 | 6.8 | 65.9 | 1.5 | 1.3 | 97.4 | 13.4 | 13.2 |
| GY1508.11 | 92.7 | 89 | 9,5 | 93.2 | 3.7 | 3.5 | 99.8 | 19.3 | 19.3 |
| Median | 99.7 | 62.5 | 25.1 | 85.2 | 8.7 | 5.3 | 98.2 | 16.4 | 16.3 |
| Range | 99.1–99.9 | 24.6–91.7 | 6.8–72.7 | 65.9–93.6 | 1.5–18.7 | 1.3–14.9 | 95.6–99.8 | 6–51.5 | 5.8–49.9 |
Analyses where performed on frozen INF PROD samples causing a discrepancy in the percentage of CD3+ CD4+ cells and CD3+ CD8+ compared to analyses performed on fresh INF PROD. The median discrepancy was within 10% which can be expected when comparing fresh and frozen samples.
Abbreviations: ID = Identification, INF PROD = Infusion product.
Figure 5.

T-cell differentiation stages and exhaustion markers in the INF PROD from the treated patients. A, the percentage of CD8+ and CD4+ TIL found to be naive- (CD45RO-CD45RA+), central memory- (CD45RO+ CCR7+) or effector memory cells (CD45RO+ CCR7-), respectively. B, the percentage of CD8+ and CD4+ TIL expressing the exhaustion markers LAG-3, PD-1 or LAG-3 and PD-1, respectively.
Specific antitumor reactivity in peripheral blood lymphocytes (PBL) was tested in the two patients with the highest overall antitumor reactivity. In general, no significant tumor reactive CD4+ or CD8 + T cells were detectable in PBL before or after treatment (Figure 3).
Figure 3.
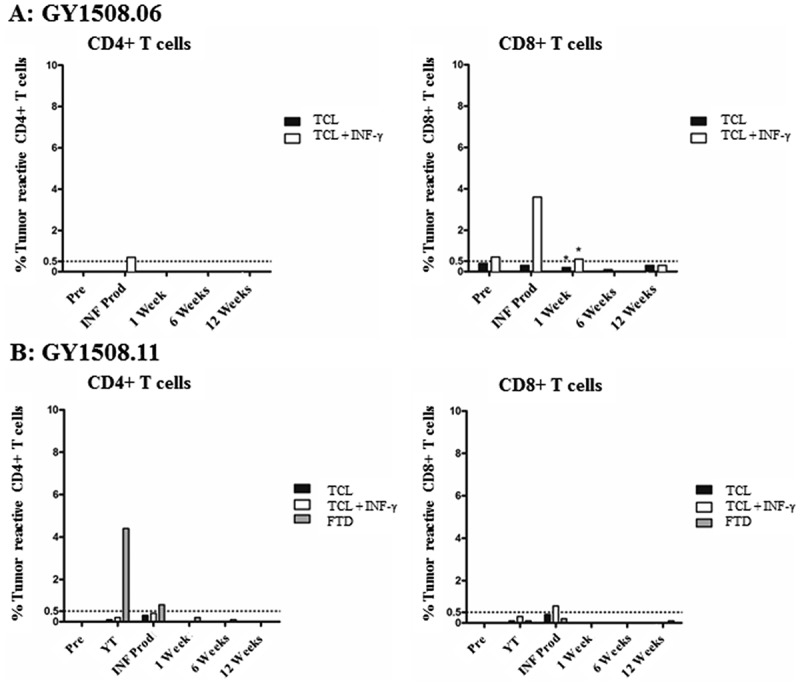
Antitumor reactivity of PBL in blood samples collected before and after TIL therapy. The figure shows the frequency of T cells expressing at least one of the T-cell functions: TNF, IFN-γ or CD107a, upon stimulation with autologous TCL (with or without low-dose IFN-γ (100 IU/ml) stimulation) or FTD with FACS analysis. A specific anti-tumor response was defined as a minimum 0.5% responding cells (horizontal line), with a minimum of 50 positive events (*). The frequency of tumor-reactive cells in stimulated samples was subtracted from un-stimulated samples.
Immune infiltrates and PD-L1 and MHCII expression in tumor tissue
The expression of lymphocyte-activation gene 3 (LAG-3) and programmed cell death protein 1 (PD-1) on infused T-cells point towards a potential risk of inhibitory signaling if the ligands major histocompatibility complex II (MHCII) and programmed death-ligand 1 (PD-L1) were expressed in the tumor. To assess this, multiplex immunohistochemistry (IHC) was performed on the tumor lesions from which TIL were expanded. Figure 4 shows an example of a patient with high and low expression of PD-L1 and MHCII, respectively. Supplementary Figure 1 shows representative examples of expression in all patients. The IHC showed a general infiltration of CD4 + T cells (detected as CD3+ CD8- cells; > 20 average counts/mm2), both cytotoxic (CD3+ CD8-TIA-1+) and non-cytotoxic/helper T cells (CD3+ CD8-TIA-1-), except in the epithelium from GY1508.11. Cytotoxic CD8 + T-cell (CD3+ CD8+ TIA-1+) infiltration was generally seen in both epithelia and stroma, except in GY1508.08, and in the epithelia of GY1508.11. Non-cytotoxic CD8 + T-cell infiltration (CD3+ CD8+ TIA-1-) was generally low and only observed in the stroma of GY1508.01 and GY1508.10). CD8/CD4 T cell ratio was generally low (< 1) except in the epithelium in GY1508.10 and in the stroma in GY1508.01 (both > 2). PD-L1 expression (> 20 average count/mm2) was found in both tumor epithelium and stroma, except in the stroma from GY1508.01 and GY1508.10. PD-L1 + macrophages (PD-L1+ CD163+) were found in both epithelium and stroma, except in the epithelium from GY1058.10. Further, MHCII expression (average H-score > 10) was shown in tumor tissue epithelium in four of six patients (except in GY1058.01 and GY1508.08).
Figure 4.

Multiplex immunohistochemistry of tumor samples used for TIL expansion. Images represent tumor slides from selected patients with a high expression/hot (top) and with a low expression/cold (bottom) for each panel. Color schemes of the selected panels were as follows. A, stained for PD-1 (blue), PD-L1 (red) and Cd163 (green). B, stained for MHCII (red) and DAPI (blue).
Discussion
ACT with TIL using standard preparative lymphodepleting chemotherapy in combination with decrescendo IL-2 stimulation was found feasible for treatment of patients with metastatic ovarian cancer. The full chemotherapy dosage and a median of 97% of the IL-2 dosage were administered. Treatment related side-effects were manageable and similar to those described in patients with metastatic malignant melanoma treated with a similar T-cell therapy regimen8.
Three of eleven patients were excluded due to deterioration of their clinical condition. Previous chemotherapeutic treatment was discontinued prior to inclusion. It is likely that the period without chemotherapeutic treatment may have led to the rapid disease progression seen in some of the patients. G-CSF-mobilized stem cell leukapheresis was strenuous on the majority of patients. The hematopoietic system of all patients recovered without stem cell support. Other studies with previously chemotherapy treated patients, using the same lymphodepleting chemotherapy regimen prior to TIL therapy, have reported similar findings12,18–20. Based on these findings and our own observations, we propose the leukapheresis procedure could be omitted in future clinical trials in a similar setting.
The best disease response observed was SD with a 23% decrease in target lesions and the longest period was five months. There were no clear differences in patient characteristics between the patients with SD for 5 months and 3 months, respectively.
We observed a difference between the RECIST and PERCIST evaluation in five of six patients with the PERCIST evaluation generally indicating response more frequently. However, as metabolic response did not correlate with clinical relevant benefit a potential role for the use of PERCIST in this patient group could not be sustained.
Since ovarian cancer is a chemotherapy-sensitive cancer, it cannot be ruled out that the lymphodepleting chemotherapy regimen might have contributed to the disease control observed. Cyclophosphamide has previously been a treatment option for patients with ovarian cancer21,22, although in a different regimen than the one used in this study. Fludarabine has only been tested in patients with ovarian cancer in a few older clinical trials showing no clinical effect23,24. In future studies, evaluation scan in close connection to the lymphodepleting chemotherapy regimen might clarify this.
Notably, only one of six patients progressed due to regrowth of target lesions while the remaining five patients progressed solely due to the occurrence of new lesion(s). During the course of tumor evolution and dissemination, different subclones might arise and give rise to tumor heterogeneity and varying degrees of resistance to anticancer therapies25. The observation that treatment failure generally was due to occurrence of new lesions indicates partial responsiveness to TIL therapy and points towards the emergence of TIL therapy resistance during continued subclonal evolution.
Although we found TIL therapy in patients with OC to be feasible and manageable with treatment-related side effects comparable to those observed in MM patients, clinical benefit were not as pronounced as in MM. TIL therapy is cumbersome and several of these heavily pretreated patients were excluded from the trial while waiting for TIL production due to clinical deterioration. This should be taken into consideration when planning future studies.
To our knowledge, TIL therapy in gynecological (cervical) cancers has only been reported with success in one study using selected TIL12. Treatment with unselected TIL has previously been successful in malignant melanoma8-10. It is possible that the lower somatic mutational load in ovarian cancer compared to malignant melanoma26 and resulting fewer potential neo-antigens for the immune system to recognize27, limits the chance of successful immune therapy.
In regards to TIL production, the REP expansion fold and total number of cells in the INF PROD were lower compared to our previous clinical trial in malignant melanoma8. A lower percentage of CD8+ cells and a higher percentage of CD4+ positive cells was also seen in the INF PROD compared to malignant melanoma8. High numbers of CD8 + T cells and high total numbers of TIL in the infusion product have previously been shown to be significantly associated with clinical response in malignant melanoma10. The lower total amount of TIL and the different composition of the INF PROD compared to malignant melanoma could therefore be part of the explanation of why only transient disease stabilization was induced.
Specific antitumor reactivity was seen more frequently in the INF PROD compared to YT for both CD4+ cells and CD8+ cells, but specific tumor reactivity could not be detected in PBL after treatment. This is in line with the observed association between lack of a persistent clinical response in malignant melanoma when antitumor reactive T cells were not established in PBL8. In the INF PROD we found a predominantly high expression of LAG3, especially on CD8+ cells, as well as a varying degree of PD-1 expression.
Tumor samples for TIL preparation were examined by IHC and we observed a general infiltration of CD4+ TIL and CD8+ cytotoxic TIL, with a CD8+/CD4+ ratio below 1. Furthermore, substantial expression of PD-L1 and MHCII was found in most tumor tissue samples.
T-cell activation, proliferation and effector functions can be regulated through several immune checkpoint pathways. We found high LAG-3 and some PD-1 expression in the INF PROD T cells and PD- L1 and MHCII expression in most tumors. PD-1/PD-L1 and LAG3/MHCII interaction is known to lead to inhibition of T cells which could potentially limit anti-tumor efficacy of the infused TIL. Surpassing negative regulation by use of checkpoint inhibitors is well known28. Thus, one way of increasing the clinical efficacy of TIL therapy in ovarian cancer could be combination therapy with checkpoint inhibitors. In support of this idea, TIL in ovarian cancer tumor models have been shown to express both surface CTLA-4 and PD-1 and blockade of both led to reversal of CD8+ TIL dysfunction and tumor regression29. CTLA-4 blockade in ovarian cancer in a clinical setting has only been scarcely investigated30,31 while a phase II study showing promising tolerability and clinical efficacy of PD-1 blockade in ovarian cancer was recently published32. Another study has found that the majority of tumor-derived CD8 + T cells from ovarian cancer patients express PD-1 and that LAG-3+ PD-1 + T cells accumulate at in ovarian tumors. Also, they found dual blockade of PD-1 and LAG-3 lead to an increased frequency and effector function of NY-ESO-1-specific CD8 + T cells33. Surprisingly, we found that the majority of TIL expressed LAG-3, but not PD-1 as could be expected. Therefore, LAG-3 could be another potential target of interest. Importantly, the findings are limited by the low number of patients in the study and observations should therefore mainly be hypothesis generating.
In conclusion, in this pilot study we have shown that TIL therapy in combination with a decrescendo IL-2 regimen in patients with metastatic ovarian cancer is feasible with manageable toxicity. Clinical indications of treatment benefits were seen, but mainly transient. Clinical efficacy might be potentiated through combination therapy targeting immune checkpoint triggered inhibition. To this end, at our center a phase I trial investigating TIL therapy in combination with Ipilimumab and Nivolumab is ongoing (NCT03287674).
Patients and methods
Patients
Patients aged 18–70 years with histologically verified high grade serous adenocarcinoma and metastatic OC, were eligible to enter the study. All patients had progressive platinum-resistant disease accessible for surgery, acceptable organ functions, an eastern cooperative oncology group (ECOG) performance status of ≤ 1. Patients with brain metastases, severe comorbidities or -allergies, active autoimmune diseases, treatment with immunosuppressive or experimental drugs or other malignancies within the past five years were excluded.
Study design
The study was conducted as a pilot study treating six patients (ClinicalTrials.gov Identifier: NCT02482090). The primary endpoints were tolerability and feasibility. Secondary endpoints were to assess objective responses (OR) and immune responses. OS and PFS were also described. The study was conducted in accordance with the Helsinki Declaration and approved by the National Committee on Health Research Ethics, the Danish Data Protection Agency and the Danish Medicines Agency. Informed consent was obtained from each patient.
Treatment
Surgical removal of tumor tissue (≥ 1 cm3) for TIL expansion was followed by G-CSF (Filgrastime 0.1 mg/kg) mobilized leukapheresis with the purpose of harvesting ≥ 2 x 106 autologous hematopoietic stem cells (HSC) for cryopreservation. TIL expansion took approximately 4–6 weeks (described below). TIL therapy consisted of lymphodepleting chemotherapy (Cyclophosphamide 60 mg/kg, day −7 and −6 and Fludarabine 25 mg/m2, day −5 to −1) as described in previous studies8,34. TIL was administered as a single intravenous infusion on day 0 and was followed by IL-2 in accordance with the decrescendo regimen (18 MIU/m2 for 6, 12 and 24 hours followed by 4.5 MIU/m2 for 24 hours for up to three days, maximum dose 135 MIU)35. IL-2 administration was terminated if unacceptable toxicities occurred. Patients were monitored daily with clinical examination and biochemical blood tests. Blood tests for immunological monitoring were collected before leukapheresis, at hospitalization (day −8), at the time of discharge and at subsequent evaluations (see below). All patients received prophylactic antibiotics and antiemetics as described in earlier studies8,34. Platelet and red blood cell transfusions were given when clinically indicated.
Evaluation
Toxicity was assessed using the common terminology criteria for adverse events (CTCAE) version 4.0. Clinical response was evaluated using the RECIST 1.1 criteria36, disease control rate was defined as non-progressive disease according to RECIST 1.1. In addition, the PERCIST 1.0 criteria37 was used exploratively to assess if there could be an additive value to the RECIST criteria, since the addition of PET to CT has previously been found to present a more detailed picture of the single lesions38. 18F-Flourodeoxyglucose positron emission tomography/computed tomography (PET/CT) scan was performed within 14 days of TIL therapy (baseline), at 6 and 12 weeks after TIL therapy, and thereafter approximately every third month for up to five years or until disease progression. Blood tests for measuring CA-125 were drawn at evaluation time points.
Generation of TIL
Tumor fragments were processed and TIL expanded according to previously published methods39,40. Briefly, 1–2 mm3 tumor fragments (48 in total) were placed into 24-well plates (Nunc, Roskilde, Denmark) in 2 mL of culture medium (90% RPMI 1640 (Invitrogen), 10% heat inactivated Human AB serum (Sigma-Aldrich, St. Louis, MO, USA) containing 6000 IU/mL of IL-2 (Aldesleukin, Novartis, Basel, Switzerland), Penicillin, Streptomycin and Fungizone (Bristol-Myers Squibb, Lyngby, Denmark). The bulked culture of YT from this initial outgrowth were then either cryopreserved or directly used in a rapid expansion protocol (REP) to generate the final infusion product. Approximately 20 × 106 TIL were used to initiate the REP using a dynamic expansion platform as previously described39. Briefly, 30 ng/ml anti-CD3 antibodies (OKT3, Miltenyi Biotec, Bergisch Gladbach, Germany) were added to the REP culture (TIL in 80/20 medium (80% CM/20% AIM-V medium)) to nonspecifically induce proliferation of the TIL. Irradiated (40 Gy) allogeneic feeder cells (peripheral blood mononuclear cells (PBMC) from at least 4 different healthy donors) at a 1:200 ratio were added to further stimulate TIL growth through secretion of stimulating cytokines. At day 14, TIL were harvested and transferred to a 400 mL infusion bag in a suspension of sodium chloride with 2.5% albumin. Sterility testing and microbiological control were performed on all TIL cultures before REP or cryopreservation and before infusion.
Generation of tumor cell lines
TCL were generated from the tumor specimens transport media or released after mechanical disaggregation of fragments. The TCL were cultured in R10 medium (RPMI-1640 with GlutaMAX, 25 mM HEPES pH 7.2, 100 U⁄mL Penicillin, 100 μg⁄mL Streptomycin and 10% fetal bovine serum (FBS) (Gibco, Nærum, Denmark). Solu-Cortef (hydrocortisone sodium succinate) (the local hospital pharmacy) 500 ng/mL was added to R10, the first month of establishing the TCL. Furthermore, enzymatically digested fresh tumor fragments, called fresh tumor digest (FTD), containing all cells present in the tumor microenvironment, were cryopreserved for later use.
Immunological analysis
Flow cytometry
Phenotype of YT and INF PROD, and antitumor reactivity of YT, INF PROD and PBL were analyzed with flow cytometry. Briefly, phenotype analysis performed on fresh cells was determined immediately after harvest by staining with fluorochrome-labeled monoclonal antibodies against CD3 FITC, CD4 PerCP, CD56 PE (all from BD bioscience, New Jersey, United States) and CD8 PB (Dako, Glostrup, Denmark). Further analysis of T cell differentiation stages and exhaustion makers were performed on cryopreserved cells. The cells rested overnight at 37°C, 5% CO2 in culture medium before staining with CD3 BV510, CD4 PerCP, CD8 BV421, CD45RA FITC, CD45RO PE, CCR7 PE-Cy7, PD-1 PE-Cy7 (all from BD Biosciences, New Jersey, United States), LAG-3 FITC (LS Bioscience, Seattle, United States) and Near-IR live/dead stain (Life Technologies, California, United States).
Tumor reactivity was tested against both autologous TCL, with- or without pretreatment with IFN-γ when available, and against FTD as described previously16. Briefly, TIL were co-cultured with target cells for 5 hours at 37°C, 5% CO2 in effector:target ratio 3:1 together with Golgiplug and CD107a BV421 antibody (BD bioscience, New Jersey, United States). Cells were then stained with extracellular antibodies Near-IR Live/Dead (Life Technologies, California, United States), CD3 FITC, CD56 PE, (BD bioscience, New Jersey, United States) CD8 QD605 (Life Technologies, California, United States) and CD4 PerCP (Biolegend, California, United States) and after fixation and permeabilization treatment (eBioscience) stained with intracellular cytokines TNF APC and IFN-γ PE-Cy7 (BD bioscience, New Jersey, United States). Tumor reactive TIL were defined as T cells expressing at least one of the following T-cell functions: tumor necrosis factor (TNF), IFN-γ or CD107a. A specific antitumor response was defined as at least 0.5% responding cells being present (subtracted from unstimulated samples and used as level of significance) with a minimum of 50 positive events.
All cells were analyzed using a FACS canto II instrument (BD bioscience, New Jersey, United States) equipped with FACS Diva 8.0 software, as previously described16.
Multi-color immunohistochemistry and multi-color immunofluorescence
Unless stated otherwise, all reagents were obtained from Biocare Medical (Pacheco, CA). To stain for the PD-L1/CD163/PD-1 combination slides were first deparaffinized through xylene and graded alcohols to water then subjected to antigen retrieval with Diva Decloaker in a Biocare decloaking chamber at 110°C for 15 minutes. Slides were then loaded into a Biocare Intellipath FLX autostainer. First Peroxidased-1 was applied for 5 minutes followed by Background Sniper for 10 minutes to block non-specific staining. Then a cocktail of PD-L1 (clone SP142, Spring Bioscience, Pleasanton, CA) and CD163 (clone 10D6, Biocare) in Da Vinci Green diluent was added for 30 minutes followed by MACH2 Double Stain # 1 Polymer for 30 minutes. The signal was then detected by adding IP Ferangi blue for 8 minutes followed by IP DAB chromogen for 5 minutes. Slides were then removed from the stainer and subjected to denaturation at 50°C for 45 minutes in a SDS-glycine pH2.0 solution as per Pirici et al (PMID: 19,223,296). Slides were then reloaded onto the Intellipath FLX and incubated with PD-1 (clone EPR4877(2), Abcam) in Da Vinci Green diluent for 30 minutes followed by MACH2 Rabbit-AP Polymer for 30 minutes then IP Warp Red Chromogen for 7 minutes. Slides were then counterstained with a 1:5 dilution of CAT hematoxylin, rinsed, air-dried and coverslipped with Ecomount.
The overall process for staining the TIA-1/CD8/CD3 panel was similar however in the first round of staining TIA-1 (clone TIA-1, Biocare) was diluted in Da Vinci Green diluent and applied to the slides for 30 minutes followed by 30 minutes with MACH2 Mouse-AP polymer and 8 minutes with IP Ferangi Blue chromogen. In the second round of staining a cocktail of CD8 (clone C8/144B, Cell Marque, Rocklin, CA) and CD3 (clone SP7, Spring Bioscience) in Da Vinci Green diluent was added for 30 minutes followed by MACH2 Double Stain #1 polymer for 30 minutes, then IP Warp Red Chromogen for 8 minutes and Hi Def Yellow Chromogen (Enzo, Farmingdale, NY) for 10 minutes. Slides were then counterstained, rinsed, air-dried and coverslipped as described.
For the MHCI/MHCII/IDO-1 panel multi-color immunofluorescence was applied using OPAL reagents from Perkin Elmer (Waltham, MA). Manufacturer instructions were followed for the overall protocol with HLA Class I A,B,C (clone EMR8-5, MBL, Woburn, MA) on OPAL520 in round 1, HLA-DP,DQ,DR (clone CR3/43, Affinity Bioreagents, Golden, CO) on OPAL650 in round 2 and IDO-1 (clone SP260, Spring Bioscience) in round 3 on OPAL570. Appropriate MACH2 polymers from Biocare were used after the primary antibody.
Imaging and staining
Slides were imaged using a Vectra 2 Multispectral Imaging system (Perkin Elmer). 10 20X images per slide were collected that had 90% of any type of tissue in the field of view. The im3 files generated were opened in inForm image analysis software (Perkin Elmer) and algorithms constructed to phenotype the various cell types of interest. For the MHCI/MHCII/IDO-1 panel, the H score function was used to score each marker. For each panel 5 algorithms were constructed and the resulting data visually validated to ensure accuracy.
Statistical analysis
Percentage, median and range were used to describe immunological and disease control rates, duration of response and patient characteristics. OS and PFS were determined as time from TIL infusion to death or progressive disease, respectively, or time to last follow-up (July 18th 2017).
Funding Statement
This work was supported by Herlev Research Council, the Danish Cancer Society, Aase and Einar Danielsen Foundation, the Anticancer Fund and Ovacure.
Acknowledgments
We would like to thank the laboratory technicians Susanne Wendt, Sandra Ullitz Færch and Betina Johansen for invaluable help with the clinical cell manufacturing; and the Department of Oncology, Herlev Hospital, physicians and nurses for their support in taking care of the patients with special thanks to project nurse Anne Kuld-Nielsen in this regard.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data can be accessed here.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A.. Global Cancer Statistics, 2012. CA a Cancer J. Clin. 2015;652:87–108. DOI: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Storm HH, Kejs AMT, Engholm G, Tryggvadóttir L, Klint A, Bray F, Hakulinen T. Trends in the overall survival of cancer patients diagnosed 1964-2003 in the Nordic countries followed up to the end of 2006: the importance of case-mix. Acta Oncol. 2010;495:713–724. DOI: 10.3109/0284186X.2010.484426. [DOI] [PubMed] [Google Scholar]
- 3.Herzog TJ, Pothuri B. Ovarian cancer: a focus on management of recurrent disease. Nat Clin Pr. Oncol. 2006;311:604–611. DOI: 10.1038/ncponc0637. [DOI] [PubMed] [Google Scholar]
- 4.Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 2013;104:211–224. DOI: 10.1038/nrclinonc.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003;3483:203–213. DOI: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Sci (80-.). 2015;3486230:62–68. DOI: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using t-cell transfer immunotherapy. Clin. Cancer Res. 2011;1713:4550–4557. DOI: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Hendel HW. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-in fi ltrating lymphocytes and an attenuated IL2 regimen. Clin. Cancer Res. 2016;1–12. DOI: 10.1158/1078-0432.CCR-15-1879. [DOI] [PubMed] [Google Scholar]
- 9.Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin. Cancer Res. 2013;1917:4792–4800. DOI: 10.1158/1078-0432.CCR-13-0380. [DOI] [PubMed] [Google Scholar]
- 10.Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Chacon J, Wu R, Lizee G, Mahoney S, Glass M, Johnson VE, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012;1824:6758–6770. DOI: 10.1158/1078-0432.CCR-12-1177.Specific. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, Kudchadkar R, Zager J, Gibney G, Sondak VK, et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J. Immunother. 2012;358:615–620. DOI: 10.1097/CJI.0b013e31826e8f5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, Dudley ME, Yang JC, Sherry RM, Kammula US, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. 2015;3314:1543–1550. DOI: 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran E, Turcotte S, Gros A, Robbins PF, Lu Y, Dudley ME, Parkhurst MR, Yang JC. Rosenberg S a. cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Sci (80-.). 2014;9May:641–645. DOI: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freedman RS, Edwards CL, Kavanagh JJ, Kudelka AP, Katz RL, Carrasco CH, Atkinson EN, Scott W, Tomasovic B, Templin S. Intraperitoneal adoptive immunotherapy of ovarian carcinoma with tumor-infiltrating lymphocytes and low-dose recombinant interleukin-2: a pilot trial. J. Immunother. Emphasis Tumor Immunol. 1994;163:198–210. DOI: 10.1097/00002371-199410000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Aoki Y, Takakuwa K, Kodama S, Cancer EO, Tanaka K, Takahashi M, Tokunaga A. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res 1991;51:1934–1939. [PubMed] [Google Scholar]
- 16.Westergaard MCW, Andersen R, Kjeldsen JW, Hasselager T, Lajer H, Donia M. Preclinical development of tumor-infiltrating lymphocytes (TILs) based adoptive cell transfer immunotherapy (ACT) for patients with advanced ovarian cancer. Ann. Oncol. 2016;27Suppl_8: DOI: 10.1093/annonc/mdw141. [DOI] [Google Scholar]
- 17.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory t cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 2004;221:745–763. DOI: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 18.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Suzanne L, Restifo NP, Royal RE, Kammula U, White DE, Sharon A, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005;2310:2346–2357. DOI: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T cell receptor: long term follow up and correlates with response. Clin. Cancer Res. 2015;215:1019–1027. DOI: 10.1161/CIRCRESAHA.116.303790.The. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Secondino S, Zecca M, Licitra L, Gurrado A, Schiavetto I, Bossi P, Locati L, Schiavo R, Basso S, Baldanti F, et al. T-cell therapy for EBV-associated nasopharyngeal carcinoma: preparative lymphodepleting chemotherapy does not improve clinical results. Ann. Oncol. 2012;232:435–441. DOI: 10.1093/annonc/mdr134. [DOI] [PubMed] [Google Scholar]
- 21.Mcguire WP, Hoskins WJ, Brady MF, Kugera PR, Partridge EE, Look KY, Clarke-Pearson DL, Davidson M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl. J. Med. 1996;3341:1–6. DOI: 10.1056/NEJM199606133342404. [DOI] [PubMed] [Google Scholar]
- 22.Piccart M, Bertelsen K, James K, Cassidy J, Mangioni C, Simonsen E, Stuart G, Kaye S, Vergote I, Blom R, et al. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: three-year results. J Natl Cancer Inst. 2000;929:699–708. [DOI] [PubMed] [Google Scholar]
- 23.Kavanagh J, Stringer A, Copeland L, Gershenson D, Phase PS. II Trial of Fludarabine in Patients With Epithelial Ovarian Cancer. Cancer Treat. Rep. 1986;703:425–426. [PubMed] [Google Scholar]
- 24.Von Hoff D, Kronmal R, O’Toole R, Surwit E, Hutton J, Phase AD. II study of fludarabine phosphate (NSC-312887) in patients with advanced ovarian cancer. Am J Clin Oncol. 1988;112:146–148. DOI: 10.1097/00000421-198804000-00011. [DOI] [PubMed] [Google Scholar]
- 25.Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol. Oncol. 2014;86:1095–1111. DOI: 10.1016/j.molonc.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawrence M, Stojanov P, Polak P, Kryukov G, Cibulskis K, Sivachenko A, Carter S, Stewart C, Mermel C, Roberts S, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;4997457:214–218. DOI: 10.1038/nature12213.Mutational. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;391:1–10. DOI: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Pennock GK, Chow LQM. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. Oncologist. 2015;207:812–822. DOI: 10.1634/theoncologist.2014-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;7312:3591–3603. DOI: 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, M V S, Davis T, Henry-Spires R, MacRae S, Willman A, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Pnas. 2003;1008:4712–4717. DOI: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodi FS, Butler M, Oble DA, Seiden MV, Fg H, Kruse A, Macrae S, Nelson M, Canning C, Lowy I, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci. U. S. A. 2008;1058:3005–3010. DOI: 10.1073/pnas.0712237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A. Safety and Antitumor Activity of Anti – PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. 2016;3334:11–13. DOI: 10.1200/JCO.2015.62.3397. [DOI] [PubMed] [Google Scholar]
- 33.Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P, et al. Tumor-infiltrating NY-ESO-1–specific CD8 + T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. 2010;10717:7875–7880. DOI: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellebaek E, Iversen TZ, Junker N, Donia M, Engell-Noerregaard L, Met O, Hölmich LR, Andersen RS, Hadrup SR, Andersen MH, et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. J. Transl. Med. 2012;101:169 DOI: 10.1186/1479-5876-10-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keilholz U, Goey SH, Punt CJ, Proebstle TM, Salzmann R, Scheibenbogen C, Schadendorf D, Liénard D, Enk A, Dummer R, et al. Interferon alfa-2a and interleukin-2 with or without cisplatin in metastatic melanoma: a randomized trial of the european organization for research and treatment of cancer melanoma cooperative group. J. Clin. Oncol. 1997;157:2579–2588. DOI: 10.1200/JCO.1997.15.7.2579. [DOI] [PubMed] [Google Scholar]
- 36.Eisenhauer EA, Therasse P, Bogaerts J, Lh S, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer. 2009;452:228–247. DOI: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 37.Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: evolving considerations for PET response criteria in solid tumors. J. Nucl. Med. 2009;50Suppl 1:122S–50S. DOI: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engell-Noerregaard L, Hendel HW, Johannesen HH, Alslev L, Svane IM. FDG PET scans as evaluation of clinical response to dendritic cell vaccination in patients with malignant melanoma. Cancer Immunol. Immunother. 2013;621:17–25. DOI: 10.1007/s00262-012-1306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donia M, Larsen SM, Met O, Svane IM. Simplified protocol for clinical-grade tumor-infiltrating lymphocyte manufacturing with use of the Wave bioreactor. Cytotherapy. 2014;168:1117–1120. DOI: 10.1016/j.jcyt.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, Robbins PF, Rosenberg SA, Dudley ME. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J. Immunother. 2008;318:742–751. DOI: 10.1097/CJI.0b013e31818403d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.