

ABSTRACT
Mutated proteins arising from somatic mutations in tumors are promising targets for cancer immunotherapy. They represent true tumor-specific antigens (TSAs) as they are exclusively expressed in tumors, reduce the risk of autoimmunity and are more likely to overcome tolerance compared to wild-type (wt) sequences. Hence, we designed a panel of long peptides (LPs, 28–35 aa) comprising driver gene mutations in TP35 and KRAS frequently found in gastrointestinal tumors to test their combined immunotherapeutic potential. We found increased numbers of T cells responsive against respective mutated and wt peptides in colorectal cancer patients that carry the tested mutations in their tumors than patients with other mutations. Further, active immunization of HLA(-A2/DR1)-humanized mice with mixes of the same mutated LPs yielded simultaneous, polyvalent CD8+/CD4+ T cell responses against the majority of peptides. Peptide-specific T cells possessed a multifunctional cytokine profile with CD4+ T cells showing a TH1-like phenotype. Two mutated peptides (Kras[G12V], p53[R248W]) induced significantly higher T cell responses than corresponding wt sequences and comprised HLA-A2/DR1-restricted mutated epitopes. However, vaccination with the same highly immunogenic LPs strongly increased systemic regulatory T cells (Treg) numbers in a syngeneic sarcoma model over-expressing these mutated protein variants and resulted in accelerated tumor outgrowth. In contrast, tumor outgrowth was delayed when vaccination was directed against tumor-intrinsic Kras/Tp53 mutations of lower immunogenicity. Conclusively, we show that LP vaccination targeting multiple mutated TSAs elicits polyvalent, multifunctional, and mutation-specific effector T cells capable of targeting tumors. However, the success of this therapeutic approach can be hampered by vaccination-induced, TSA-specific Tregs.
KEYWORDS: Long-peptide vaccination, tumor-specific mutated antigens, tumor mutation specific T cell responses, regulatory T cells, Treg, common driver mutations, p53, Kras
Introduction
The expression of antigens (Ags) that discriminate malignant from non-transformed cells and their recognition by T cells form the basis of targeted cancer immunotherapy.1, 2 Tumor antigens (TAs) are grouped according to their tumor specificity. The first group of tumor-associated Ags (TAAs) can also be expressed by non-transformed tissue and includes over-expressed and differentiation Ags of the tissue the tumor originates from3, Ags resulting from abnormal posttranslational modifications4, and re-expressed cancer germline Ags.5,6 In contrast, the second group of strictly tumor specific antigens (TSAs) comprises mutated and oncoviral Ags7, which are exclusively expressed by the tumor and are not shared with normal tissue. Therefore, TSAs reduce the risk of autoimmune adverse effects and increase the chance to overcome immune tolerance compared to non-mutated (self-) Ags.
Mutated TSAs can be of different quality for the tumor.8 Mutations in genes which are essential for oncogenesis are known as driver mutations.9 Passenger mutations are expendable to sustain a malignant phenotype, which can result in Ag-loss variants of the tumor once a Darwinian selection pressure is imposed by therapeutic intervention.10,11 Therefore, targeting driver mutations in a TSA-specific therapy is indicative. Important driver genes in colorectal9,12 and pancreatic13 tumorigenesis are mutated alleles of the oncogene KRAS and the tumor suppressor gene/oncogene TP53. Indeed, mutated Kras and p53 proteins can comprise TSAs, as mutated Kras epitopes around the hot-spot mutation site G1214-18 and also mutated p53 epitopes comprising hot-spot variants have previously been described.19,20
We and others proved the existence of both TA-specific CD4+ and CD8+ memory T cells (Tmem) in cancer patients prior to any immunotherapeutic intervention.2,21–25 This polyvalent T cell pool consists of effector and central Tmem cells, which produce IFN-γ upon Ag re-encounter and can eliminate autologous tumor cells.26–28 However, there is increasing evidence that tumor-associated immune suppression can be mediated in an Ag-specific manner, too. Our group has previously shown that TA-specific Treg cells are present in cancer patients, which can suppress the proliferation of polyclonally activated conventional T cells (Tcon cells) and inhibit TA-specific effector T cell responses in vitro.29–31 The origin of TA-specific Treg cells and the extent to which natural, thymic-derived Treg cells (nTreg) and/or induced Treg cells (iTreg) are involved in the suppression of anti-tumor T cell responses is not clarified yet. Further, it is not well understood whether TA-specific Treg cells are solely induced and expanded by a suppressive tumor-microenvironment or possibly also by therapeutic intervention.
Vaccinations are targeted cancer-immunotherapies predestined to employ TAAs/TSAs with the goal to sensitize the patient to recognize TAs de novo or boost preexisting immune responses. Peptide vaccination allows for several TAs and adjuvants to be readily combined in one formulation. Herein, the use of peptide vaccines that are longer than minimal MHC class I ligands (8–10 aa) has major advantages.32 First, they need to be processed ensuring effective (cross)-presentation by professional antigen-presenting cells (APCs). This process is indispensable for proper priming and activation of TSA-specific naïve T cells.33, 34 Second, long peptides can provide several MHC class I alleles with ligands, thus permitting a broader cohort of patients to benefit from a vaccine. Third, long peptides can comprise both MHC class I and II epitopes. Therefore, both cytotoxic CD8+ T cells (CTLs) as well as helper CD4+ T cells (TH) can be activated. Particularly, TSA-specific T helper 1 cells (TH1) assure important roles in the tumor setting by licensing dendritic cells (DCs) for effective cross-priming of naïve CTLs.34 In addition, TH cells can exert direct tumor-eradicating functions.35 Moreover, combining several TSAs in one vaccine might broaden the responses towards sub-dominant epitopes36,37 and thereby prevent or delay the tumor’s escape from immune surveillance through emergence of Ag-loss variants.11
Following this line of thought, cancer vaccination with long synthetic peptides33, presents a versatile and easily applicable therapeutic platform. Indeed, peptide vaccination was effective in eliciting tumor-protective immunity in animal studies.38 Unfortunately, clinical translation has been considerably less successful. Although TA-specific T cell responses could broadly be elicited, they were of only little or no therapeutic benefit. One possible explanation for this failure is attributed to the fact that early trials mostly included late-stage patients, generally displaying severe systemic immune suppression that in the ‘pre-immune checkpoint inhibitor’ era of immunotherapy could hardly be overcome.39 Then small clinical pilot studies (phase I/II) were launched exploring vaccination with mutated Kras and p53 peptides for their clinical benefit.40,41 Vaccination trials with mutated Kras peptides in advanced-stage pancreatic cancer patients resulted in longer survival of immune responders compared to non-responding patients.40,42 In a second study, immune responses against mutated peptides were detected in the majority of the patients.43 Other patients were immunized using autologous peripheral blood mononuclear cells (PBMCs) loaded with a single long peptide harboring either a p53 or a Kras mutations found in the patients’ tumors. Half of the patients in that study showed TSA-specific immune responses after vaccination.44
Subsequently, recent studies focus on combining cancer peptide vaccination with other cancer therapeutic interventions, including surgically de-bulking of tumor masses, chemotherapy, radiotherapy, small molecule inhibitors, immune checkpoint blockade, and other concepts of immune modulation.45 In combinatorial approaches several peptide vaccines have entered phase III clinical trials.46 Rammensee and colleagues, for example, showed in a phase II trial for metastatic renal cell carcinoma that overall survival was associated with T-cell responses against IMA901 (a multi-epitope peptide vaccine)47. This led to a phase III study combining IMA901 with sunitinib (a small molecule receptor tyrosin kinase inhibitor). However, in this carefully designed randomized multi-center study IMA901 did not prolong overall survival in the IMA901 co-treated patient cohort.48
It is evident that more research is required in order to fully understand the underlying mechanisms that hamper the potential of TA-specific (peptide) vaccination. Our goal was to gain more insight into vaccination-induced T cell responses towards mutated oncogene/tumor suppressor gene derived Ags. In this study we combined the most frequent mutations in KRAS and TP53 found in gastrointestinal cancers and explored preexisting immune responses against these sequences in colorectal cancer (CRC) patients. We tested their cancer immunotherapeutic potential in a multiple-epitope long-peptide vaccination setting by utilizing HLA-I/II double transgenic mice together with a syngeneic tumor model, and assessed the tumor protective capacity of immunogenic mutated long peptides in a preventive vaccination setting. Furthermore, we aimed to investigate possible caveats of this therapeutic approach, foremost immunosuppressive counter-reactions through the induction and expansion of Treg cells.
Results
Pronounced T cell responses against common hotspot mutations in TP53 and KRAS in the blood and bone marrow of CRC patients
To test for T cell responses against driver gene mutations of GI tumors we assembled a panel of long peptides comprising the most frequent KRAS49-51 and TP5352 hotspot mutations found in CRC and pancreatic cancer. We also included the BRAF V600E mutation53, which is a frequent driver mutation in melanoma but is also found to a lower percentage (10.7%) in GI tumors51. For the structural design of the 19 long mutated peptides, a respective oncogene mutation was placed in the middle of a 28–35-mer peptide. As a result, the point mutations were flanked by roughly 15 aa on either side taken from the respective non-mutated, adjacent, natural human protein backbone (Supplementary Table 1). As references, corresponding wt peptides (eight in total) for each mutation site displaying the unaltered protein sequences were also tested (Supplementary Table 1). We analyzed a cohort of 26 CRC patients (Supplementary Table 2) to investigate the relevance of mutation specific T cell responses in human GI cancer patients who did not undergo previous immunotherapeutic interventions. The enrolled patient cohort comprised 10 female and 16 male individuals in an age range from 40 to 85 years at sample collection and spanning tumor stages 1 to 4. Peripheral blood (PB) samples and bone marrow (BM) samples were collected on the day of surgery prior the procedure. The patients’ PB and BM were screened for memory T cell reactivity against the panel of long peptides. Purified T cells were in vitro re-stimulated on autologous DCs pulsed with either mutated peptides or their wt counterparts. T cell recall responses were monitored by IFN-γ ELISpot analysis. Along with T cell reactivity, paraffin-embedded tissue samples of the same patients’ primary tumors and metastases (24 out of 26 patients, Supplementary Table 3) were sequenced for the abundance of TP53, KRAS, and BRAF mutations in relevant hotspot mutation site-comprising exons employing the QIAsymphony technology.
The results of ELISpot assays performed with PBMCs of representative CRC patients are shown in Figure 1A and Supplementary Figure 1. Notably, the patient presented in Figure 1A carried the point mutation p53 R175H in his primary tumor and, indeed, T cells purified from PB were significantly more responsive against the peptide harboring the mutation R175H than towards the corresponding wt peptide. Compared to the negative assay control (human IgG) the patient showed T cell recall responses against several other wt (p53 R273/R282 wt, Braf V600 wt) and mutated (p53 R273C, p53 R248W) long peptides.
Figure 1.
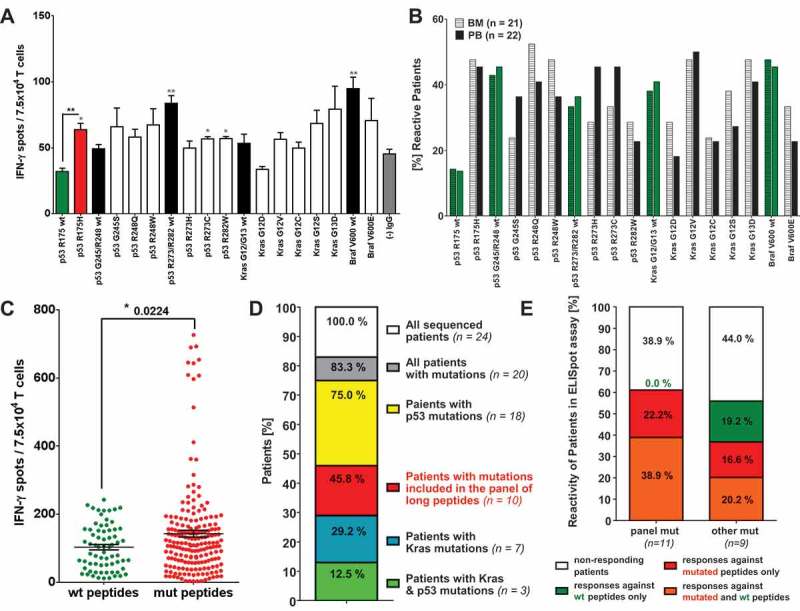
Relevance of mutation specific T cell responses in CRC patients. IFN-γ ELISpot analysis with T cells derived from of peripheral blood (PB) and bone marrow (BM) samples of 26 CRC patients. (A) IFN-γ-ELISpot analysis of peripheral blood (PB) derived T cells from a representative (HLA-A*0201 negative) CRC patient carrying the mutation p53 R175H in their primary tumor. Mutated peptide responses were tested against wt peptide responses (black *) and the negative assay control ((-) IgG, gray *). Wt peptide responses were tested against the negative assay control ((-) IgG, gray *) applying unpaired Student’s t-test. The assay was conducted in triplicates. (B) Percentage of patients responding to tested mutated and wt peptides in INFγ-ELISpot analysis. Peptide responses were considered specific in case they were significantly higher than responses towards the negative assay control (IgG, see Figure 1 (A)) in unpaired t-test. (C) Frequency of peptide-specific T cell responses against wt and mutated peptides. Accumulated ELISpot data from blood and BM samples of 26 CRC patients (unpaired t-test). Peptide responses were considered specific in case they were significantly higher than responses towards negative assay control ((-) IgG, see Figure 1 (A)) in unpaired t-test. (D) Distribution of KRAS and TP53 mutations in the tumors and/or metastasis of the CRC patient analyzed previously by ELISpot assay. DNA for exon sequencing of TP53 and KRAS genes was derived from paraffin embedded tissue of primary tumors and metastases of 24 CRC patients. (E) T cell reactivity of patients against wt and mutated peptides correlated with the mutations detected in the patients’ tumors and/or metastasis. panel mut = patients carrying mutations included in the panel of long peptides, other mut = patients carrying TP53 and KRAS mutations different from the mutations included in the panel of long peptides.
The response rates against all peptides tested within the whole patient cohort are shown in Figure 1B. Anti-peptide T cell responses were considered positive in case they showed a significantly higher response than the IgG negative assay control applying the one-sided t-test. According to the CIP (CIMT Immunoguiding Program) recommendations ELISpot responses smaller than 6 spots per 100’000 PBMCs should be viewed with caution, which was the case in 1 out of the 43 ELISpot assays included in the presented data. Following recent CIP suggestions we also used a nonparametric statistical distribution-free resampling (DFReq) test method together with the recommended R based web tool (http://www.scharp.org/zoe/runDFR/) suitable to detect moderate immune response magnitudes (Moodie et al.54–56) for assessing immune responses in the 42 samples meeting the CIMT criteria. Response rates obtained with the DFR(eq) method are shown in Supplementary Figure 2A. The response pattern using the DFR(eq) method vastly resembles the results obtained using the one-sided t-test (Figure 1B).
The resulting response rates for wt and mutated peptides are comparable using t-test as well as the DFR(eq) method, ranging from roughly 15% for the p53 R175 wt peptides to around 50% for some of the mutated (p53 R248W/Q, Kras G12V) and wt (p53 G245/R248 wt, Braf wt) peptides. Interestingly, the frequencies of patients responding against only one, two, three or more wt peptides were equally distributed throughout the patient cohort (Supplementary Figure 2C). However, 70% of patients responded against three or more mutated peptides simultaneously and only 30% against less than three. Cumulative analysis of peptide-specific T cell recall responses (BM- and PB-derived responses pooled) demonstrated that IFN-γ production and therewith T cell reactivity against mutated peptides was generally higher than against wt peptides (Figure 1C). The same was true for the accumulated paired analysis of all wt and mutated peptide pairs tested (Supplementary Figure 2E, including all PB and BM ELISpot results of the whole patient cohort). Further, responses against mutated peptides were considered mutation-specific if they elicited significantly higher IFN-γ spot counts than corresponding wt counterparts. The mutation-specific response rates for the mutated peptides (Supplementary Figure 2D) revealed, that the peptide representing the mutation p53 R175H was selectively recognized by 40% of patients and peptide p53 R248W by 20% of BM T cells and 10% of patients’ PB T cells, respectively. Of note, the peptides p53 R175H and p53 R248Q/W were possibly recognized in an HLA-A2-restricted context.
As indicated by the abundance of wt peptide reactivity it is likely that the IFN-γ T cell responses observed, are partially directed against epitopes within the non-mutated, peptide backbone sequences flanking the mutation sites. To address the question, which epitopes within the long peptide sequences (mutated and non-mutated), could elicit T cell responses in the analyzed patients, epitope prediction was performed employing the NetMHC57,58 algorithms. Within the present patient cohort HLA typing was solely performed for the HLA-A2 allele and 5 five of the 26 patients were tested positive (Supplementary Table 3). To draw a more representative picture of which epitopes could be recognized by the patient cohort, the most frequent HLA class I and II alleles within the German population were retrieved from Germany pop8 (n = 39689, www.allelefrequencies.net). Employing the NetMHC version 4.0 for HLA class I and NetMHCII version 2.3 algorithm for the HLA class II alleles all strong and weak HLA binding peptides were predicted and categorized (Supplementary Figure 3). Interestingly, all of the tested long peptides contain WT sequences around the mutation site that have the potential to be weak and strong MHC class I binders and therewith could elicit CTL responses. According to NetMHC predictions the number of potential wt epitopes comprising the mutation site is lower as the number for potential epitopes not comprising the mutation. However, the acquisition of the respective mutations can give rise to neo-epitopes for most of the peptides (indicated in light and dark red, Supplementary Figure3A), some of which are strong binders, as it is the case for the mutated peptides p53 R248W and Kras G12V (red parts of the bars, Supplementary Figure 3A). A similar pattern of potential epitopes within the long peptides can be observed for MHC class II binders (Supplementary Figure 3B). The number of potential epitopes, however, is substantially lower than for MHC class I epitopes. Surprisingly, there were no strong binding neo-epitopes predicted for the mutated peptide p53 R175H, which is most frequently responded to peptide in the investigated patient cohort.
Sequencing results revealed that 20 of the 24 tested patients (83%) carried KRAS and/or TP53 mutations in their primary tumors and/or metastasis (Figure 1D, Supplementary Table 3), whereas no BRAF mutations could be detected. TP53 (75%, 18 patients) mutations were more abundant than KRAS mutations (29%, 7 patients). Moreover, 11 patients’ tumors and/or metastases (46% of all patients analyzed) harbored mutations that were included in the panel of long peptides. Correlation of the ELISpot results with the abundance of TP53 and KRAS mutations identified in the patients’ primary tumors and metastases revealed that patients carrying a mutation that was tested for by ELISpot assay, were more likely to be responsive against the respective mutated peptide and sometimes showed a concomitant response against the respective wt peptide, than patients carrying other mutations in their tumors (Figure 1E). Instead, the latter patients showed a higher proportion of responses against respective wt peptides only (19% compared to 0 % for patients carrying panel mutations). Taken together, our data provide evidence for spontaneous immunogenicity of Tp53 and Kras hotspot mutations expressed in patients with primary and metastasized colorectal cancer.
Mutation-specific T cell responses induced by multiple long-peptide vaccination against mutated TP53 and KRAS sequences in HLA-transgenic mice
Since we identified the existence of memory T cell responses in CRC patients against wt and mutated peptide sequences derived from hotspot mutation sites of KRAS, TP53 and BRAF genes, we were encouraged to investigate the therapeutic potential of these sequences. For active vaccination, mixes of four to five mutated peptides or mixes of corresponding wt peptides (Figure 2A) were tested in HLA-I/II double transgenic A2.DR1 dtg (Figure 2) as well as C57BL/6 mice (Supplementary Figure 4). To initially screen for the immunogenicity of all peptides in the whole panel, animals were immunized with PBS-based formulations of peptide mixes and the adjuvant CpG ODN1668 twice on a bi-weekly basis. Groups of three gender and age-matched mice were vaccinated with one out of six groups of peptides and analyzed on day 28 after the first vaccination. Purified T cells from immunized mice were Ag-specifically re-stimulated for 16 h on peptide-pulsed DCs. Ag-specific T cell responses were assessed via the production of the cytokines IFN-γ, IL-2 and/or TNF-α by FACS-based cytokine secretion assays and/or intracellular cytokine staining.
Figure 2.
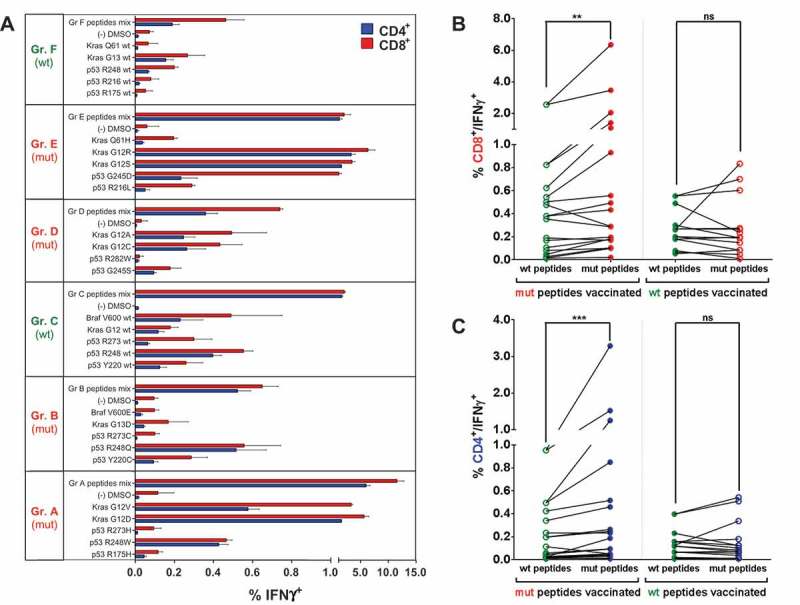
Mutation-specific, polyvalent CD4+ and CD8+ T cell responses after vaccination with mutated and wt p53 and Kras derived peptides. (A) Overview of recall responses against all peptides tested in HLA-humanized A2.DR1 dtg mice immunized with different long peptide cocktails. Six cohorts of mice were immunized with one group of mutated (mut) or wild-type (wt) peptides (A to F), each, in PBS-based formulations including 50 μg of CpG ODN 1668 as an adjuvant, twice on a bi-weekly bases. In vitro recall responses were obtained from combined IFN-γ secretion assays and intracellular cytokine stainings performed with CD90+ purified T cells from immunized mice. IFN-γ secretion of CD8+ and CD4+ T cells upon in vitro recall against single peptides of each respective mix (single) and against whole peptide mixes (mix) presented by CD11c+ DCs are displayed. Each peptide, peptide mix and control sample was tested in triplicates. Results are plotted as means of triplicate assays ± SEM. Data obtained from one of two identical lines of experiments are shown. (B)/(C) Mutation specific responses after vaccination of A2.DR1 dtg mice with mutated peptides. Percentages of IFN-γ secreting splenic CD8+ (B) and CD4+ (C) T cells upon in vitro recall against mutated (mut) and wild-type (wt) peptides represented on CD11c+ DCs. Results were accumulated from six cohorts of mice immunized with one group of mutated (mut) or wild-type (wt) peptides (A to F, see Figure 2 (A)). Means of mutated and wt peptide triplicates are plotted and groups were tested for differences by applying two-tailed Wilcoxon signed rank test. Representative data obtained from one of two identical lines of experiments are shown. Filled dot symbols: peptides used for vaccination and in vitro recall response testing, open dot symbols: peptides used for in vitro recall response testing only, ns: not significant.
In vitro IFN-γ recall responses of vaccination-induced splenic T cells against each of the individual wt and mutated peptides tested, as well as responses towards the whole peptide mixes used for immunization, are displayed in Figure 2A for MHC-humanized A2.DR1 dtg mice. The same analysis was performed with immunized C57BL/6 wt mice (Supplementary Figure 4). We detected in vitro recall responses against several peptides in the same mix, indicating the induction of T cell responses against multiple epitopes. These observations proved to be true for both mutated and wt peptide mixes as well as for both mouse strains, demonstrating the processing and presentation of the tested long peptide Ags in a human and murine MHC context. Furthermore, some of the long peptides were highly immunogenic in A2.DR1 dtg (and C57BL/6) mice, and thus efficiently induced CD8+ as well as CD4+ T cell cytokine responses. Interestingly, the strongest responses were elicited by similar peptides in both, the human and the murine MHC context. In C57BL/6 mice the mutated peptides p53 R248W/Q, p53 G245S, and Kras G12V/D/wt were repeatedly highly stimulatory (Supplementary Figure 4A). Similarly, the peptides p53 R248W/Q and Kras G12V/D/R excelled in the human MHC context of A2.DR1 dtg mice (Figure 2A). Since the used wt Kras sequences are identical in human and mouse, murine immune responses against long Kras peptides likely represented a true breaking of self-tolerance.
We next addressed whether there was a general difference between the immunogenicity of mutated and wt peptides when employed for active vaccination. Therefore, a cumulative analysis of in vitro recall responses towards wt and mutated peptides after vaccination with mutated or wt peptide cocktails was carried out. The readout of in vitro recall responses after vaccination included the peptides used for vaccination and the testing of T cell responsiveness towards their corresponding wt or mutated peptide counterparts. The same testing was conducted vice versa with T cells from wt peptides vaccinated mice. All T cell responses from mutated and wt peptide vaccinated mice were accumulated in two groups, respectively, namely cytokine production induced by mutated (Figure 2B and C – column labeled with ‘mut peptides’) peptides and corresponding wt (Figure 2B and C – column labeled with ‘wt peptides’) peptides. The analysis was done for IFN-γ secretion of splenic CD8+ (Figure 2A) and CD4+ (Figure 2B) T cells, which resulted in the same tendencies for both MHC contexts (for C57BL/6 data see Supplementary Figures 4 B and C). Upon vaccination with mutated peptides the cumulative CD4+ and CD8+ T cell responses against mutated peptides were significantly higher than those towards wt peptides in A2.DR1 dtg mice (CD4+ p = 0.0015, and CD8+ p = 0.0040), whereas there were no significant differences between the percentages of IFN-γ positive T cells towards wt and mutated peptides after vaccination with wt peptides. The same results were obtained in a murine MHC context with C57BL/6 mice (CD4+ p = 0.0002, and CD8+ p < 0.0001).
In addition to splenic T cells, T cells purified from the vaccination site draining lymph nodes (LNs) were analyzed (exemplarily shown for CD4+ T cells in Supplementary Figure 5A). Pooled IFN-γ secretion responses of LN T cells of A2.DR1 dtg mice also indicate a mutation specificity after vaccination with mutated peptides (p = 0.0166). The same trend was found when analyzing intracellular TNF-α accumulation after vaccination with mutated peptides for splenic CD4+ T cells (Supplementary Figure 5B, (p = 0.0451)) and CD8+ T cell responses. Taken together, active vaccination with oncogene-mutation harboring long peptides generated polyvalent, mutation-specific CD4+ and CD8+ T cell responses in both a murine and a human MHC context.
Along with several direct tumor-eradicating functions, CD4+ T cells are prone to interact with and provide help to CD8+ T cells. To assess the significance of CD4+ TH cell help, differentially purified T cell subsets from immunized mice were tested in IFN-γ secretion assays. Data were obtained from vaccination of six cohorts of either A2.DR1 dtg or C57BL/6 mice with the wt and mutated peptide cocktails. Splenic T cells were either purified for CD8+ or CD90+ T cells (comprising CD4+ and CD8+ T cells) and tested for in vitro recall responses against respective wt and mutated peptides in IFN-γ secretion assays. Percentages of IFN-γ positive T cell subsets responsive against wt and mutated peptides presented by CD11c+ BM-derived DCs were cumulated for this analysis (data obtained from A2.DR1 dtg mice and C57BL/6 mice are shown in Supplementary Figures 4D and E, respectively). CD4+ T cells were the most responsive T cell subset tested, showing the highest percentages of IFN-γ secretion compared to those of co-cultured splenic CD8+ T cells (p = 0.0041 for A2.DR1 dtg, p = 0.0080 for C57BL/6) and CD8+ T cells re-stimulated alone (p = 0.0019 for A2.DR1 dtg, p = 0.0115 for C57BL/6). Furthermore, the presence of CD4+ T cells enhanced the in vitro responsiveness of co-cultured CD8+ T cells compared to CD8+ T cells re-stimulated on DCs in the absence of CD4+ T cells. This was indicated by a significantly higher IFN-γ secretion of CD8+ T cells derived from the co-culture of CD90+ CD4/CD8 T cells as compared to CD8+ T cells re-stimulated in the absence of CD4+ in CD57BL/6 mice (p = 0.0301) as well as in A2.DR1 dtg mice (p = 0.0415). Taken together, the presence of CD4+ T cells in in vitro cultures enhanced CD8+ T cell in vitro IFN-γ secretion and argues for the significance of Ag-specific T helper cells to sustain an efficient CTL response towards TSAs.
Vaccination-induced, mutation-specific T cells are poly-functional
After screening the peptide panel for immunogenicity, we investigated in more detail whether mutated peptide-specific T cell responses showed signs of poly-functionality and cytotoxic properties. Therefore, A2.DR1 dtg mice were vaccinated with previously identified, highly immunogenic long mutated peptides (p53 R248W/Q and Kras G12V/D/R). T cells from vaccinated mice were purified and re-stimulated in vitro for 16 h on peptide pulsed DCs, and analyzed in combined IFN-γ/IL-2 cytokine secretion assays. CD4+ and CD8+ T cells secreted high levels IFN-γ and IL-2 after re-stimulation with mutated peptides (Figure 3A). Importantly, vaccination with highly immunogenic peptides elicited cytokine double-positive, poly-functional CD4+ as well as CD8+ T cells (Figure 3A, quadrant Q2 and Figure 3D). In accordance with results shown above, the proportions of cytokine positive cells reacting towards the mutated peptides used for vaccination were higher than towards corresponding wt peptides as exemplarily shown for the whole peptide mixes and the p53R248W/p53R248wt peptide pair. Figure 3B to D summarize the response patterns towards the highly immunogenic peptides from triplicate assays analyzed for IFN-γ single positive (Figure 3B), IL-2 single positive (Figure 3C) and IFN-γ/IL-2 double positive (Figure 3D) T cells. Notably, peptides (see e.g. p53 R248W) eliciting a strong IL-2 secretion relative to other peptides also displayed higher proportions of IFN-γ secreting and cytokine double positive T cells. Similar observations were made in vaccination experiments employing C57BL/6 mice (Supplementary Figure 6).
Figure 3.
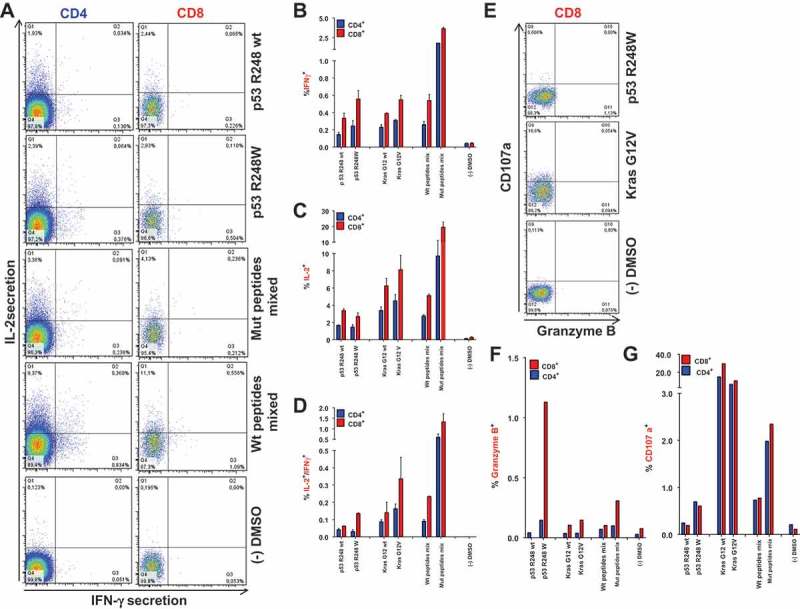
Vaccination-induced, mutation-specific T cells show poly-functionality. (A) IFNγ/IL-2 two-color cytokine secretion assay with splenic T cells purified from A2.DR1 dtg mice immunized with the most-immunogenic peptides (p53 R24Q/W, Kras G12V/D/R, vaccine regimen: peptides in PBS-based formulations including 50 μg CpG ODN 1668 as an adjuvant, twice on a bi-weekly basis). Cell surface markers were stained with fluorescently labeled monoclonal antibodies against IFN-γ (detection Ab), IL-2 (detection Ab), CD4, CD8, CD11c and NK1.1 (samples shown: T cells re-stimulated on DCs pulsed with wt peptide p53 R248 wt, mutated peptide p53 R248W, most immunogenic mutated peptides mixed; negative control: dendritic cells pulsed with peptide solvent (DMSO) only) . (B)-(D) Quantification of IFNγ/IL-2 two-color cytokine secretion assay shown in (A). In vitro recall responses were obtained from two-color cytokine secretion assays (IL-2, IFN-γ) with pan-T cells purified from immunized A2.DR1 dtg mice. Percentages of IFN-γ (B), IL-2 (C) and IFN-γ/IL-2 (D) double-positive of CD8+ and CD4+ T cells upon in vitro recall against single peptides, corresponding wt peptides and against whole peptide mixes (mix) presented by CD11c+ DCs are displayed. Results are plotted as means of triplicate assays ± SEM. (E) CD8+ effector T cells derived from Kras G12V and p53 R248W mutated peptides vaccinated mice express markers of cytotoxicity upon in vitro antigen-specific re-stimulation. Effector pan-T cells derived from A2.DR1 dtg mice vaccinated with Kras G12V/p53 R248W mutated peptides were in vitro re-stimulated for 12 h on different peptide pulsed DCs. Fluorescently-labeled anti-CD107a mAb and GolgiStop reagent were added to the co-culture. Thereafter, live-dead staining was performed and samples were fixed. Fixed cells were stained for markers granzyme B, IFN-γ, CD11c, CD4, and CD8 with fluorescent-labeled mAbs. CD107a plotted against granzyme B in CD8+ T cells are shown. Samples displayed: T cell re-stimulated on DCs pulsed with mutated peptide p53 R248W and mutated peptide Kras G12V, negative control: DCs pulsed with peptide solvent (DMSO) only. Percentages of granzyme B (F) and CD107a (G) positive of CD8+ and CD4+ T cells from all different peptide samples tested are shown.
To estimate the tumor eradicating properties of vaccination-induced CTLs, we also analyzed the expression of cytotoxicity and degranulation markers granzyme B, perforin and CD107a in T cells derived from multi-peptide vaccinated A2.DR1 dtg mice. When T cells were re-stimulated for 12 h on peptide-pulsed DCs, CD107a was highly expressed and to a lesser degree also granzyme B, whereas perforin was hardly detectable. Interestingly, re-stimulation with Kras G12 peptides (wt and mutated) led to relatively high proportions of CD107a positive cells (Figure 4G) in both CD8+ and CD4+ T cell populations. The p53 R248W peptide elicited the highest induction of granzyme B in CD8+ T cells (Figure 3E and F). Moreover, the proportions of granzyme B and CD107a positive CD4+ and CD8+ T cells were higher when they were re-stimulated with mutated mixed peptides and single mutated p53 R248W peptide pulsed DCs compared to re-stimulation with corresponding wt peptides, underlining once more the preponderance for mutation-specific CTL responses. Hence, CD8+ and CD4+ T cell responses, elicited by mutated long-peptide vaccination with Kras G12V and p53 R248W peptides, showed a poly-functional cytokine response pattern and cytotoxic features by in vitro analysis.
Figure 4.
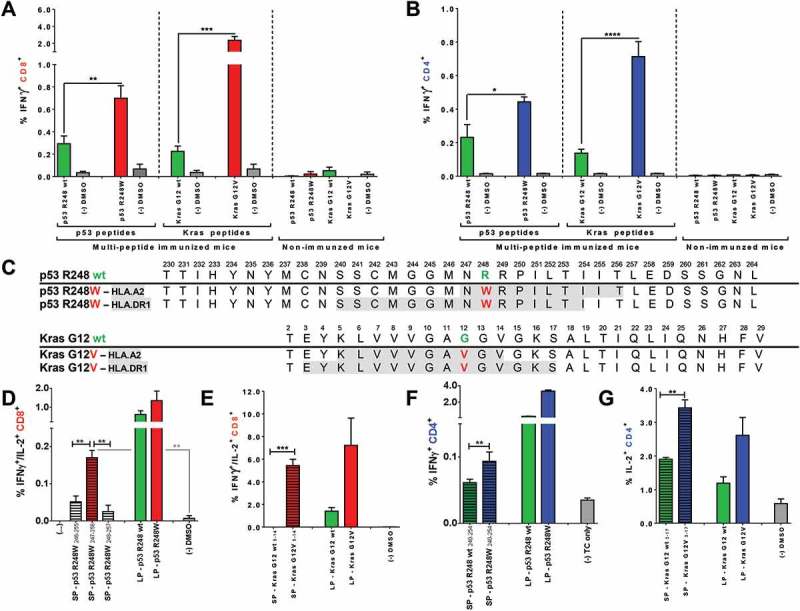
Mutated p53 R248W and Kras G12D/V peptides are more immunogenic than corresponding wt peptides when used for vaccination of A2.DR1 dtg mice and comprise HLA class I and II restricted epitopes. CD8+ (A) and CD4+ (B) T cell recall responses against mutated and wt peptides tested in A2.DR1 dtg mice immunized with group A and C long peptide cocktails (vaccination regimen: peptides in PBS-based formulations including 50 μg CpG ODN 1668 as an adjuvant, twice on a bi-weekly basis) or untreated mice are shown. In vitro recall responses were obtained from combined IFN-γ secretion assays and intracellular cytokine stainings performed with splenic CD90+ purified T cells from immunized mice. IFN-γ secretion of CD8+ T cells upon in vitro recall against respective single peptides presented by CD11c+ DCs are displayed. Each peptide and control sample was tested in triplicates. Results are plotted as means ± SEM. Differences were tested for by unpaired, two-tailed t-test. (C) Putative class I and II epitopes (highlighted in gray) within the long p53 R248W and Kras G12V peptide sequences. (D) – (G) T cell recall responses against mutated and wt Kras G12(V) and p53 R248(W) peptides tested in A2.DR1 dtg mice immunized with the long peptides Kras G12V or p53 R248W. In vitro recall responses were obtained from two-color cytokine secretion assays performed with splenic pan-T cells from immunized mice. Percentages of IFN-γ/IL-2 (D, E) double-positive CD8+ (C, D) and IFN-γ (F), IL-2 (G) positive CD4+ T cells upon in vitro recall against short and long Kras or p53 peptides presented by CD11c+ DCs are displayed. Each peptide and control sample was tested in triplicates. Results are plotted as means of triplicate assays ± SEM. Differences were tested for by unpaired, two-tailed t-test. ns: not significant.
Highly immunogenic mutated peptides carrying the mutations p53 R248W and KRAS G12V comprise HLA-A2- and HLA-DR1-restricted CD8+ and CD4+ T cell epitopes
In order to identify and validate the most immunogenic mutated oncogene-derived sequences within the panel of long peptides, the responses towards mutated peptides were directly compared to those of corresponding wt counterparts. In the HLA-A2/HLA-DR1 restricted context this comparison identified the peptides p53 R248W and Kras G12V to be more immunogenic than their wt counterparts when used for vaccination as the proportions of IFN-γ secreting CD8+ (Figure 4A) and CD4+ (Figure 4B) T cells upon in vitro re-stimulation were significantly increased in this context. Spontaneous responses against the peptides p53 R248W and Kras G12V were negligible, as IFN-γ secretion of T cells purified from non-immunized animals did not exceed negative assay controls (depicted on the right-hand side panel of each graph in Figure 4A and 4B). After identification of the two most immunogenic mutated Kras and p53 derived peptide sequences (2 out of the total 19 mutated peptide sequences tested), we next determined possible epitopes that could be processed from the long peptides and presented in the employed HLA context. To identify HLA binding minimal epitopes, the two long peptide sequences were subjected to in silico prediction algorithms of the NetMHC57,58 and the SYFPEITHI59 databases. Affinity scores for all overlapping 9-mer, 10-mer and 15-mer small peptides possibly processed from the long peptides and binding to HLA-A*0201 and HLA-DRB1*0101 were predicted and affinity scores for wt and mutated sequences were compared. Subsequently, high affinity predicted short peptide candidates were synthesized and tested in in vitro re-stimulation assays with T cells derived from respective long peptide vaccinated mice.
In silico algorithms predicted higher HLA-A*0201 (HLA-A2) binding affinity values for 10-mer minimal epitopes than for 9-mer peptides of the long Kras G12V peptide. The 10-mer peptides with the highest affinities within the wt and the mutated Kras G12V long peptide sequence encompassing the mutation site are listed in Supplementary Table 4. Although both wt and mutated 10-mers were not predicted to be strong binders, the mutated 10-mer peptide scored with a higher affinity for HLA-A2 than the corresponding wt 10-mer (NetMHC scores: 300.18 nM versus 506.91 nM, SYFPEITHI scores: 24 versus 22). In contrast to the moderate predicted binding affinities of potential class I epitopes, strong 15-mer MHC class II binders for the HLA-DRB*0101 (HLA-DR1) alloform within the long wt and mutated Kras peptides were predicted (Supplementary Table 4). The four peptides were synthesized and tested in in vitro re-stimulation assays. T cells purified from mice immunized with the two highly immunogenic mutated long peptides p53 R248W and Kras G12V were re-stimulated on DCs pulsed with putative wt and mutated short epitopes or respective long peptides. T cell responses after in vitro re-stimulation were assessed by combined IL-2/IFN-γ secretion assays. While the wt 10-mer peptide elicited a weak CD8+ T cell cytokine response, the corresponding mutated Kras G12V 10-mer peptide generated a strong response by IFN-γ/IL-2 double-positive CD8+ T cells (Figure 4E). Moreover, the strength of the cytokine response towards the short, mutated peptides was comparable to that of the original mutated long peptide. Additionally, the mutated predicted MHC class II 15-mer binder elicited significantly higher levels of IL-2 secretion in CD4+ T cells than the respective 15-mer wt peptide (Figure 4G). Unlike the minimal class I wt epitope, the 15-mer wt peptide, albeit of lower immunogenicity than the mutated peptide, was able to restimulate cytokine responses similar to those observed against the 28 aa long Kras G12 wt peptide. This suggests that the C-terminal portion of the peptide was not important for binding to HLA-DR1. On the other hand, A2-restricted responses against the long Kras wt peptide, that were not mirrored in responses against the short peptide, may have been directed against C-terminal cleavage products of the long peptide, e.g. the high-scoring 10-mer Kras 20–29. Our findings collectively suggest that the predicted Kras G12V 10-mer and 15-mer peptides are indeed immunogenic HLA-I and II restricted epitopes within Kras.
Like for the Kras peptide, in silico prediction was performed for the long, highly-immunogenic p53 R248W peptide. The resulting predicted strongest minimal MHC class I and II binders, however, failed to elicit T cell responses in vitro re-stimulation experiments. As an alternative approach epitope mapping with overlapping short peptides was performed. The HLA-DRB*0101 and HLA-A*0201 affinity scores of all possible overlapping, MHC class II and I binding wt and mutated peptides are listed in Supplementary Table 5. Overlapping 9-mer, 10-mer and 15-mer p53 R248(W) wt and mutated peptides were tested in vitro for their potential to re-stimulate T cells purified from A2.DR1 dtg mice immunized with the mutated, long peptide p53 R248W and were compared to recall responses against the respective long peptides. Data obtained from IFN-γ/IL-2 two color cytokine secretion assays revealed that none of the overlapping 9-mer peptides elicited a cytokine response above background controls as exemplary shown in Supplementary Figure 7A. However, 10-mer peptide SP-p53 R248W247-256 elicited significantly higher percentages of IFN-γ/IL-2 double positive (Figure 4C) CD8+ T cells when compared to background controls (for whole 10-mer epitope mapping see Supplementary Figure 7B) although it did not score as a putative HLA-A2 ligand (Supplementary Table 5). Testing of overlapping mutated 15-mer peptides comprising the mutation site p53 R248W did not yield a clear candidate peptide in two consecutive, identically performed experiments. Five mutated 15-mer peptides, however, elicited stronger cytokine responses than all other 15-mers tested and were therefore chosen for revalidation in a third experiment in which they were tested against their corresponding wt 15-mer counterpart peptides (see Supplementary Figure 7C and D). Among them, the 15-mer p53 R248240-254, elicited the strongest response when tested against its corresponding minimal epitope wt counterpart (Figure 4E).
Figure 4C highlights the potential minimal HLA-A2 and HLA-DRB1 ligands identified within the sequences of the long peptides p53 R248W and Kras G12V. HLA class I and II potential minimal epitope sequences are overlapping for the p53 peptide, whereas the HLA class I restricted Kras G12V 10-mer is nested within the putative longer 15-mer HLA class II epitope. Nested and overlapping MHC class I and II epitopes have previously been suggested as a general phenomenon to provide efficient Ag presentation to both CD4+ and CD8+ T cells by the same DC and thereby ensure proper priming of naïve T cells.38,60
Vaccination with highly immunogenic mutated peptides induces antigen specific Treg cells and accelerates tumor growth
To investigate the tumor protective capacity of the mutated oncogene-derived long-peptide vaccine in vivo we generated syngenic A2.DR1 dtg sarcoma lines by carcinogen-induced tumorigenesis35 and subsequent passaging in vitro and in vivo within immune deficient NOD/SCID mice and immunocompetent A2.DR1 mice (see Supplementary Figure 8A). Several A2.DR1 dtg sarcoma cell lines were established and characterized by histology to be fibrosarcomas displaying a spindle-like morphology, expression of the mesenchymal marker vimentin, and the endothelial marker CD29/integrin β1 (exemplarily shown for sarcoma line 2277-NS35 in Supplementary Figures 8B and 5C, respectively). CD31 expression of in vivo established tumors suggested their micro-vascularization. Furthermore, the sarcoma cell lines expressed differing levels of HLA class I, whereas no HLA class II expression was detected or could be induced through IFN-γ treatment. Mouse Tp53 and/or Kras mutations were found in numerous tumor cell lines but none of them expressed the hotspot mutations of interest (Figure 4, Supplementary Table 6). We therefore stably introduced a doxycycline (DOX)-inducible transgene for KRAS G12V and TP53 R248W joined by a linker and fused to an HA-tag into the Rosa26 locus of the cell line 2277-NS35 by site-directed recombination (Supplementary Figure 8D-G, Supplementary Figure 9).
Prior to launching tumor challenge experiments, different clinically relevant adjuvant combinations were tested to maximize vaccine-induced, anti-tumor T cell responses. Therefore, we tested two clinically relevant incomplete Freund’s adjuvants (IFAs), the mineral oil based Montanide ISA 51 and the non-mineral oil Montanide ISA 720, in different combinations with TLR ligands CpG or poly(I:C) (Supplementary Figue 10A). CpG and poly(I:C) have been pre-dominantly used as adjuvants in clinical trials, because they not only ensure optimal DC priming but were also shown to increase the Teff/Treg ratios.61 Moreover, water-in-oil emulsions known as IFAs protect peptide antigens from degradation, facilitate a slow and lasting release depot, and mediate immune-activating effects. Although previously found to lead to fatal sequestration and deletion of TAA-specific CTLs when used for vaccination with minimal epitopes, IFAs are superior adjuvants when used in combination with long synthetic peptides, that require processing by APCs.62 In our experimental system the formulation of peptides emulsified in Montanide ISA 720 together with 50 µg of CpG ODN 1668 (Supplementary Figure 10B and C) elicited the highest effector T cell responses and was therefore used for preventive vaccination prior tumor challenge.
For tumor challenge experiments groups of age- and gender-matched A2.DR1 dtg mice were vaccinated with either mutated (Kras G12V, p53 R248W), wt (Kras G12 wt, p53 R248 wt), or irrelevant (murine IgG sequences comprising peptides IgG47-81, IgG273-304) peptides formulated in Montanide ISA720/CpG ODN 1668 in a preventive setting three times prior tumor inoculation. During the challenge experiments, mice were boosted twice with peptide/CpG ODN 1668 vaccinations, two weeks and four weeks after tumor inoculation (Figure 5A).
Figure 5.
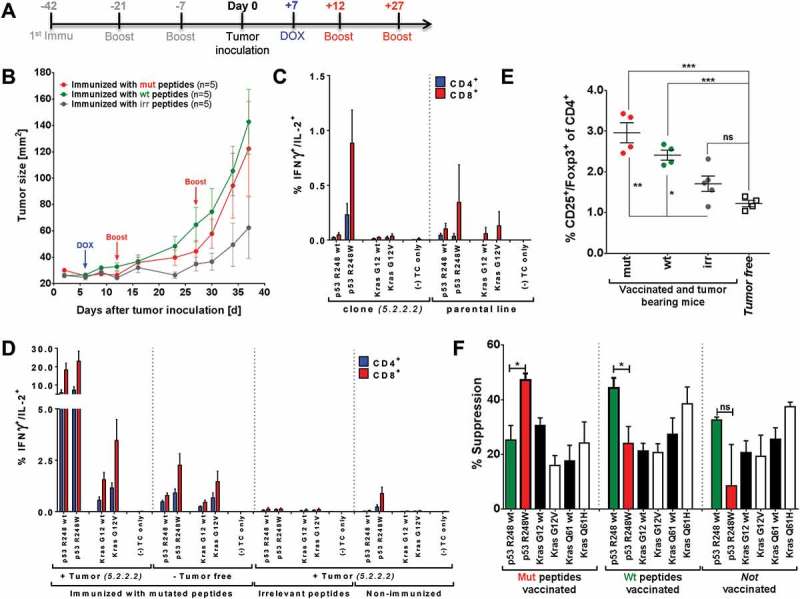
Tumor protection after vaccination with highly immunogenic peptides can be hampered by induction of mutation-specific regulatory T cells. (A) Vaccination schedule for tumor challenge experiments. Depot: IFA-based formulation, Boost: boost vaccination containing CpG ODN 1668 and peptides in PBS (water-based formulation). (B) Tumor growth curves of differently vaccinated A2.DR1 dtg mice inoculated with a 1:1 mixture of 2277-NS clones 5.2.2.1 and 5.2.2.2. In total 5 × 105 tumor cells were subcutaneously administered in 100 µl of Matrigel on the right flank of each animal on day 0. Three groups of 5 A2.DR1 dtg mice each (n = 5), have been vaccinated prior to the challenge according to the vaccination schedule shown in (A) with either mutated (‘mut’ group: p53 R248W, Kras G12V), wt (‘wt’ group: p53 R248 wt, Kras G12 wt), or irrelevant (‘irr’ group: IgG47–81, IgG273-304) peptides. Beginning on day 7 after tumor inoculation all groups of mice were fed 2 g/l DOX in the drinking water ad libitum until the end of the challenge experiment. Mice were boosted twice with respective water-based peptide/CpG formulations during the challenge. n: number of biological replicates; error bars: mean ± SEM (C)/(D) T cell responses against mutated and wt peptide sequences tested for in A2.DR1 dtg mice immunized with p53 R248W and Kras G12V mutated peptides or non-vaccinated mice either challenged with 2277-NS clone 5.2.2.2, parental line NS-2277 or from tumor-free mice are shown. In vitro recall responses were obtained from two-color cytokine secretion assays (IL-2, IFN-γ) with pan-T cells purified from immunized or non-vaccinated animals Percentages of IFN-γ or IFN-γ/IL-2 positive CD8+ and CD4+ T cells upon in vitro recall against single wt and mutated peptides presented by CD11c+ DCs are displayed. Each peptide and control sample was tested in triplicates. Results are plotted as means of triplicate assays ± SEM. (E) Percentage of splenic Treg cells in differentially vaccinated tumor bearing mice compared to non-tumor bearing mice on the day of sacrifice (day 38 of the tumor challenge experiment). Treg cells were stained for in whole splenocyte suspensions with fluorescent-labeled mAbs as CD4+CD3+CD25+Foxp3+ living cells in FACS. P values as per unpaired, two-tailed t-test are shown. irr: irrelevant, mut: mutated. (F) Mutated peptide p53 R248W and the corresponding wt peptide p53 R248 wt enhance antigen-specific Treg cell activity when used for vaccination. The antigen-specific activity of Treg cells purified from the spleen and lymph nodes of C57BL/6 mice vaccinated with mutated or wt peptides was analyzed in Treg specificity assays. Groups of n = 3 mice were vaccinated with either wt or mutated peptide mixes. Purified Treg cells were re-stimulated on CD11c+ DCs pulsed with single peptides of the same mix used for vaccination and corresponding wt or mutated peptides. The next day OVA323-339 antigen-specifically activated, proliferating OT-II CD4+ conventional T cells were added to the culture and proliferation was measured additional 2 days later via the uptake of radioactive labeled 3H-thymidine employing a scintillation counter. Respective control samples containing only peptide pulsed DCs and activated conventional OT-II T cells but no purified regulatory T cells were handled identically. This leads to two sets of triplicate data per tested peptide: one with Treg cells added and one without Treg cells. Percentages of specific suppression were calculated by using c.p.m. values of the different assay plates. Differences were tested for by unpaired, two-tailed t-test. ns: not significant.
Preventively vaccinated mice were inoculated with 5 × 105 double mutant Kras/Tp53-HA-transgenic 2277-NS sarcoma cells subcutaneously into the flank of each animal. One week after tumor inoculation (day 0) transgene expression was induced followed by two boost vaccinations (peptides + 50 µg CpG ODN 1668 in PBS – week 2, week 4 after tumor inoculation).
The tumor growth curves of the differentially vaccinated groups of mice reveal an unexpectedly accelerated tumor outgrowth starting day 14 post tumor-inoculation after vaccination with mutated (red curve) and wt (green curve) peptides (Figure 5B). To elucidate the mechanism that facilitates the unexpected vaccine-induced accelerated tumor growth, we assessed the functionality of effector CD4+ and CD8+ T cells in late-stage tumor bearing mice on the day of sacrifice by two-color IFN-γ/IL-2 cytokine secretion assay. When testing for mutation-specific IFN-γ responses in non-vaccinated tumor bearing mice (inoculated with either the parental cell line or the 2277-NS clone expressing specific mutations), recall responses especially against the mutated p53 R248W peptide were detected (Figure 5C, left panel; of note, the parental cell line used for genetic engineering harbors a mutation at position 248 of Tp53 (R248H), which might explain a p53 R248W cytokine response towards the parental cell line.) This result suggests spontaneous immunogenicity of the introduced mutations in the tumor in vivo and argues for a natural processing of mutated tumor Ags. Further evidence was provided by the observation that vaccination-induced preexisting polyfunctional T cells were highly active after in vitro re-stimulation (Figure 5D, very left panel). Importantly, the percentages of tumor-induced IL-2/IFN-γ double positive, mutated TA-specific T cells in non-vaccinated mice or in mice vaccinated with irrelevant peptides were much lower than those after vaccination in non-tumor bearing mice (Figure 5D, right panels and middle left panel, respectively), clearly indicating the effect of vaccination. The highest percentage of restimulated T cells reactive against p53 R248(W) or Kras G12(V) peptides was found in vaccinated tumor-bearing mice (Figure 5D, very left panel), suggesting that vaccination-induced effector T cell responses in tumor-bearing mice were not hampered but rather boosted in the presence of the tumor. Moreover, in vaccinated tumor bearing mice mutation specificity of T cell responses against each mutated epitope were preserved. In addition, the vaccination with long peptides also induced robust CD8+ and CD4+ T cell responses specific for MHC-I/II-restricted wild type epitopes since they could be restimulated using the p53 R248 wt long peptide.
As the in vitro effector T cell responses were not hampered, but boosted in tumor bearing mice, we next assessed the number of regulatory T cells (Treg) in the periphery of challenged animals on the day of sacrifice (see Figure 5E). Notably, the number of splenic Treg cells was significantly increased in tumor-bearing animals vaccinated with mutated (red group) and wt (green group) peptides compared to tumor bearing mice vaccinated with irrelevant peptides (grey group) or to completely naïve mice (square symbols group). Therefore, we wondered whether Ag-specific effector T cells were induced by the vaccination regimen along with Ag-specific Treg cells. To address this question, we established a Treg cell specificity assay for murine T cells which is based on an increased suppressive activity of Treg cells after antigen specific TCR stimulation.63 In this assay, Treg cells purified from untreated C57BL/6 mice or from mice vaccinated with mutated or wt peptides were re-stimulated with peptide-pulsed DCs in vitro and their capacity to suppress the proliferation of activated ovalbumin-specific CD4+ effector T cells (OT-II Teff cells) was determined. Representative data from one out of three suppression assays are shown in Figure 5F. Whereas there were no obvious differences in suppression after re-stimulation with Kras peptides, an increased suppression of effector T cells by Treg cells derived from mutation vaccinated mice were detected after re-stimulation with mutated peptide p53 R248W. A similar observation was made for the corresponding p53 R248 wt sequence and Treg cells purified from wt peptide-vaccinated mice.
We therefore conclude that vaccination with highly immunogenic wt and mutated long peptides like p53 R248W, harboring CD4+ T cell epitopes, might comprise the risk to induce a dominant, immune-inhibitory response by induction of Treg cells during preventive vaccination. These Ag-specific Treg cells may expand as a consequence of systemic immune suppression elicited by the growing tumor, which accelerates tumor outgrowth rather than delaying it due to a very efficient Ag-specific inhibition of TSA-specific effector T cells.
Vaccination with low immunogenic mutated tumor peptides delays tumor growth without Treg cell expansion
Based on the observations above, we returned to our bank of A2.DR1 tumor cell lines and screened for lines with intrinsic Kras and Tp53 mutations that may be less immunogenic. One cell line (sarcoma line 39) carrying an MCA-induced intrinsic Kras mutation at position G12 (Kras G12C) and a p53 mutation (p53 C176F) was chosen for further tumor challenge experiments (Figure 6A). Consequently, long peptides were synthesized carrying these two mutations (Figure 6B) and tested for their potential for active vaccination. Both of the mutated peptides elicited mutation-specific responses when compared to their wt counterparts in in vitro cytokine secretion assays with T cells from mutated peptides vaccinated mice (Figure 6C). The strength of the cytokine response towards the p53 peptide comprising the mutation C176F, however, was considerably lower than the response towards the strongly immunogenic peptide p53 R248W (Supplementary Figure 11).
Figure 6.
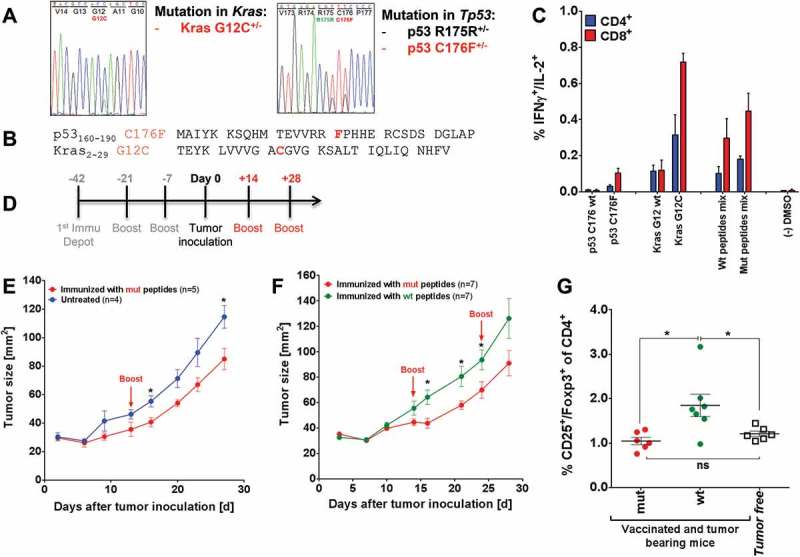
Mutated peptide vaccination induced T cell responses against intrinsic mutations in A2.DR1 dtg sarcoma and was superior to wt vaccination in controlling in vivo tumor growth (A) Sequencing histogram analysis of sarcoma cell line 39. Chromosomal DNA as well as cDNA of line 39 were used as templates for amplification with gene specific primers (Kras and Tp53). Amplified PCR products were analyzed by Sanger sequencing to detect mutations in Kras and Tp53 genes and mRNAs. (B) Amino acid sequences of the mutated peptides carrying intrinsic mutations used for vaccination. (C) Recall responses against mutated and wt peptides sequences tested in A2.DR1 dtg mice immunized with p53 R176F (mur) and Kras G12C mutated peptides (vaccination regimen: peptides in PBS-based formulations including 50 μg CpG ODN 1668 as an adjuvant, twice on a bi-weekly basis). In vitro recall responses were obtained from two-color cytokine secretion assays (IL-2, IFN-γ) with pan-T cells. Percentages of IFN-γ/IL-2 double positive CD8+ and CD4+ T cells upon in vitro recall against single wt and mutated peptides presented by CD11c+ DCs are displayed. Each peptide and control sample was tested in triplicates. Results are plotted as means of triplicate assays ± SEM. (D) Vaccination schedule for tumor challenge experiments. Depot: IFA-based formulation, Boost: boost vaccination containing CpG ODN 1668 and peptides in PBS (water-based formulation). (E) Tumor growth curves of vaccinated and untreated A2.DR1 dtg mice inoculated with sarcoma line 39. 2.5 x 105 tumor cells were subcutaneously administered in 100 µl of Matrigel on the right flank of each animal at day 0. One group of A2.DR1 dtg mice (n = 5) was vaccinated prior to the challenge according to the vaccination schedule shown in Figure 5 (D) with mutated (‘mut’ group: p53 C176F, Kras G12V) peptides. Mice were boosted only once with respective water-based peptide/CpG formulation during the challenge on day 14. n: number of biological replicates; error bars: mean ± SEM. Significances per unpaired two-tailed t-test are shown. (F) Tumor growth curves of differently vaccinated A2.DR1 dtg mice inoculated with sarcoma line 39. 2.5 x 105 tumor cells were subcutaneously administered in 100 µl of Matrigel on the right flank of each animal at day 0. Two groups of 7 A2.DR1 dtg mice each (n = 7), have been vaccinated prior to the challenge according to the vaccination schedule shown in (D) with either mutated (‘mut’ group: p53 C176F, Kras G12V) or wt (‘wt’ group: p53 C176 wt, Kras G12 wt) peptides. Mice were boosted twice with respective water-based peptide/CpG formulations during the challenge. n: number of biological replicates; error bars, mean ± SEM. (G) Number of splenic Treg cells of differentially vaccinated, tumor bearing mice of the experiment shown in (E) compared to non-tumor bearing mice on the day of sacrifice. Treg cells were stained for in whole splenocyte suspensions with fluorescent-labeled mAbs as CD4+CD3+CD25+Foxp3+ living cells in FACS. Significance per unpaired two-tailed t-test are shown. vac: vaccinated, mut: mutated.
When challenging preventively vaccinated mice (peptides/Montanide ISA 720/CpG ODN 1668 vaccine formulations) with sarcoma line 39 a growth delay in mutated-peptide vaccinated animals was observed compared to non-vaccinated (Figure 6E) and also wt peptide vaccinated mice (Figure 6F). Analyzing the frequency of peripheral Treg cells revealed that Treg levels were not elevated in tumor-bearing mice after vaccination with mutated peptides, only after vaccination with wt peptides, when compared to non-tumor-bearing mice (Figure 6G). This observation supports our conclusion that vaccination with highly immunogenic TSAs can lead to an induction of Ag-specific Treg cells as a natural counter-regulatory mechanism of the immune system.
Discussion
In the present study we investigated the cancer immunotherapeutic potential of synthetic long peptide sequences comprising common driver gene mutations at hot-spot sites of GI cancers in the oncogene KRAS and the tumor suppressor gene TP53. We identified mutation-specific T cell responses in non-vaccinated CRC patients, demonstrating their immunogenicity in vivo. Furthermore, we found in a humanized mouse model that vaccination with mixtures of long peptides comprising MHC class I and – II epitopes derived from mutant Kras and p53 protein variants simultaneously induce robust cytotoxic and T helper cell responses against multiple mutations. However, we also encountered an important caveat of this therapeutic approach by unraveling the induction of TSA specific Treg cells upon vaccination with strongly immunogenic wt and mutated MHC class II epitopes that could drastically hamper vaccine effectiveness.
To assess preexisting memory T cell responses towards the targeted TSAs, we analyzed Ag-specific IFN-γ responses in the BM and PB of CRC patients who had previously not undergone immunotherapeutic intervention. In line with our previous findings23,24,26, we detected both wt and mutated peptide-specific memory T cell responses that were polyvalent in the majority of patients. The responses against mutated peptides were overall higher than those against wt peptides arguing for mutation specificity of the tumor Ag specific T cell pool in CRC patients and for immunogenicity of the chosen mutations and long peptide sequences. Our observations are in accordance with investigations defining the preexistence of intrinsic and vaccination-induced TC responses against wt and mutated Kras15,40 and p5364,65 sequences in patients with gastrointestinal cancers.
Vaccination of HLA-transgenic A2.DR1 dtg mice with mixes of up to five different long peptides resulted in polyvalent CD4+ and CD8+ T cell responses against mutated Kras and p53 peptides. Hence, the multi-epitope vaccine targeting driver gene mutations met state-of-the-art requirements for TSA-based vaccines. Most of the latest and more advanced clinical trials for TSA-specific vaccines are based on the administration of Ag cocktails47,66. Another recent approach delivers multiple Ags as an individualized neo-antigen mRNA-polytope vaccine67-69 which represents a promising alternative to administration of antigens via synthetic peptides. Eliciting a broad immune response against several tumor-specific CD4 and CD8 epitopes has major advantages. It circumvents the problem of selecting for tumor Ag-loss variants11, broadens the immune response towards sub-dominant epitopes36, and induces CD4+ T cell responses that are indispensable to prime and sustain an effective anti-tumor CTL response34,70. We were able to re-validate the latter point in in vitro co-culture experiments. CD8+ T cells from vaccinated mice re-stimulated on peptide-pulsed DCs showed significantly higher IFN-γ production when co-cultured with CD4+ T cells than being re-stimulated on DCs in the absence of helper T cells.
Mutated peptides generally elicited higher CD8+ and CD4+ T cell responses than the corresponding wt peptides implying that the mutation-comprising peptides were recognized as foreign Ags endowed with higher immunogenicity than wt sequences. Vaccination induced CD8+ but also CD4+ T cells displayed a poly-functional profile, characterized by simultaneous IFN-γ and IL-2 expression and signs of cytotoxic activity in vitro. Moreover, according to their cytokine profile, CD4+ T cells presented a TH1-like phenotype. A related study focusing on the glioma antigen IDH-1 also showed the importance of vaccine induced TH1 cell responses for effective tumor control. In that study vaccination with a single mutated IDH-1 HLA-DRB1-restricted epitope elicited a TH1 effector CD4+ T cell response that was sufficient to delay the outgrowth of IDH-1 mutant sarcomas in A2.DR1 dtg mice.35
We further identified Kras G12V and p53 R248W as highly immunogenic mutated peptides in a human HLA-A2/HLA.DRB1 restricted context when used for active vaccination. Memory T cell responses against the very same mutations were also prominent in human CRC patients. However, p53 R175H is another highly immunogenic mutation in CRC patients but was only weakly immunogenic after active immunization in the two mouse strains. A possible explanation for this discrepancy might be attributed to differences in the antigen processing machinery or to differences in tolerance induction to intracellular self-antigens between humans and mice.
As strongly immunogenic mutated peptides elicited CD8+ as well as CD4+ TC responses, we concluded that these peptides harbor HLA-A2 and HLA-DRB1-restricted mutated epitopes that are naturally processed by APCs. Within the long Kras G12V peptide we validated the 10-mer Kras5-14 G12V to be HLA-A2-restricted18 and nested within the HLA-DRB1-restricted 15-mer Kras3-17 G12V that we herewith add to the list of class II alleles being able to present the Kras G12V mutation71. Conclusively, these promiscuous mutated Kras epitopes are naturally processed and presented by a wide range of mouse and human MHC class I and II alleles, which highlights them as distinguished Ag targets for vaccination. After in vitro epitope mapping we further suggested the short peptides 10-mer p53246-256 R248W and 15-mer p53240-254 R248W as novel, overlapping HLA-A2 and HLA-DRB1-restricted mutated epitopes. Together with C. Melief’s findings64, our results suggest that the region spanning amino acids 190 to 260 (center of the DNA binding domain) within the p53 protein seems to represent an immunogenic stretch.
To investigate the protective capacity of long peptide vaccination on in vivo tumor growth we developed an A2.DR1 dtg syngenic sarcoma model. We anticipated that a long peptide multi-epitope vaccine, being able to elicit polyvalent, mutation-specific, multi-functional CTL and TH1 responses would delay or prevent the outgrowth of sarcoma lines over-expressing the respective mutations when used in a preventive treatment setting. Unexpectedly, vaccination with wt and mutated peptides in the engineered highly immunogenic TSA system rather accelerated tumor outgrowth instead of delaying it compared to mice vaccinated with irrelevant peptides. Even though peripheral effector T cell responses of vaccinated mice were not hampered but boosted in tumor bearing mice (arguing for natural processing of mutated epitopes in vivo), they did not result in protection at the tumor site. In addition to increased numbers of cytokine responsive effector T cells, however, we detected significantly increased numbers of peripheral Treg cells in animals vaccinated with wt and mutated peptides during tumor challenge. Treg cell numbers in tumor challenge experiments were, however, not elevated after vaccination against lower immunogenic mutations. Here, mutation-specific vaccination delayed tumor outgrowth compared to vaccination with wt peptides. It was previously shown that vaccines are critically hampered by increased pre-treatment levels of Treg cells and that depletion of Treg cells can restore T cell function and vaccination success47,72. We previously demonstrated that TAA-specific Treg cells in the PB of cancer patients were able to suppress polyclonally activated T cells in an Ag-specific manner. Moreover, after depleting the PBMC T cell pool of Treg cells, effector T cell responses towards some of the tested TAAs increased29.
We here demonstrate that vaccination with peptides containing strongly-immunogenic MHC class II epitopes can induce and expand TSA-specific Treg cells and is associated with overall strongly increased Treg cell numbers in tumor bearing mice. In contrast, the number of Treg cells in non-tumor-bearing mice vaccinated with highly immunogenic peptides compared to mice vaccinated with lower immunogenic or non-vaccinated mice was not elevated. This suggests that vaccination induced TSA-specific Treg cells only detrimentally expand in the context of an established tumor environment. The tumor growth curves support this notion. Accelerated growth of highly immunogenic tumors started not before 15 d post tumor inoculation, since tumor growth did not differ between the groups during the first 14 d after challenge, while the tumor was establishing. Induction of Ag-specific Treg cells by vaccination was previously observed. In the field of autoimmune research, for example, induction of Ag-specific Treg cells is discussed as a therapeutic option73. In regard to cancer, P. Romero and colleagues reported increased Treg numbers in mice vaccinated against both a foreign (ovalbumin) and non-mutated model-tumor antigens61. Another study showed that ovalbumin-specific vaccine-induced Treg cells were able to home to the tumor microenvironment and suppress conventional CD4+ T cells, specific for the same model Ag74. We here demonstrate that TSA specific Treg cell expansion and activity was associated with high immunogenicity of the TSAs, as TSAs of lower immunogenicity did not induce Ag-specific Treg cells. However, we cannot exclude the possibility that in the tumor model with lower immunogenic, endogenous mutations the systemic Ag load may have been lower than in the tumor model with over-expressed highly, immunogenic mutations which may have potentially influenced the degree of Treg cell stimulation and recruitment to the tumor. Thus, our data does not allow a generalization of this observation but one can speculate that the induction of Ag-specific Treg cells may be a naturally occurring physiological consequence to counterbalance very strong effector T cell responses. Indeed, the levels of IFN-γ producing Tcon cells in our experimental system reaches the levels of T cell responses during viral infections.75–77 However, such a potentially beneficial effect converts into a disadvantage in a situation of tumor growth, as the tumor preferentially attracts and expands Treg cells in its microenvironment in an Ag-specific manner.
In conclusion, we here showed that poly-functional CD8+ and CD4+ helper type 1 effector T cells responses can simultaneously be induced against multiple mutated TSAs in the context of long peptide vaccines. Our data further suggest that increased immunogenicity of antigenic peptides can be associated with induction of deleterious TSA specific Treg cell responses, if the peptide sequences chosen contain highly immunogenic MHC class II epitopes. This phenomenon might be avoided by the use of less immunogenic TSAs, especially when targeting a TH cell response, or by therapeutic interventions that reduce the priming and expansion of Treg cells such as cyclophosphamide or anti-CTLA4 antibodies.
Materials & methods
Patient samples
Heparinized peripheral blood (PB) and bone marrow (BM) samples from metastasized 26 CRC patients were obtained after informed consent from the Surgical Clinic of Heidelberg University. The protocol was approved by the local Ethical Committee of the University of Heidelberg: ethic vote #323/2004.
Antigens
The most relevant mutations in TP53, KRAS and BRAF genes for colorectal and pancreatic cancer were chosen using the IARC TP53 Database (version R17, November 2013)52 and COSMIC database for somatic mutations in cancer (from the Sanger Institute, COSMIC v69 Release)51, respectively. Peptides were produced by the DKFZ core facility for peptide synthesis. After solid phase synthesis peptides were HPLC purified and lyophilized. Lyophilized peptides were either stored at −20°C or directly reconstituted in 100% DMSO to stock concentrations of either 25 mM (short and long peptides) or 10 mM (long peptides). Stocks were aliquoted (25 µl, 50 µl) and stored at −20°C. Possible HLA-A*0201 and HLA-DRB1*0101 ligands that might be processed from the long peptides were predicted using the SYFPEITHI59 and NetMHC57,58 algorithms. Lists of peptides used can be found in the Supplementary Tables 1, 4 and 4.
Exon sequencing from paraffin-embedded tissue
Exon sequencing was performed to screen for mutations in the TP53, KRAS and BRAF genes in CRC patients’ tumors and metastasis. Sequencing was performed by W. Weichert and R. Penzel at the Institute for Pathology of the University of Heidelberg. Briefly, slices from formalin-fixed and paraffin-embedded tissues were deparaffinized via xylol-ethanol-treatment. Tumor tissue-rich segments of the slices were selected to purify genomic DNA (QIAsymphony DNA Mini Kit, QIAGEN, 937236). Samples were sequenced in a fully-atomized fashion employing QIAsymphony SP/AS instruments (QIAGEN, 9001297/9001301).
Cell culture with primary human cells
Mononuclear cells were directly isolated by density gradient (Biocoll, Biochrom, L6115) employing 50 ml LeucosepTM tubes (Greiner, REF 227290). 20 ml sample volume was thereby diluted with 10 ml RPMI-1640 medium (Sigma-Aldrich, R8758). For adherence, cells in X-Vivo-20 medium (Lonza, BE04-448Q) were plated on Petri dishes for 30 min. Afterwards, the non-adherent cell fraction was removed by repeated washing with RPMI-1640 and further cultured in X-Vivo-20 supplemented with 100 IU/ml interleukin 2 (rHu IL-2 (proleukine), Novartis, DA1318BG) and 60 IU/ml IL-4 (rHu IL-4, Miltenyi, 130–093-924). Adherent monocyte precursors were matured to dendritic cells by culture in X-Vivo-20 medium containing 560 IU/ml of granulocyte macrophage colony-stimulating factor (GM-CSF, Sanofi, NDC0024-5843–05) and 1000 IU/ml IL-4 for 7 d.
Cell purification
DCs were purified from adherent cell cultures by depletion of negative cells via magnetic separation with Dynabeads Pan Mouse IgGs (Invitrogen, 110.42) coated with anti-CD3, anti-CD19 and anti-CD56 antibodies according to manufacturer’s instructions. From non-adherent cells cultures T cells were purified employing the Dynabeads Untouched Human T-Cell kit (Invitrogen, 113.44D).
INF-γ elispot assay
Interferon-γ (IFNγ) ELISpot assay for the detection of IFNγ secreting T cells were carried out as described previously27,30. Accordingly, total T cells were cultured on peptide-pulsed DCs in a 5:1 ration for 40 h. Assays were conducted in triplicates for each of the peptides tested. IFNγ secreting T cells were detected with the Human INFγ ELISPOT kit (Mabtech, 3420-2A) according to manufacturer’s instructions. Substrate reaction was stopped by washing three times with ddH2O. ELISpot plate’s membranes were allowed to air dry for 72 h in the dark. Plates were read facilitating a CTL ImmunoSpot reader (CTL Technologies) and analyzed with the ImmunoSpot software (CTL Technologies). As negative controls served wells with DCs pulsed with human IgGs (Sandoglobulin, CSL Behring, PZN-0571760).
Mice
H2 class I-/class II-knockout (β2m-deficient), HLA-A*0201/Dbα3 chimera (HHD) and HLA-DR1 double transgenic (A2.DR1 dtg) mice,78 C57BL/6J mice, OTII TCR tg mice, and NOD/SCID mice were bred locally under specific pathogen-free conditions in the animal core facility of the German Cancer Research Center (DKFZ), Heidelberg, Germany. A2.DR1 dtg were generated by F.A. Lemonnier and purchased from the Institute Pasteur, Paris. Mice of both genders (male and/or female, 6–10 weeks of age) were used in all experiments. During experimental procedures mice were kept in individually ventilated cage systems (IVC racks). The protocol containing all experimental (in vivo) procedures performed with mice in the present work was formally approved by the national animal care committee (Regierungspräsidium) Karlsruhe, Germany.
Vaccination procedure
Mice were vaccinated with different mixtures of two to five long peptides, always resulting in a total peptide amount of 50 nM per animal and vaccination shot. 5 µg of TLR9 ligand CpG ODN 1668 (5’- TCC ATG ACG TTC CTG ATG CT – 3’, ordered as HPLC-purified primer at Eurofins) per animal and shot was used as an adjuvant. CpG and peptides were either administered in water based (PBS) or in IFA (Incomplete Freund’s Adjuvant; MONTANIDETM ISA 720 VG sterile, Seppic, 36059V) based formulation. Mice were injected with 100 and 50 µl of vaccines subcutaneously either in the neck and the tail base (water-based formulations) or in the neck and the flanks (IFA-based formulations). Vaccinations were performed in two (water-based) or three (IFA-based) week intervals. Boost vaccination were always carried out by subcutaneous injection of water-based formulations into the neck with only half the amount of antigens and CpG every second week.
Tumor challenges
Six to eight weeks old mice were vaccinated in a preventive setting with a prime-boost regimen. One IFA-depot was set in the beginning followed by two to three water-based boost immunizations. Mice were inoculated with tumor cells one week after the last boost immunization. 2,5 x 105 cells of Line 39 or 5,0 x 105 of cells of 2277-NS clones were injected in 100 µl MatrigelTM Matrix (BD, 354234) subcutaneously into the previously shaved right flank of each animal. Every second week after tumor inoculation mice received additional boost vaccinations. In case of challenges with 2277-NS clones mice were fed with 2 g/l doxycycline (Doxycycline hydrochloride, Sigma-Aldrich, D9891-25G) in the drinking water starting on day 7 after tumor inoculation until the end of the challenge ad libitum. For better compliance of the bitter antibiotic, sugar was added to the drinking water. Tumor growth was monitored every three to four days by measuring tumor sizes with a digital caliper.
Generation and engineering of MCA-induced tumor cell lines
Several syngenic tumor cell lines for the A2.DR1 dtg mouse strain were generated with carcinogen-induced tumorigenesis through injection of the carcinogen 3-methylcholanthrene (MCA) as described previously35. Cells were characterized by H&E stainings with the kind help of Dr. Herpel from the tissue bank at the NCT, Heidelberg, Germany. Cell line 2277-NS was engineered to express p53 and Kras mutations in a transgene construct (scheme see Supplementary Figure 8). Transgenes were synthesized and introduced in pDonor221 Gateway entry vectors by the company Geneart (Life technologies).
Generation of 2277-NS acceptor cell lines
To generate stable 2277-NS acceptor cell lines, a vector containing a Flp recombinase target site, an antibiotic selection marker (neomycin, Sigma-Aldrich, 1405–10-3), an eGFP fluorescence marker as wells as homologous recombination sites for mRosa26 was stably integrated into the Rosa26 locus of the 2277-NS cell line by ZNF targeting (CompoZr® Targeted Integration Kit – mRosa26, Sigma-Aldrich, CTIM-1KT). Targeting of the mRosa26 locus was confirmed by junction PCR according to the manufacturer’s recommendations. The 2277-NS/Flp isogenic acceptor lines were further validated for single-copy integration of the FRT site.
Generation of 2277-NS transgenic clones expressing chimeric mutant Tp53/Kras transgenes
For the generation of 2277-NS mutant Tp53/Kras transgene expressing sublines, FRT expression vectors containing a hygromycin (InvitrogenTM, Life technologies, 10687–010) resistance gene, tet repressor gene (tetR) as well as a tetracycline-responsive CMV promoter were cloned upstream of the wt and mutant chimeric mutant Tp53/Kras transgenes’ ORFs, respectively, for inducible transgene expression. After sequence verification, the wt and mutated constructs were individually co-transfected together with a Flp recombinase expression vector (pOG44, InvitrogenTM, Life technologies, V6005-20) to integrate the respective expressions constructs into the mouse Rosa26 locus of the 2277-NS acceptor cell line by site specific recombination. After selection (hygromycin positive/eGFP negative), single colonies were picked and analyzed.
Preparation of tumors and organs from mice
At the end of tumor challenges tumors were excised from sacrificed mice. Tumors were either snap-frozen for preparation of protein extracts for Western blot, embedded in Tissue-Tek® (O.C.T.TM compound, Sakura, REF 4583) and frozen at −80°C, or used for preparation of single cell suspension for FACS staining. Spleens and lymph nodes were removed from vaccinated and tumor bearing mice. For preparation of bone marrow (BM), femur and tibia from hind limbs were resected from untreated mice.
Cell culture and purification of murine cells
All A2.DR1 dtg cell lines were cultured in DMEM (Sigma-Aldrich, D6429) supplemented with 10% FCS (Biochrom, S0415), 10 mM HEPES (Sigma-Aldrich, H0887) buffer 0.05 mM 2-Mercaptoethanol (Gibco, 31350–010) and 0.25 mg/ml Gentamicin (Sigma-Aldrich, G1397-100ML) (referred to as mouse medium). B16-F1 melanoma cells were in cultured DMEM supplemented with 10% FCS, 10 mM HEPES, 1.0 mM sodium pyruvate (Sigma-Aldrich, S8636), 100 U/ml penicillin/streptomycin (Gibco, G1397-100ML) and 0.05 mM 2-Mercaptoethanol. SW480 cells were gown in RPMI-1640 medium containing 10% FCS, 10 mM HEPES, 1.0 mM sodium pyruvate and 100 U/ml penicillin/streptomycin. All cells grew adherent and were split via trypsinization.
Single BM cell suspensions were prepared from bones using mortar and pistil. Bone suspension was filtered to 100 µm cell strainer to remove bone splinters. Afterwards, BM cell suspension was cultured for 6 d in mouse medium supplemented with 10 ng/ml murine GM-CSF (mouse GM-CSF, Miltenyi Biotec, 130–094-043) in petri dishes. Single cell suspensions from spleen and LN samples were made by mincing the cell through 40 and 100 µl cell strainers (BD Falcon, REF352340 and REF352360), respectively. Erythrocytes in spleen samples were lysed with 2.5 ml ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0,1 mM EDTA, pH = 7.2–7.4) per spleen for 1 min. Reaction was stopped with PBS and spun down.
DCs from BM cultures were purified positively with CD11c (N418) MicroBeads (Miltenyi, 130–052-001). T cells were purified either positively employing CD90.2 MicroBeads (Miltenyi, 130–049-001) or negatively with the Pan-T Cell Isolation Kit II (Miltenyi, 130–095-130) from spleen and LN of mice. CD8+ T cell subpopulation was gained from purified T cells using the CD8a (Ly-2) MicroBeads (Miltenyi, 130–049-401). Regulatory T cells were enriched from bulk T cell populations with the CD4+CD25+ Regulatory T cell Isolation Kit (Miltenyi, 130–042-201).
CD11c+ dendritic cells were always pulsed with 10 pmol of respective long peptides or 25 pmol of short peptides in X-Vivo-20 medium overnight. The next day purified T cells were added in X-Vivo-20 medium in a 1:5 ratio for short term in vitro restimulation for up to 20 h at 37°C and 5% CO2. For detection of intracellular cytokines or degranulation markers samples were treated with BD GolgiStopTM (BD Biosciences, containing monensin, 554724) in a 1:1500 dilution for 8 to 12 h.
Cytokine secretion assays
Cytokine secretion assays (Miltenyi, Mouse IL-2 Secretion Assay Detection Kit (APC*) 130–090-987 Mouse IFN-γ Secretion Assay Detection Kit (PE) 130–090-516) were downscaled to a 96-well plate format and carried out in triplicates for each tested peptide. 7.5 x 104 pulsed DCs were seeded in X-Vivo-20 medium per well and incubated overnight. The next day 3,75 x 105 purified T cells in X-Vivo-20 medium were added (resulting in 1:5 DC:TC-ratio) and in vitro re-stimulated for exactly 16 h. Afterwards an Fc receptor blocking was performed for 20 min on ice. Therefore 10 µl of a culture supernatant from hybridoma cell line 2.4G2 producing an antibody against the murine Fc gamma receptor (FcRII, CD32) was mixed with 90 µl of FACS buffer (1:10 dilution) per samples. Next the cells were labeled with 5 µl IL-2 and/or 5µl IFNγ Catch Reagents in a total volume of 100 µl X-Vivo-20 medium for 20 min on ice. After labeling the samples were resuspended in 200 µl fresh X-Vivo-20 each for a cytokine secretion period of 3 h at 37°C and 5% CO2. (Assay controls were resuspended in 200 µl X-Vivo-20 supplemented with PMA (50 ng/ml) and Ionomycin (500 ng/ml).) Then, samples were stained with surface marker antibodies for FACS analysis.
FACS analysis
Following Fc receptor blocking (see above) a viability staining was performed employing the LIVE/DEAD® Fixable Yellow Dead Cell Stain Kit (Life Technologies, L-34959). The reconstituted dye was diluted 1:500 in FACS buffer (PBS with 1% v/v of FCS) to achieve working concentration. A maximum of one million cells was resuspended in 200 µl working solution and incubated in the dark on ice for 20 min. Afterwards samples were washed twice.
For cytokine secretion assays cell surface was stained for 20 min on ice in the dark with the following directly fluorescent-labeled antibodies: anti-CD4-FITC (BD, 553047), anti-CD8-Pacific Blue (BD, 558106), IFNγ-detector-PE (Secretion assay kit, Miltenyi, 130–090-516), IL-2-detector-APC (Secretion assay kit, Miltenyi, 130–090-987), anti-CD11c-PE-Cy7 (BD, 558079).
In case of combinations of secretion assay and intracellular cytokine stainings the surface markers were stained for with anti-CD4-V500 (BD, 560782), anti-CD8-Pacific Blue (BD, 558106), IFNγ-detector-PE (Secretion assay kit, Miltenyi, 130–090-516) anti-CD11c-PE-Cy7 (BD, 558079). Before viability staining employing ethidium bromide monoazide (EMA, Sigma-Aldrich, E1374) was performed. Therefore, samples were resuspended in FACS buffer containing 150 µg/ml EMA and incubated on ice exposed to bright light for 10 min. Then samples were washed twice and fixed with BD Cytofix/CytopermTM Fixation/Permeabilization Kit (BD Biosciences, 554715) according to manufacturer’s instruction. Intracellular cytokines were stained for with anti-IL-2-FITC (BD, 554427), anti-TNF-APC-Cy7 (BD, 560658) and IFNγ-A647 (BD, 557735) for 30 min on ice in the dark.
When staining for degranulation markers anti-CD107a-FITC (BD, 553793) antibody was added to in vitro restimulation cultures. After fixation of samples with BD Cytofix/CytopermTM samples were stained with anti-granzyme B-PE (eBioscience, 12–8898), anti-perforin-APC (eBiosience, 17–9392), CD11-cPE-Cy7 (BD, 558079), anti-CD4-V500 (BD, 560782) and anti-CD8-Pacific Blue (BD, 558106).
Regulatory T cells were stained for after Fc blocking and viability staining by first labeling the cell surface with anti-CD4-FITC (BD, 553047), anti-CD8-Pacific Blue (BD, 558106), anti-CD25-PE (Miltenyi, 120–001-294) for 20 min on ice in the dark. Cells were washed and fixed with the Foxp3/Transcription Factor Staining Buffer Kit (e-Bioscience, REF 00–5523-00). Fixed cells were stained with anti-Foxp3-A647 (BD, 560401) on ice for 30 min in the dark.
MHC molecules were stained for after Fc blocking and viability staining using altering combinations of the following antibodies: anti-HLA-A2-APC (BD, 561341), anti-HLA-DR-APC (BD, 559866), anti-H-2Kb-PE (BD, 553570), anti-H-2Db-PE (BD, 553574), anti-I-Ad/I-Ed-PE (BD, 558593) and anti-I-Ab-FITC (BD, 553551).
All fluorescent antibody labeled cells were analyzed facilitating a FACS Canto II flow cytometer (being operated with DIVA software FCS 3.0 (BD Biosciences)) and FlowJo software (FlowJo, LLC).
Immunohistochemistry
Four micrometer consecutive cryostat sections were mounted on Superfrost Ultra Plus® slides (Thermo Scientific, 1014356190), dried for 10 min at RT and fixed in −20°C cooled acetone for 10 min. Slides were washed three times with PBS and then blocked with protein block serum free, ready to use blocking solution (DAKO, X0909) for 1 h at RT. In case of a streptavidin-biotin-coupled staining, slides were additionally blocked with the Avidin/Biotin Blocking Kit (Invitrogen, Life Technologies, REF 004303) according to manufacturer’s instruction. Slides were stained over night at 4°C with the following primary antibodies: anti-CD31 (BD, 550274), anti-CD29/ITGB1 (eBioscience, 14–0291-81), anti-vimentin (Cell Signaling), pan-HLA-A,B,C (clone B9.12.1, a kind gift of Dr. G. Moldenhauer), pan-HLA-A,B,C (clone W6/32, a kind gift of Dr. G. Moldenhauer) and anti-HLA-A2-Biotin (Abcam, ab31564). The next day slides were washed with PBS and incubated with either secondary antibodies (chicken anti-rat-IgG-AlexaFlour® 594 (Invitrogen, A21471), chicken anti-rabbit-IgG-AlexaFlour® 594 (Invitrogen, A21442) or goat anti-hamster-IgG-Dylight 549 (ABD Serotec, STAR104D549GA)) or streptavidin-AlexaFlour® 488 (Invitrogen, S-11223) for 1 h at RT in the dark. Slides were washed and mounted with VectaShield® mouting medium with DAPI (Vecta Laboratories, H-1200) and a cover slip. A Leica DM IRB (Leica) microscope was facilitated for slide imaging and resulting images were RGB merged using ImageJ software (Wayne Rasband, NIH).
Treg specificity assay
Treg specificity assays were conducted similar as described elsewhere29,30 with some necessary modifications for the transfer into the murine system. 1 × 104 DCs were pulsed in triplicates for each tested peptide overnight. The next day purified Tregs (5 x 104) were added in a 5:1 ratio and restimulated overnight. The following day antigen-specifically pre-activated OT-II T cells were added to each well, resulting in a 1:1 ratio of Treg:Tcon. (OT-II T cells were in vitro activated for one night on OVA-peptide pulsed DCs.) After 48 h of culture 1 µCi 3H tritium was added and samples were incubated for another 24 h. Harvesting of assay plates was performed as described. The specific suppression by antigen-specific Tregs was calculated as percent reduction in the proliferation of OT-II Tcons compared to values from mirror plates (plates with DCs and Tcons but without Tregs) according to the following formula:
Western blot
Total protein was isolated from tumor cell lysates or harvested, previously-cultured cells with M-PER® Mammalian Protein Extraction Reagent (Thermo Scientific, 78501) supplemented with one tablet of proteinase inhibitors (Complete ULTRA Tablets, Mini, EDTA free; Roche, 05892791001) according to manufacturer’s instructions. Protein concentrations form cell lysates were estimated photospetroscopically. Each well of the gel was loaded with the same amount and volume of protein. 4 X denaturing loading buffer (NuPAGE® SDS sample buffer (4X), Novex® by Life technologiesTM, REF NP0007) supplemented with DTT (100 mM) was added to each sample. For denaturation samples were boiled for 10 min at 95°C and afterwards spun down. NuPAGE® 10% or 4–12% Bis-Tris Precast Gels (1.0 mm, 10 or 15 wells; Novex® by Life TechnologiesTM, NP0302BOX or NP0321BOX) were used for SDS-PAGE. PAGE was run for roughly 1 h at constant voltage of 120 V. Proteins were wet-blotted on Hybond-P PVDF Transfer Membrane (0.45 µm, Amersham – GE Healthcare Life Sciences, RPN2020F) at constant amperage of 400 mA for 45 min in the 4°C facilitating a Mini Trans-Blot® Cell (Bio-Rad Laboratories, 170–3930). Blocking with either performed in 2.5% BSA (Sigma-Aldrich, A9418-50G) in PBS-T (PBS plus 0.1% Tween-20) or 5% milk powder in PBS-T depending on primary antibodies. Transgene expressing was stained for with anti-HA tag monoclonal antibody (1:1500 in 2.5% BSA/PBS-T, O/N, 4°C, Sigma-Aldrich, H3663-200UL) and equal loading was shown with an anti-β-actin antibody (1:10000 in 5% blocking milk, 1 h, RT, MP Biomedicals, 691001). Both primary antibodies were stained for with HRP-conjugated goat-anti mouse antibody (1:3000, 1h at RT, Santa Cruz Biotechnology, sc-2306) followed by substrate reaction using AmershamTM ECLTM Prime (GE Healthcare, RPN2232) according to manufacturer’s instruction.
Sanger sequencing for mutation screening
RNA was purified from cell pellets using the RNeasy® Mini Kit (QIAGEN, 74104) according the manufacturer’s protocol. To eliminate possible genomic DNA contaminations the RNA samples were treated with DNA-freeTM DNA Removal Kit (Ambion®, Life Technology, AM1906). Complementary DNA (cDNA) was synthesized from RNA templates using the RETROscript® Kit (Ambion®, Life technology, AM1710). To amplify Tp53 and Kras sequences form cDNA and genomic DNA for sequencing as templates gene specific primers were designed (see Supplementary Table 6) and purchased from Eurofins, Martinsried, Germany. PCRs were carried out using Platinum Taq High Fidelity DNA polymerase (Invitrogen, 11304–029) and 10 mM dNTP Mix (PCR Grade, Invitrogen, Life Technologies, 18427–088). Trp53 and Kras PCR products were sent for Sanger sequencing to external companies (GATC Biotech, Constance, Germany and LGC Genomics, Berlin, Germany). Received sequences were screened for mutations by alignment with corresponding wild-type sequences (Serial Cloner, SerialBasics and BLAST, NCBI) and analysis of sequencing histograms employing Chromas Lite software (Technelysium).
Statistical analysis
Statistical analysis and graphs were made employing GraphPad Prism software. Cytokine secretion assays, ELISpot assays, Treg specificity assays and in vitro cytotoxicity assays were conducted in triplicates. T cell responses against mutated and wt peptides were either tested against each other or against respective assay (negative) controls employing two-tailed Student’s t-test. Cumulative results were compared using two-tailed Mann-Whitney test if not indicated elsewise. Differences were considered statistically significant in case p ≤ 0.05.
Supplemental Metrial
Supplemental data for this article can be accessed here.
Acknowledgments
The authors want to thank Dr. Herpel from the tissue bank at the NCT, Heidelberg, Germany for the histopathological analysis of the generated sarcoma cell lines and Rita Schatten and Birgit Kaiser of the DKFZ Genomics and Proteomics Core Facility for excellent services.
We are grateful to Prof. Dr. Wilko Weichert and Dr. Roland Penzel at the Institute for Pathology of the University of Heidelberg for genomic sequencing of CRC tissue samples.
Abbreviations
| Aa | amino acids |
| Ag | antigen |
| BM | bone marrow |
| CRC | colorectal cancer |
| DC | dendritic cell |
| Foxp3 | forkhead box P3 |
| IFNγ | interferon γ |
| IL | interleukin |
| GI | gastrointestinal |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| PB | peripheral blood |
| TAA | tumor-associated antigen |
| TSA | tumor-specific antigen |
| Tcon | conventional T cells |
| Treg | regulatory T cell |
| Wt | wild-type |
References
- 1.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T.. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14:135–146. doi: 10.1038/nrc3670. [DOI] [PubMed] [Google Scholar]
- 2.Reissfelder C, Stamova S, Gossmann C, Braun M, Bonertz A, Walliczek U, Grimm M, Rahbari NN, Koch M, Saadati M, et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. J Clin Invest. 2015;125:739–751. doi: 10.1172/JCI74894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J Exp Med. 1995;181:2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vlad AM, Kettel JC, Alajez NM, Carlos CA, Finn OJ. MUC1 immunobiology: from discovery to clinical applications. Adv Immunol. 2004;82:249–293. doi: 10.1016/S0065-2776(04)82006-6. [DOI] [PubMed] [Google Scholar]
- 5.Gnjatic S, Nishikawa H, Jungbluth AA, Gure AO, Ritter G, Jager E, Knuth A, Chen Y-T, Old LJ. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006;95:1–30. doi: 10.1016/S0065-230X(06)95001-5. [DOI] [PubMed] [Google Scholar]
- 6.van der Bruggen P, Traversari C, Chomez P, Lurquin C, de Plaen E, van den Eynde B, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. [DOI] [PubMed] [Google Scholar]
- 7.Ressing ME, Sette A, Brandt RM, Ruppert J, Wentworth PA, Hartman M, Oseroff C, Grey HM, Melief CJ, Kast, WM. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A*0201-binding peptides. J Immunol. 1995;154:5934–5943. [PubMed] [Google Scholar]
- 8.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 9.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Gronroos E, Martinez P, Matthews N, Stewart A, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen Y-S, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- 13.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 14.Fossum B, Gedde-Dahl T 3rd, Hansen T, Eriksen JA, Thorsby E, Gaudernack G. Overlapping epitopes encompassing a point mutation (12 Gly-->Arg) in p21 ras can be recognized by HLA-DR, -DP and -DQ restricted T cells. Eur J Immunol. 1993;23:2687–2691. doi: 10.1002/eji.1830231045. [DOI] [PubMed] [Google Scholar]
- 15.Gjertsen MK, Saeterdal I, Saeboe-Larssen S, Gaudernack G. HLA-A3 restricted mutant ras specific cytotoxic T-lymphocytes induced by vaccination with T-helper epitopes. J Mol Med. 2003;81:43–50. doi: 10.1007/s00109-002-0390-y. [DOI] [PubMed] [Google Scholar]
- 16.Abrams SI, Stanziale SF, Lunin SD, Zaremba S, Schlom J. Identification of overlapping epitopes in mutant ras oncogene peptides that activate CD4+ and CD8+ T cell responses. Eur J Immunol. 1996;26:435–443. doi: 10.1002/eji.1830260225. [DOI] [PubMed] [Google Scholar]
- 17.Abrams SI, Khleif SN, Bergmann-Leitner ES, Kantor JA, Chung Y, Hamilton JM, Schlom J. Generation of stable CD4+ and CD8+ T cell lines from patients immunized with ras oncogene-derived peptides reflecting codon 12 mutations. Cell Immunol. 1997;182:137–151. doi: 10.1006/cimm.1997.1224. [DOI] [PubMed] [Google Scholar]
- 18.Khleif SN, Abrams SI, Hamilton JM, Bergmann-Leitner E, Chen A, Bastian A, Bernstein S, Chung Y, Allegra CJ, Schlom J. A phase I vaccine trial with peptides reflecting ras oncogene mutations of solid tumors. J Immunother. 1999;22:155–165. [DOI] [PubMed] [Google Scholar]
- 19.Ciernik IF, Berzofsky JA, Carbone DP. Human lung cancer cells endogenously expressing mutant p53 process and present the mutant epitope and are lysed by mutant-specific cytotoxic T lymphocytes. Clin Cancer Res. 1996;2:877–882. [PubMed] [Google Scholar]
- 20.Mayordomo JI, Loftus DJ, Sakamoto H, De Cesare CM, Appasamy PM, Lotze MT, Storkus WJ, Appella E, DeLeo AB. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J Exp Med. 1996;183:1357–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feuerer M, Rocha M, Bai L, Umansky V, Solomayer EF, Bastert G, Diel IJ, Schirrmacher V. Enrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer patients. Int J Cancer J Int Du Cancer. 2001;92:96–105. [PubMed] [Google Scholar]
- 22.Muller-Berghaus J, Ehlert K, Ugurel S, Umansky V, Bucur M, Schirrmacher V, Beckhove P, Schadendorf D. Melanoma-reactive T cells in the bone marrow of melanoma patients: association with disease stage and disease duration. Cancer Res. 2006;66:5997–6001. doi: 10.1158/0008-5472.CAN-04-0484. [DOI] [PubMed] [Google Scholar]
- 23.Schmitz-Winnenthal FH, Escobedo LV, Beckhove P, Schirrmacher V, Bucur M, Ziouta Y, Volk C, Schmied B, Koch M, Antolovic D, et al. Specific immune recognition of pancreatic carcinoma by patient-derived CD4 and CD8 T cells and its improvement by interferon-gamma. Int J Oncol. 2006;28:1419–1428. [DOI] [PubMed] [Google Scholar]
- 24.Sommerfeldt N, Schutz F, Sohn C, Forster J, Schirrmacher V, Beckhove P. The shaping of a polyvalent and highly individual T-cell repertoire in the bone marrow of breast cancer patients. Cancer Res. 2006;66:8258–8265. doi: 10.1158/0008-5472.CAN-05-4201. [DOI] [PubMed] [Google Scholar]
- 25.Sommerfeldt N, Beckhove P, Ge Y, Schutz F, Choi C, Bucur M, Domschke C, Sohn C, Schneeweis A, Rom J, et al. Heparanase: a new metastasis-associated antigen recognized in breast cancer patients by spontaneously induced memory T lymphocytes. Cancer Res. 2006;66:7716–7723. doi: 10.1158/0008-5472.CAN-05-2363. [DOI] [PubMed] [Google Scholar]
- 26.Beckhove P, Feuerer M, Dolenc M, Schuetz F, Choi C, Sommerfeldt N, Schwendemann J, Ehlert K, Altevogt P, Bastert G, et al. Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J Clin Investig. 2004;114:67–76. doi: 10.1172/JCI20278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feuerer M, Beckhove P, Bai L, Solomayer EF, Bastert G, Diel IJ, Pedain C, Oberniedermayr M, Schirrmacher V, Umansky V. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat Med. 2001;7:452–458. doi: 10.1038/86523. [DOI] [PubMed] [Google Scholar]
- 28.Schmitz-Winnenthal FH, Volk C, Z’Graggen K, Galindo L, Nummer D, Ziouta Y, Bucur M, Weitz J, Schirrmacher V, Büchler MW, et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005;65:10079–10087. doi: 10.1158/0008-5472.CAN-05-1098. [DOI] [PubMed] [Google Scholar]
- 29.Bonertz A, Weitz J, Pietsch DH, Rahbari NN, Schlude C, Ge Y, Juenger S, Vlodavsky I, Khazaie K, Jaeger D, et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest. 2009;119:3311–3321. doi: 10.1172/JCI39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt HH, Ge Y, Hartmann FJ, Conrad H, Klug F, Nittel S, Bernhard H, Domschke C, Schuetz F, Sohn C, et al. HLA Class II tetramers reveal tissue-specific regulatory T cells that suppress T-cell responses in breast carcinoma patients. Oncoimmunology. 2013;2:e24962. doi: 10.4161/onci.24962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rathinasamy A, Domschke C, Ge Y, Bohm HH, Dettling S, Jansen D, Lasitschka F, Umansky L, Gräler MH, Hartmann J, et al. Tumor specific regulatory T cells in the bone marrow of breast cancer patients selectively upregulate the emigration receptor S1P1. Cancer Immunol Immunother. 2017;66:593–603. doi: 10.1007/s00262-017-1964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melief CJ, Van Der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8:351–360. doi: 10.1038/nrc2373. [DOI] [PubMed] [Google Scholar]
- 33.Bijker MS, Van Den Eeden SJ, Franken KL, Melief CJ, Offringa R, Van Der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033–5040. doi: 10.4049/jimmunol.179.8.5033. [DOI] [PubMed] [Google Scholar]
- 34.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 35.Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324–327. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- 36.Tian J, Gregori S, Adorini L, Kaufman DL. The frequency of high avidity T cells determines the hierarchy of determinant spreading. J Immunol. 2001;166:7144–7150. [DOI] [PubMed] [Google Scholar]
- 37.Lustgarten J, Dominguez AL, Cuadros C. The CD8+ T cell repertoire against Her-2/neu antigens in neu transgenic mice is of low avidity with antitumor activity. Eur J Immunol. 2004;34:752–761. doi: 10.1002/eji.200324427. [DOI] [PubMed] [Google Scholar]
- 38.Van Hall T, Van Der Burg SH. Mechanisms of peptide vaccination in mouse models: tolerance, immunity, and hyperreactivity. Adv Immunol. 2012;114:51–76. doi: 10.1016/B978-0-12-396548-6.00003-2. [DOI] [PubMed] [Google Scholar]
- 39.Hale DF, Clifton GT, Sears AK, Vreeland TJ, Shumway N, Peoples GE, Mittendorf EA. Cancer vaccines: should we be targeting patients with less aggressive disease? Expert Rev Vaccines. 2012;11:721–731. doi: 10.1586/erv.12.39. [DOI] [PubMed] [Google Scholar]
- 40.Gjertsen MK, Gaudernack G. Mutated Ras peptides as vaccines in immunotherapy of cancer. Vox Sang. 1998;74(Suppl 2):489–495. [DOI] [PubMed] [Google Scholar]
- 41.Rahma OE, Hamilton JM, Wojtowicz M, Dakheel O, Bernstein S, Liewehr DJ, Steinberg SM, Khleif SN. The immunological and clinical effects of mutated ras peptide vaccine in combination with IL-2, GM-CSF, or both in patients with solid tumors. J Transl Med. 2014;12:55. doi: 10.1186/1479-5876-12-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, Soreide O, Eriksen JA, Møller M, Baksaas I, Lothe RA, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer J Int Du Cancer. 2001;92:441–450. [DOI] [PubMed] [Google Scholar]
- 43.Weden S, Klemp M, Gladhaug IP, Moller M, Eriksen JA, Gaudernack G, Buanes T. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int J Cancer J Int Du Cancer. 2011;128:1120–1128. doi: 10.1002/ijc.25449. [DOI] [PubMed] [Google Scholar]
- 44.Carbone DP, Ciernik IF, Kelley MJ, Smith MC, Nadaf S, Kavanaugh D, Maher VE, Stipanov M, Contois D, Johnson BE, et al. Immunization with mutant p53- and K-ras-derived peptides in cancer patients: immune response and clinical outcome. J Clin Oncol. 2005;23:5099–5107. doi: 10.1200/JCO.2005.03.158. [DOI] [PubMed] [Google Scholar]
- 45.Arens R, Van Hall T, van der Burg SH, Ossendorp F, Melief CJ. Prospects of combinatorial synthetic peptide vaccine-based immunotherapy against cancer. Semin Immunol. 2013;25:182–190. doi: 10.1016/j.smim.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11:509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 47.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich P-Y, Mendrzyk R, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18:1254–1261. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 48.Rini BI, Stenzl A, Zdrojowy R, Kogan M, Shkolnik M, Oudard S, Weikert S, Bracarda S, Crabb SJ, Bedke J, et al. IMA901, a multipeptide cancer vaccine, plus sunitinib versus sunitinib alone, as first-line therapy for advanced or metastatic renal cell carcinoma (IMPRINT): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2016;17:1599–1611. doi: 10.1016/S1470-2045(16)30408-9. [DOI] [PubMed] [Google Scholar]
- 49.Burmer GC, Loeb LA. Mutations in the KRAS2 oncogene during progressive stages of human colon carcinoma. Proc Natl Acad Sci U S A. 1989;86:2403–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312:71–75. [DOI] [PubMed] [Google Scholar]
- 51.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 53.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 54.Moodie Z, Huang Y, Gu L, Hural J, Self SG. Statistical positivity criteria for the analysis of ELISpot assay data in HIV-1 vaccine trials. J Immunol Methods. 2006;315:121–132. doi: 10.1016/j.jim.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 55.Moodie Z, Price L, Gouttefangeas C, Mander A, Janetzki S, Lower M, Welters MJP, Ottensmeier C, Van Der Burg SH, Britten CM. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother. 2010;59:1489–1501. doi: 10.1007/s00262-010-0875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moodie Z, Price L, Janetzki S, Britten CM. Response determination criteria for ELISPOT: toward a standard that can be applied across laboratories. Methods Mol Biol. 2012;792:185–196. doi: 10.1007/978-1-61779-325-7_15. [DOI] [PubMed] [Google Scholar]
- 57.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24:1397–1398. doi: 10.1093/bioinformatics/btn128. [DOI] [PubMed] [Google Scholar]
- 58.Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M. NetMHCIIpan-3.0. A Common Pan-Specific MHC Class II Prediction Method Including All Three Human MHC Class II Isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013;65:711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. [DOI] [PubMed] [Google Scholar]
- 60.Perez SA, Von Hofe E, Kallinteris NL, Gritzapis AD, Peoples GE, Papamichail M, Baxevanis CN. A new era in anticancer peptide vaccines. Cancer. 2010;116:2071–2080. doi: 10.1002/cncr.24988. [DOI] [PubMed] [Google Scholar]
- 61.Perret R, Sierro SR, Botelho NK, Corgnac S, Donda A, Romero P. Adjuvants that improve the ratio of antigen-specific effector to regulatory T cells enhance tumor immunity. Cancer Res. 2013;73:6597–6608. doi: 10.1158/0008-5472.CAN-13-0875. [DOI] [PubMed] [Google Scholar]
- 62.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465–472. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmidt AM, Lu W, Sindhava VJ, Huang Y, Burkhardt JK, Yang E, Riese MJ, Maltzman JS, Jordan MS, Kambayashi T. Regulatory T cells require TCR signaling for their suppressive function. J Immunol. 2015;194:4362–4370. doi: 10.4049/jimmunol.1402384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lauwen MM, Zwaveling S, de Quartel L, Ferreira Mota SC, Grashorn JA, Melief CJ, Van Der Burg SH, Offringa R. Self-tolerance does not restrict the CD4+ T-helper response against the p53 tumor antigen. Cancer Res. 2008;68:893–900. doi: 10.1158/0008-5472.CAN-07-3166. [DOI] [PubMed] [Google Scholar]
- 65.Speetjens FM, Kuppen PJ, Welters MJ, Essahsah F, Voet van den Brink AM, Lantrua MG, Valentijn ARPM, Oostendorp J, Fathers LM, Nijman HW, et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin Cancer Res. 2009;15:1086–1095. doi: 10.1158/1078-0432.CCR-08-2227. [DOI] [PubMed] [Google Scholar]
- 66.Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP, Essahsah F, Fathers LM, Offringa R, Drijfhout JW, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–1847. doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
- 67.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 68.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081–1091. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 70.Bijker MS, Van Den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol. 2008;38:1033–1042. doi: 10.1002/eji.200737995. [DOI] [PubMed] [Google Scholar]
- 71.Gjertsen MK, Bjorheim J, Saeterdal I, Myklebust J, Gaudernack G. Cytotoxic CD4+ and CD8+ T lymphocytes, generated by mutant p21-ras (12Val) peptide vaccination of a patient, recognize 12Val-dependent nested epitopes present within the vaccine peptide and kill autologous tumour cells carrying this mutation. Int J Cancer J Int Du Cancer. 1997;72:784–790. [DOI] [PubMed] [Google Scholar]
- 72.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.von Herrath MG, Harrison LC. Antigen-induced regulatory T cells in autoimmunity. Nat Rev Immunol. 2003;3:223–232. doi: 10.1038/nri1029. [DOI] [PubMed] [Google Scholar]
- 74.Schreiber TH, Wolf D, Bodero M, Podack E. Tumor antigen specific iTreg accumulate in the tumor microenvironment and suppress therapeutic vaccination. Oncoimmunology. 2012;1:642–648. doi: 10.4161/onci.20298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tilton JC, Luskin MR, Johnson AJ, Manion M, Hallahan CW, Metcalf JA, McLaughlin M, Davey RT, Connors M. Changes in paracrine interleukin-2 requirement, CCR7 expression, frequency, and cytokine secretion of human immunodeficiency virus-specific CD4+ T cells are a consequence of antigen load. J Virol. 2007;81:2713–2725. doi: 10.1128/JVI.01830-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Betts MR, Nason MC, West SM, de Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Darrah PA, Patel DT, de Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 78.Pajot A, Michel ML, Fazilleau N, Pancre V, Auriault C, Ojcius DM, Lemonnier FA, Lone Y-C. A mouse model of human adaptive immune functions: HLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur J Immunol. 2004;34:3060–3069. doi: 10.1002/eji.200425463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.