

ABSTRACT
This proof-of-concept study investigates the immune effects in metastatic breast cancer (MBC) patients after “vaccination” with activated T cells (ATC) armed with anti-CD3 x anti-HER2 bispecific antibody (HER2 BATs) followed by immune consolidation with immune ATC “boost” after high dose chemotherapy (HDC) and autologous stem cell transplant (SCT). Approximately 2 weeks after completion of vaccination portion of the study, immune T cells were obtained by leukopheresis, activated and expanded ex vivo and re-infused after HDC and SCT to test the hypothesis that transfer of immune unarmed ATC would accelerate reconstitution of anti-tumor activity after SCT. Eight metastatic breast cancer (MBC) patients received 8 infusions of HER2 BATs, low dose IL-2, and GM-CSF in the first part of the protocol to induce adaptive cellular and humoral responses. In the “boost” portion of the protocol, 6 of 8 patients received multiple infusions of unarmed ATC post SCT. There were no dose-limiting toxicities or delays in engraftment. Four of 6 patients tested for the immune correlative studies exhibited increases in anti-breast cancer (BrCa) cytotoxicity, antigen specific IFN-γ Elispots, anti-BrCa antibodies and increased IL-12 and Th1 serum cytokine levels after HER2 BATs infusions. Anti-BrCa tumor responses were seen as early as 2 weeks after SCT and persisted up to 2 years post-SCT. One out of 6 patients’ rapidly progressed and showed poor immune responses and high Th2 cytokine levels. There was a significant correlation (p < 0.002) between time to progression (TTP) and anti-BrCa cytotoxicity by immune T cells. This is the first study to show that adoptive transfer of immune T cells after SCT accelerates reconstitution of anti-BrCa specific immunity and correlates with delay TTP.
KEYWORDS: Vaccination, transfer of tumor specific immunity, specific cytotoxic T lymphocytes, in vitro tumor specific antibody synthesis, breast cancer, bispecific antibody, HER2/neu, immunotherapy
Introduction
Arming activated T cells (ATC) with bispecific antibodies (BiAb) provides a non-toxic approach to enhance T cell killing of breast cancer (BrCa) cells1. In a recent phase I study, infusions of HER2 bispecific antibody armed activated T cells (BATs) in women with metastatic breast cancer (MBC) induced specific anti-breast cancer immunity and increased IL-12 and Th1 cytokines2. Infusions of anti-CD3 x anti-HER2 BiAb armed ATC (BATs) were safe, induced anti-BrCa cytotoxic T lymphocytes (CTL), anti-BrCa antibodies and induced a Th1 cytokine pattern with encouraging clinical results.2 In another phase I study3, after infusions of unprimed and unarmed ATC in 23 MBC patients after autologous stem cell transplant (SCT), 50% of the patients were stable and 70% were alive whereas 10% of those who received SCT alone were stable and 50% alive at 32 months3. Although the differences were not significant (p = 0.09), the data suggested that a prime and boost strategy would augment anti-BrCa immunity. While SCT for the treatment of BrCa remains controversial, a recent meta-analysis of 15 randomized high-risk primary BrCa trials (n = 6102) showed a 13% event-free survival benefit for SCT (P = 0.001) over standard of care with a 6 year median follow-up.4
This proof-of-concept study was designed to investigate whether cellular and humoral anti-breast cancer immunity induced by infusions BATs can be transferred after HDC and SCT by immune T cells obtained after BATs infusion. This study takes advantage of SCT to reduce tumor burden, create immune space, and augment transfer of anti-tumor immunity. We present evidence that BATs induce BrCa-specific cellular, humoral, and innate immunity that can be transferred with infusions of immune ATC and stem cell product.
Results
Clinical status
Table 1. summarizes patient age, HER2 status, prior therapies, doses of BATs and ATC, days to myeloid and lymphoid engraftment, time to progression (TTP), overall survival (OS) from enrollment or SCT, and disease status. Total eight patients were enrolled, 7 patients had visceral disease. The median TTP and OS for the 6/8 evaluable patients who received BATs and ATC was 14.6 and 37.3 months, respectively; whereas the median TTP and OS for all 8 patients (including 2 patients who did not receive a SCT and Boost) are 11.2 and 32.0 months, respectively. In contrast, the other 17 patients in the phase I clinical trial who received BATs alone had a median TTP and OS of 2.7 and 27.5 months, respectively. Treatment schema is shown in Figure 1(a).
Table 1.
Shows patient demographics, HER2 status, cell doses, engraftment, OS and disease status prior to IT and post SCT.
| Engraftment |
Clinical Features |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Enrollment (months) |
Post SCT (months) |
||||||||||||
| Patients | Age | HER2 (IHC) |
Total HER2 BATs Infused (x 109) | Total ATC Infused after SCT (x 109) | Chemotherapy | ANC≥ 500 | ALC≥ 500 | TTP | OS | TTP | OS | Status Pre IT | Status Post IT**/SCT |
| FH1699 | 58 | Neg | 160 | 43.52 | FAC, R-CHOP#, D, Ex, Ful, Cap | 11 | 13 | 4.8 | 18.9 | 2.7 | 16.8 | SD | PD |
| FH1702 | 31 | 3+ | 160 | 109.96 | D+ Carbo+ H, H, RT, T+ Carbo, Nav+ H, Abrax+ H, Cap | 19 | 13 | 14.6 | 84.7 | 12.03 | 82.1 | SD | SD |
| IT20007 | 48 | Neg | 133 | 39.84 | AC, TC | 16 | 11 | 31.2 | 38 | 28.7 | 35.5 | SD | SD |
| IT20017 | 44 | Neg | 63.7 | 59.92 | ACD, Cap, B, Carbo, Abrax, B, C, H, C | 16 | 8 | 7.9 | 9.9 | 3.9 | 5.9 | PD | PD |
| IT20020 | 47 | 3+ | 47.2 | 16.4 | ACT, H | 14 | 16 | 14.8 | 36.6 | 12.5 | 34.3 | PD | SD |
| IT20031 | 55 | Neg | 61.8 | 41.6 | ACD, Abrax | 9 | 17 | 10.7 | 64.2 | 6.3 | 59.8 | SD | SD |
| IT20001* | 38 | Neg | 83.2 | NA* | ATC, Cap, B, Nav, G, Tipi+ Abrax+ G, Carbo, Carbo+ Abrax | NA* | NA* | 2.03 | 9.2 | NA | NA | PD | PD* |
| IT20038* | 39 | Neg | 90.0 | NA* | ACT, B | NA* | NA* | 1.03 | 27.5 | NA | NA | PD | PD* |
A: adriamycin; Abrax: Abraxane; B: bevacizumab (Avastin); C: cyclophosphamide (Cytoxan); Cap: capecitabine (Xeloda); Carbo: carboplatin; Ex: exemestane (Aromasin); Ful:fulvestrant (Faslodex); FAC: 5-fluorouracil, A, and C; H: Herceptin (trastuzumab); RT: radiation therapy; T: paclitaxel; D: docetaxel; R-CHOP: Rituximab, cyclophosphamide, doxorubicin, vincrtistine, and prednisolone; Nav: navelbine; Tipi: Tipifarnib. **:Clinical status after IT or SCT; SD: stable disease; PD: progressive disease. *Patients did not receive SCT.
Figure 1.
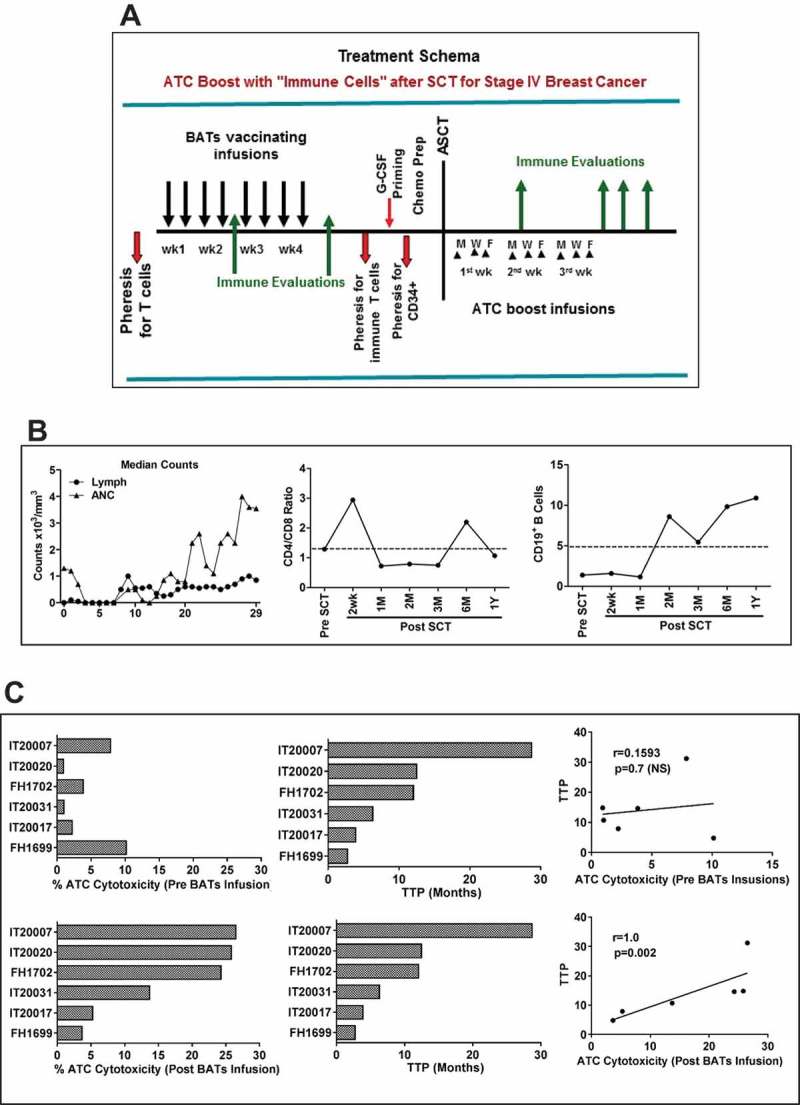
a) Treatment schema shows leukapheresis to obtain T cells for expansion and immunization with BATs. BATs were administered twice weekly for four consecutive weeks. A second leukapheresis was done to obtain immune T cells prior to G-CSF priming for collecting stem cells. PBMC were activated with OKT3 and expanded in IL-2 (100 IU/ml) to generate ATC after 12–14 days of culture. After a 3rd pheresis to collect G-CSF primed CD34+ cells, patients received cyclophosphamide, thiotepa, and carboplatin (CTC) as the preparative regimen for chemosensitive disease and ICE for resistant disease followed by autologous SCT. Immune ATC were infused day + 4 after SCT thrice (n = 2) or twice (n = 4) a week for total 8–15 infusions and immune testing was performed at indicated time points after ATC infusions. b) Shows average daily Lymphocyte (Lymph) and Absolute Neutrophil Counts (ANC) of all 6 evaluable patients (Left panel). Middle and right panels show CD4/CD8 ratio and CD19 + B lymphocytes monitored up to 12 months post SCT (n = 6). C) Shows the bar graph of cytotoxicity (Left) mediated by ATC prior to BATs infusions (top panel) and expanded immune ATC (lower panel) of 6 patients against breast cancer cells (SK-BR-3) measured by the chromium (51Cr) release assay and their corresponding TTP. There were no significant correlation of ATC cytotoxicity prior to BATs infusions with TTP, but there was a significant correlation (p = 0.002) between immune ATC (ATC expanded after BATs infusions) cytotoxicity and TTP (right), suggesting that higher cytotoxicity, may improve progression free survival after BATs infusion contribute to TTP prolongation.
Correlation between immune T-cell cytotoxicity and time to progression (TTP)
Spearman correlation coefficient was calculated and tested for the correlation between anti-BrCa cytotoxicity by immune T cells and TTP. The results show that TTP had significant correlation with percent anti-BrCa cytotoxicity of immune T cells (Spearman correlation coefficient r = 1, p < 0.002) (Figure 1(c), lower panel). Patients with high ATC cytotoxicity had significantly longer TTP (Log-rank test p = 0.025). There was no correlation between OS and ATC cytotoxicity (Figure 1(c), upper panel).
Neutrophil and lymphocyte recovery is shown in Figure 1(b) (Left panel). Phenotyping for T and B lymphocytes show that early post SCT infusions of ATC transiently normalized the CD4/CD8 ratio (Figure 1(b), Middle and Right panels). The phenotypes and cytotoxicity of harvested products (BATs and ATC) are shown in Table S1.
Transfer of tumor specific T-cell immunity by immune ATC after SCT
Immune testing was performed in 4 patients who received 8 infusions without IL-2 and GM-CSF. The PBMC from all four patients showed increased cytotoxicity during and after infusions of BATs. After SCT, cytotoxic activity appeared between 2 weeks and 2 months post SCT and recovered to the levels seen after BATs infusions by 3–6 months (Figure 2(a), Left panel). IFNγ EliSpots to BrCa stimulation were restored by 1 month and persisted up to 3–12 months post SCT (Figure 2(a), Right panel). Three of 4 patients with stable disease for at least 6 months or more after SCT showed enhanced anti-tumor T cell responses. Patient IT20007 who had TTP of 28.7 months, showed peak anti-BrCa cytotoxicity of 37% at 3 months, and a 2 fold increase from 550 at pre SCT to 1100 after SCT in IFN-γ EliSpots/106 PBMC at 6 months post SCT. Patient IT20020 had a peak cytotoxicity of 12% at 2 months post-SCT compared to less than 3% pre SCT and restored IFN-γ EliSpot responses at 6–12 months; this patient was progression free at 12.5 months. Patient IT20031 who had a TTP of 6.3 months had more than a 2 fold increase in specific anti-BrCa cytotoxicity at 1 month post SCT that persisted up to 3 months and reached remarkably high cytotoxicity of 45% at 6 months post SCT. Likewise, IFN-γ EliSpots were 2–5 fold higher than the pre SCT IFN-γ EliSpot activity as early as 1 month that persisted up to 3 months after SCT. The immediate non-MHC restricted specific cytotoxicity and IFN-γ secretion mediated by specific TCR clones has been described by Simpson-Abelson et al.5
Figure 2.
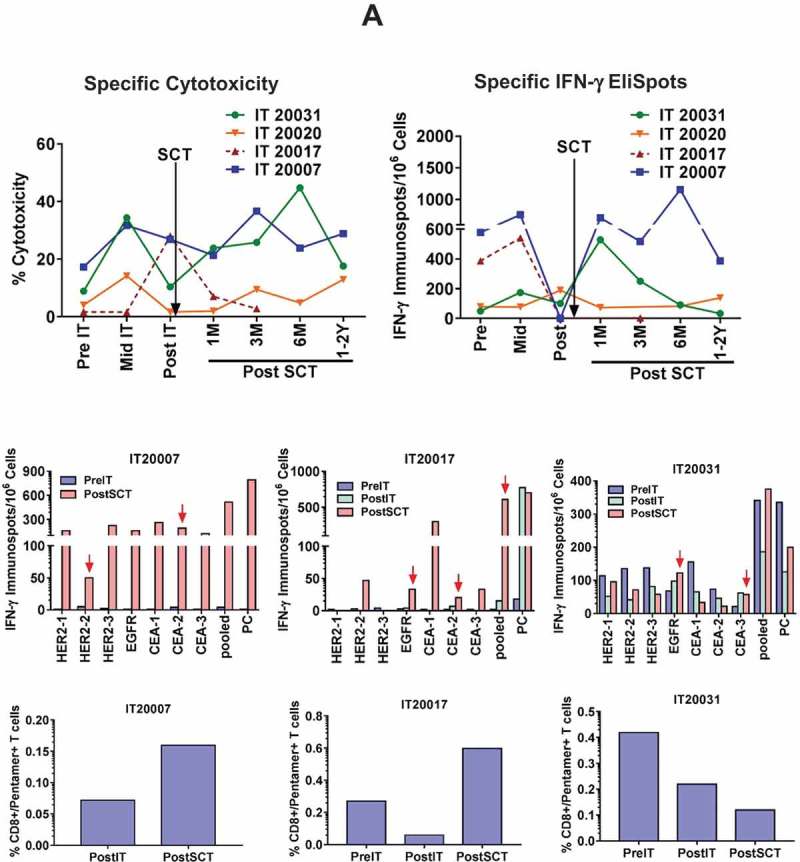
Transfer and reconstitution of T cell responses. a) Left Upper panel shows the cytotoxicity by PBMC (n = 4) against breast cancer cell line SK-BR-3 at pre IT, mid IT (infusion #4 or #5), post IT at 25:1 E/T ratio measured by the chromium (51Cr) release assay. Increase in cytotoxicity between 1–6 months post SCT indicate transfer and reconstitution of T cell responses. Right Upper panel shows T cell IFN-γ EliSpots (n = 4) directed at SK-BR-3 at pre IT, mid IT, post IT and multiple time points post SCT. Middle panel shows the epitope-specific IFN-γ EliSpot responses by overnight stimulation of PBMCs with indicated 9-mer peptide preloaded HLA-A2-pentamers from three patient at pre IT, post IT and post SCT time points (PC = Positive Control). Bottom panel shows the percentages of pentamer/CD8 double positive T cells stained with HLA-A2-pentamers-R-PE and anti-CD8-FITC antibodies and analyzed by flow cytometry. b) Upper panel shows the persistent and transient expansion of tumor specific Vβ repertoire after multiple infusions of BATs and transfer of breast cancer specific T cells after SCT. Middle and Bottom panels show CD4+ and CD8 + T cell clones in immune ATC secreting IFN-γ upon stimulation (S) with breast cancer cells compared to nonstimulated (NS) immune ATC.
It is noteworthy that one out of four patients (IT20017, TTP 3.9 months) who progressed rapidly after SCT had high specific cytotoxicity (30%) pre SCT that sharply decreased at 1 month after SCT and continued to decrease thereafter, while IFN-γ EliSpots could not be detected after SCT (indicated in dotted red line).
Frequency of antigen specific CD8+ T cells after SCT
Next we showed that the frequency of HLA-A2 restricted IFN-γ producing CD8+ T cells specific to HER2, EGFR and CEA epitope (peptide listed in Table S2) was higher post IT and post SCT compared to pre IT PBMC (Figure 2(a), Middle panel). The pentamer staining using pooled 9-mer peptides of HER2, EGFR and CEA showed increased frequency of HLA-A2 restricted epitope-specific CD8+ T cells after SCT in two out of three patients tested by flow cytometry (Figure 2(a), Bottom panel). Despite the low frequency of pentamer+/CD8+ T cells, total epitope-specific IFN-γ producing CD8+ T cells was much higher compared to pentamer positive CD8 + T cells.
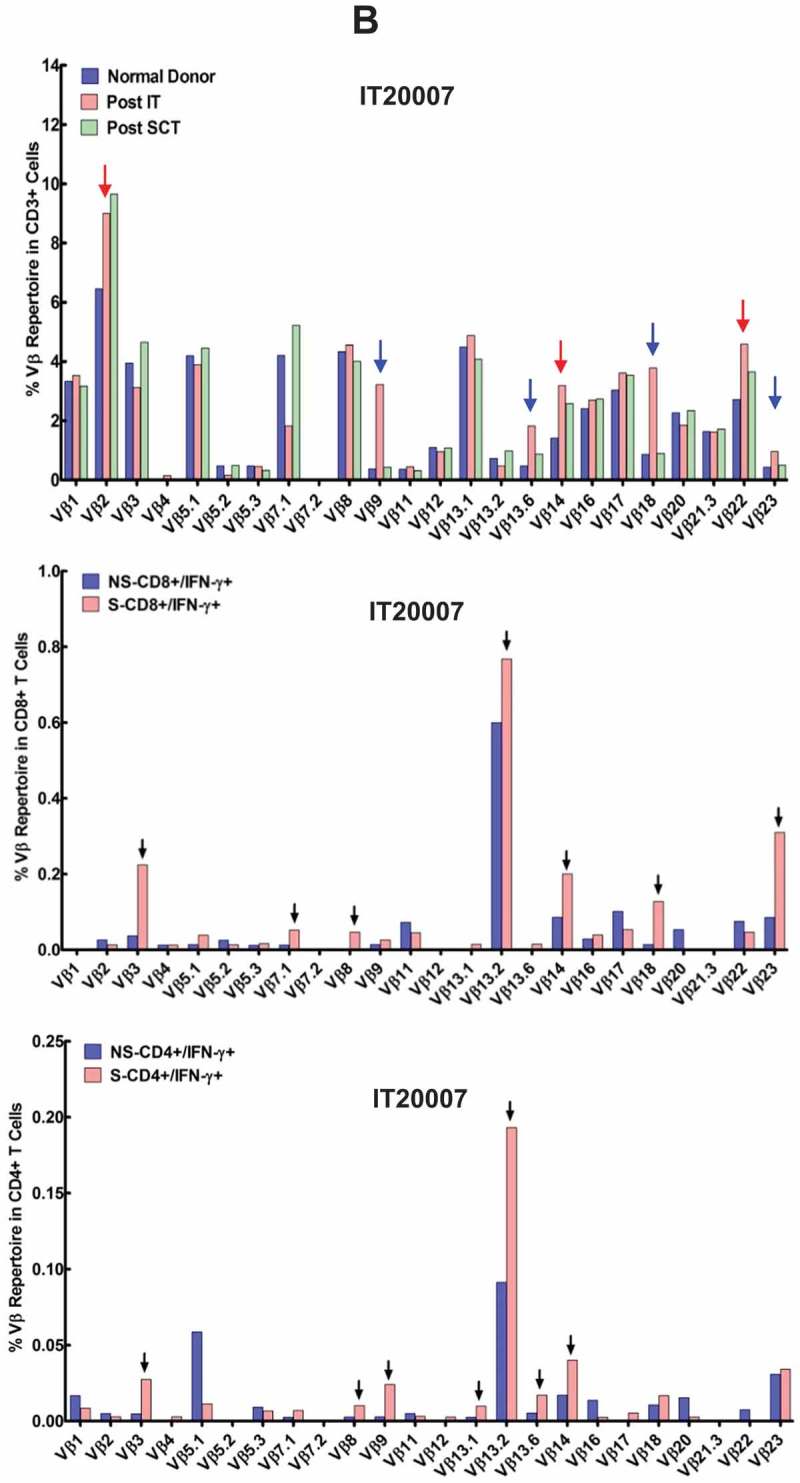
Selected vβ repertoire pattern post SCT mirrored post IT pattern
The predominant T cell receptor (TCR) Vβ clones expanded in vivo after BATs infusions and persisted after SCT in the PBMC of patient detected by flow cytometric analysis as a result of transfer of immunity via stem cell product and/or boost using immune ATC. A representative data from IT20007 at post IT and 1 year post SCT is presented in Figure 2(b). The three distinct patterns were observed for Vβ repertoire post IT and post SCT (Figure 2(b), Upper panel). Pattern 1) the proportions of Vβ expression were similar in 17 of 24 Vβ repertoire post IT and post SCT relative to normal donor (ND); Pattern 2) the proportions that were high after IT and persisted post SCT (Vβ2, Vβ14, and Vβ22) relative to ND (red arrows); Pattern 3) the proportions that were high after IT but did not persist post SCT (Vβ9, Vβ13.6, Vβ18, and Vβ23) relative to ND (blue arrows). The TCR pattern 1 shows transfer and persistence of most pre-existing Vβ clones. The higher proportions of TCR clones (Vβ2, Vβ14, and Vβ22) in pattern 2 provide evidence for expansion, transfer, and persistence induced by IT. In contrast, pattern 3 TCR clones (Vβ9, Vβ13.6, Vβ18, and Vβ23) may have expanded after IT but did not persist (Figure 2(b), Upper panel). Next, we determined the frequency of IFN-γ expressing CD4+ and CD8+ Vβ repertoires in ATC expanded from immune PBMC after stimulated (S) or in non-stimulated (NS) cells. Intriguingly, 10/24 CD4+ and/or CD8+ clones (Vβ3, Vβ5.1, Vβ7.1, Vβ8, Vβ13.1, Vβ13.2, Vβ13.6, Vβ14, Vβ18, and Vβ23 chains) produced IFN-γ upon stimulation with breast cancer cells, providing validation that vaccination with BATs may have induced breast cancer specific memory T cell clones that were transferred after SCT and persisted up to 1 year post SCT (Figure 2(b), Middle and Bottom panels).
Figure 2.
(Continued).
Transfer of humoral immunity
Next, we determined whether BATs infusions would induce anti-tumor humoral immunity, and whether humoral immunity could be transferred and augmented with multiple infusions of immunized T cells after SCT. Figure 3(a) (Left panel) shows serum anti-SK-BR-3 antibody levels at pre immunotherapy (IT), post IT, and post SCT. Right panel of Figure 3(a) shows in vitro anti-SK-BR-3 antibody synthesis. Given low precursor frequency of antigen-specific B cells, the antibody synthesis by B cells is quite remarkable. About 25 ng/ml of specific IgG were produced by PBMC at pre IT and up to 200 ng/ml (8 fold increase) of specific IgG after IT. Antibody synthesis increased from low levels in the first 2–3 months after SCT and persisted in 3 of 4 patients (IT20007, IT20020, and IT 20031) up to 6 months post SCT. In one patient with PD (IT20017), antibody synthesis and memory B cell responses were lower suggesting that IT20017 lacked robust cellular and humoral immune responses. Our data suggest that booster ATC infusions after SCT provided T cell help to enhance specific B cell recovery and sustain serum antibody levels post SCT.
Figure 3.
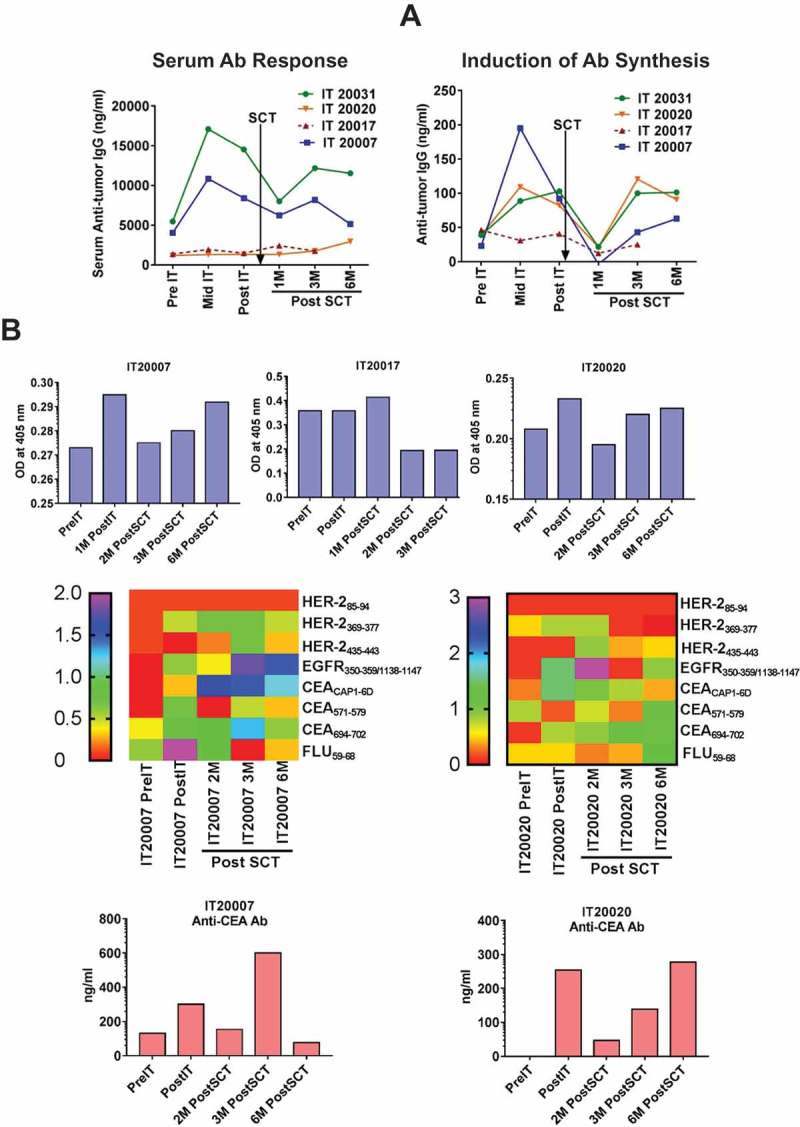
The epitope-specific B-cell responses. a) Left panel shows anti-BrCa antibody levels in patient serum (n = 4). Right panel shows the anti-SK-BR-3 IgG antibody synthesis in 4 patients at pre IT, post IT and post SCT time points. The gradual increase in anti-SK-BR-3 IgG antibody synthesis after SCT indicate transfer and reconstitution of B cell responses. b) Top panel shows specific antibody titers by ELISA (1:100 dilution) in 4 patients at pre IT, post IT and multiple time points post SCT against pooled peptide. Serum antibody steadily increase up to 6M post SCT in 3 out of 4 patient serum samples indicating transfer and reconstitution of B cell responses. Middle panel shows specific serum antibody responses against indicated individual peptides by semi quantitative dot-blot assay at 1:100 dilution in two serum samples from patients IT20007 and IT 20020 at various time points. Bottom panel shows validation of dot blot assay by ELISA to quantitate anti-CEA specific antibody levels in the serum samples of IT20007 and IT 20020.
Antigen specific IgG antibody levels after IT and SCT
Specific antibody responses were screened in serum for all 4 patients at pre IT, post IT and post SCT against pooled 9-mer peptides of HER-2, -EGFR and -CEA. Interestingly, 3 of 4 responder patients (IT20007, IT20020, IT20031) showed gradual increase in antibody levels against pooled peptides while a non-responder patient (IT20017), who had high levels up to 1M post SCT, showed two fold decrease at 2 and 3M post SCT (Figure 3(b), Top panel). Next, we screened the individual 9-mer peptide specific antibody levels in serum using modified dot blot assay, 2 of 3 responder patients showed reactivity to EGFR and all 3 CEA peptides post SCT (Figure 3(b), Middle panel). To further validate the dot blot results, we performed ELISA using anti-CEA peptides that showed most reactivity with IT20007 and IT20020 serum samples, corroborating the pattern seen with dot blot (Figure 3(b), Bottom panel).
Serum cytokine/chemokine levels
Immunokine levels were detected in 4 of 6 patients. Th1 (IL-2, TNF-α, IFN-γ) and Th2 cytokines (IL-4 and IL-10) increased sharply from baseline to post IT. After SCT, levels of IL-2, TNF-α, and IL-12 remained high up to 2 months (Figure 4(a)). MIP-1β and IFN-γ-induced chemokines IP-10 and MIG (Figure 4(c)) showed the same pattern after IT and levels decreased to near baseline by 2 months except for IT20017 (dotted red line). The IL-6 levels (Figure 4(b)) which were higher pre SCT disappeared post SCT in 3 patients (IT20007, IT20020 and IT20031) who were stable for 6 months or more post SCT. Patient IT20017 (dotted red line in Figure 4(a)-(c)) who progressed stood out with elevated levels of IL-6, IL-4, and IL-10 post SCT. The Type 1/Type 2 cytokines ratio shifted towards anti-tumor Type 1 cytokine profile during therapy as shown Figure 4(d), dotted black arrow represent SCT.
Figure 4.
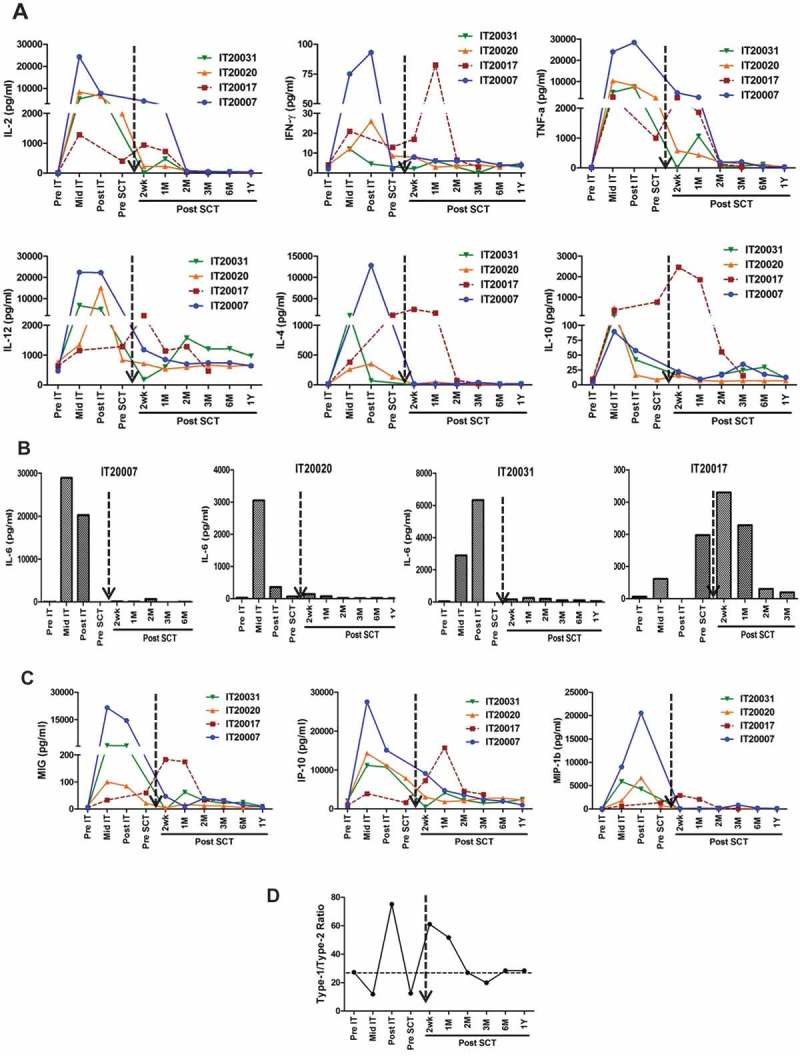
a)Shows profile of serum cytokines. Analysis of serum samples at pre-immunotherapy (Pre IT), mid IT, post IT and post SCT. Infusions of HER2 BATs induce increase in both type 1 cytokines IL-2, IFN-γ, TNF-α, IL-12, and type 2 cytokines IL-4, IL-6 and IL-10. Levels and remained high up to 2 months post SCT. b) Shows IL-6 levels in 4 patients during IT and Post SCT. It is noteworthy that one patient (IT20017) who had rapid progression of disease had high levels of not only IL-6 but also the IL-4 and IL-10. c) Shows the chemokine profile at Pre IT, mid IT, post IT and multiple time points post SCT. d) Shows the mean ratio of Th1/Th2 = [IL-2+ IFNγ]/[IL-4+ IL-10] at pre-, mid- post-IT and indicated time points post SCT.
Discussion
This is the first study that reports that anti-tumor immunity of T and B cells induced by BATs infusions can be adoptively transferred after SCT to enhance anti-tumor immunity in MBC patients. In our earlier phase I study in MBC patients3, infusions of unprimed ATC after HDC and SCT suggested that unprimed ATC can accelerate the reconstitution of anti-tumor responses by providing polyclonal helper, CTL, and LAK-like cytotoxicity after lymphodepletion.6,7
This proof of principal study uses a “vaccinate” with BATs with low dose IL-2 and GM-CSF, and “boost” with ex vivo expanded immune ATC after SCT to maximize anti-tumor immune activity. We use the low dose IL-2 for the therapeutic efficacy of BATs infusions to support their survival and in vivo proliferation without causing IL-2 mediated toxicity that is associated with high dose IL-2 therapy. While both high and low dose IL-2 can induce regulatory T cell expansion, we routinely phenotype peripheral blood mononuclear cells (PBMC) to monitor T regulatory cells (Tregs) status, and have not seen any increase in Tregs during or after completion of the therapy compared to the pre-treatment Tregs (< 1%). HDC was used to create immune space by depleting T regulatory cells and myeloid derived suppressor cells. The boost infusions permitted immune cells to expand after transfer and accelerated reconstitution of anti-tumor T and B cell responses. All 4 patients tested for immune studies, exhibited non-MHC restricted cytotoxicity and tumor specific IFN-γ EliSpots by PBMC obtained after SCT, clinical and immune correlates are shown in Table 2. These data show that the transferred tumor-specific immune responses were mediated by endogenous immune cells.2,8 Our findings are consistent with earlier studies showing that adoptive transfer of vaccine-primed and anti-CD3/anti-CD28 co-stimulated T cells boosts cellular and humoral responses to tumor antigens after SCT in multiple myeloma.10–12
Table 2.
Immune responses at Pre IT, Pre SCT and 3–6 Months Post SCT.
| Immune Response | Time Intervals | IT20007 | IT20017 | IT20020 | IT20031 | Total # (%) of positive responses in boosted patients |
|---|---|---|---|---|---|---|
| CTL | PreIT | + | - | - | ± | 1/4 (25) |
| PreSCT | ++ | ++ | + | +++ | 4/4 (100) | |
| 3–6 M PostSCT | ++ | +/- | + | +++ | 3/4(100) | |
| IFNγ | PreIT | ++ | ++ | +/- | +/- | 2/4 (50) |
| PreSCT | ++ | ++ | + | + | 4/4 (100) | |
| 3–6 M PostSCT | +++ | - | + | ++ | 3/4 (75) | |
| Serum Ab | PreIT | + | +/- | +/- | + | 2/4 (50) |
| PreSCT | ++ | +/- | +/- | ++ | 2/4 (50) | |
| 3–6 M PostSCT | ++ | +/- | + | ++ | 3/4 (75) | |
| In Vitro Ab Synthesis | PreIT | +/- | + | + | + | 3/4 (75) |
| PreSCT | +++ | + | ++ | ++ | 4/4 (100) | |
| 3–6 M PostSCT | ++ | +/- | ++ | ++ | 3/4 (75) | |
| Th1 | PreIT | + | + | + | + | 4/4 (100) |
| PreSCT | +++ | +++ | +++ | +++ | 4/4 (100) | |
| 3–6 M PostSCT | +++ | +++ | ++ | ++ | 4/4 (100) | |
| Th2 | PreIT | + | + | +/- | +/- | 2/4 (100) |
| PreSCT | +++ | ++ | + | + | 4/4 (100) | |
| 3–6 M PostSCT | + | +++ | +/- | +/- | 2/4 (100) | |
| Cell Dose (x 109) | 39.8 | 59.9 | 16.4 | 41.6 | ||
| Survival (months) | 35.5 | 5.9 | 34.3 | 59.8 | ||
There were no DLTs in patients given BATs2 or ATC after SCT. One patient developed sepsis after SCT. She fully recovered and is alive at 61 months from enrollment for BATs (IT 20031). The absolute lymphocyte counts recovered to 500/mm3 between 8 and 17 days after SCT without lymphocytosis, cytokine storm or impairment of neutrophil engraftment after SCT for all patients.
The immediate killing exhibited by fresh PBMC from patients directed at allogeneic SK-BR-3 cells shows non-MHC restricted cytotoxicity mediated by in vivo primed T cells.5 The CTL activity persisted up to 2 years after SCT. Since fresh PBMC and ATC expanded from the PBMC before infusions of BATs did not exhibit high levels of anti-BrCa cytotoxicity, the post IT and post SCT responses are most likely to be due to the infusions of BATs. Patients IT2007 and IT20031 had robust CTL and IFN-γ responses detected at 2 weeks after SCT. Serum anti-BrCa antibody reached levels comparable to pre SCT levels within 2–3 months after SCT in all 4 patients. Antibody synthesis after SCT gradually increased after IT.13 For specific antibody production to be detected, immune B cells not only have to be present but they had to be transferred in the stem cell product. The transfer of immune T and B cells in the stem cell product is not likely to be sufficient to provide the robust CTL activity detected 2–4 weeks after SCT since there are intrinsic T and B cell defects in the first 3 months after SCT14. In vitro antibody synthesis and circulating antibodies to BrCa detected in the first 3 months after SCT show that boosting with immune ATC can provide antigen specific helper and CTL activity that not only overcomes the T and B cell defects seen in the first 3 months after SCT,15–20 but also augments the transfer and persistence of the anti-tumor immunity for up to 2 years.
Our rationale to target HER2 low to negative patients were based on our previous studies1,2. Specific cytotoxicity mediated by HER2 BATs is non-MHC-restricted and is independent of HER2 receptor-mediated signaling mechanisms, only a few HER2 receptors on the tumor cells for T cell engagement are required and HER2 negative targets are routinely killed1. In our earlier studies, we have shown that HER2 BATs were able to target HER2 negative MCF-7 cell line which has only fewer HER2 receptors compared to HER2 positive SKBR-3 cell line1. Furthermore, evaluation of immune responses in our phase I clinical trial patients suggests that infusions of HER2 BATs induce robust immune responses regardless of the patient’s HER2 status2. Indeed, 6/8 patients enrolled in this study were HER2 negative, evaluation of immune responses in this study showed that infusions of HER2 BATs induce robust anti-BrCa immune responses regardless of the patient’s HER2 status.
Furthermore, we verified the specificity of immune responses by multiple assays that provide evidence of T cell and serum antibody reactivity to multiple breast cancer antigens (9-mer peptides; three HER-2, two pooled EGFR and three CEA peptides) in post IT and post SCT samples. The quantitation of peptide-specific CD8+ T cells demonstrated that there were T cell clones that could bind to the HLA-peptide complex in higher frequencies in post IT and memory T cell clones post SCT (transferred in ATC boost infusions). Likewise, there were epitope specific antibody induction after infusions of HER2 BATs and transfer of memory B cells in ATC boost after SCT. Our findings suggest that multiple BATs infusions may result in immunogenic epitope spreading and development of the broad and durable T- and B-cell memory responses and “transfer of immunity” by immune ATC after SCT that parallels with the clinical responses.
A significant correlation (r = 1.0; p < 0.002) between immune ATC cytotoxicity directed at BrCa cells and TTP suggests that more robust vaccinations with a Th1 shift in cytokine profiles can lead to clinical benefit. There were no obvious differences in clinical responses in those who received IL-2 and GM-CSF and the patients who did not receive the cytokines. Although, we did not have a negative control arm in this study, our previously published study that showed that the PFS was 10% and survival was 50% at 32 months in patients who received SCT without immunotherapy3; however, immune monitoring studies were not done to make any comparisons. A randomized trial comparing BATs before SCT and immune ATC after SCT with infusions of non-immune ATC after SCT will be needed to validate the our observations.
In summary, this study shows that BATs induced endogenous anti-BrCa specific cellular, humoral immunity that could be detected after SCT and may have provided clinically meaningful anti-tumor immunity. There was robust reconstitution of T and B cell functions early after SCT as evidenced by CTL and NK activity, IFN-γ EliSpots, in vivo/in vitro antibody synthesis, and Th1 cytokine responses. Furthermore, these responses persisted up to 2 years after SCT. On the other hand, elevated Th2 cytokines and low CTL activity may predict poor anti-tumor responses and clinical outcomes.
Materials and methods
Study population, patient enrollment and eligibility
The phase I trial was registered on clinicaltrial.gov as NCT00027807. Women, 18 years of age or older, with histologically documented MBC of the breast with 0 – 3+ HER2/neu expression with no measurable disease were eligible. Specific details of organ specific function are in the supplemental information. Eight of 23 patients in the phase I were treated in the second part if the study registered at clinicaltrial.gov as NCT00020722. All protocols were approved by protocol review committee, Institutional Review Boards at Roger Williams Hospital and Wayne State University, and the FDA. All patients signed informed consent forms.
Immunization with BATs and expansion of pre-immunized ATC
Patients received a total of 8 twice weekly BATs infusions1 for 4 weeks and subcutaneous injections of 3.0 x 105 IU of IL-2/m2/day and 250 µg granulocyte-macrophage colony stimulating factor (GM-CSF)/m2 twice per week starting 3 days before the 1st BAT infusion and ending 7 days after the last BATs infusion (Figure 1(a), Treatment Schema). Seven to 14 days after the last BATs infusion, the patients were leukapheresed for the expansion of immune T cells. Immune ATC were harvested and cryopreserved after 14 days for infusions after SCT.
G-CSF primed PBMC collection for SCT
Following PBMC collection for generating pre-immunized ATC, PBMC were collected by 1–2 leukapheresis to obtain a minimum of 1 x 106/kg of CD34+ cells after 4 days of priming with granulocyte-colony stimulating factor (G-CSF at a dose of 15 μg/kg/day) for SCT.
HDC to reduce tumor burden
Patients received cyclophosphamide, thiotepa, and carboplatin (CTC; Stamp V) as the preparative regimen for chemosensitive disease and ifosfamide, carboplatin, and etoposide (ICE) for chemoresistant disease. For the CTC regimen (chemosensitive disease): Cyclophosphamide (2000 mg/m2 i.v.) was given on days −4, −3, and −2 (total = 6000 mg/m2); Thiotepa (167 mg/m2 IV) on days −4, −3, −2 (total = 500 mg/m2); Carboplatin (267 mg/m2 IV) on days −4, −3, and −2. For the ICE regimen (chemoresistant disease): Ifosfamide (2,500 mg/m2 IV) was infused on days −8 to −3 (total dose = 15,000 mg/m2) with Mesna (1,000 mg/m2 IV) given beginning 30 min before the start of each ifosfamide dose and then as a continuous IV infusion (1,500 mg/m2) over next 12 hours; Carboplatin (250 mg/m2) on days −8, −7, −6, −5, −4, and −3 (total dose = 1500 mg/m2); Etoposide (VP-16; 200 mg/m2 IV) on days −8, −7, −6, −5, −4 and −3 (total dose = 2,400 mg/m2).
Stem cell transplant
On day 0, the cryopreserved peripheral blood stem cells (SC) were thawed and infused at the bedside.
Immune ATC transfer after SCT
Two patients received infusions of up to 1010 ATC three times per week beginning day + 4 after SCT for three weeks and then once weekly infusions of up to 20 × 109 for 6 weeks. Daily subcutaneous injections of IL-2 (3.0 × 105 IU/m2/day) were given starting on day + 4 after SCT and ending on the day of the last infusion. GM-CSF (250 µg/m2 twice per week) was given for 3 weeks starting day + 5. A second group of patients (n = 4) received ATC twice per week for 4 weeks in a row without IL-2 or GM-CSF, samples from this group of patients were used for immune correlative studies.
Infusion related toxicities
All vital signs and side effects were recorded on the patient’s chart using the NCI CTC v3.0 toxicity table. Patients were observed up to 6 hours after their infusions. Patients with grade 4 non-hematologic toxicity or persistent grade-3 toxicity would be removed from the study. Infusions were held until toxicity improved to grade 0 or 1. There were no dose limiting toxicities observed.
Immune correlative studies
Specific cytotoxicity was performed using fresh PBMC plated with 51Cr labeled SK-BR-3 cells at effector:target (E/T) of 25:1 unless otherwise indicated.16 Cytotoxicity was calculated by using following formula: [(experimental release – spontaneous release)/(maximum release- spontaneous release) x 100]. The IFN-γ Elispots were used to measure of CD8-mediated memory CTL activity and CD4-mediated helper responses.12 Cytokines were measured by Luminex Array.17 Phenotyping was performed by multicolor flow cytometry.17 Pentamer staining and TCR Vβ repertoire of human T cells were determined by multi-color flow cytometry (see details in supplemental information). An assay for in vitro anti-SK-BR-3 specific antibody synthesis was performed as described.9,21 Specific antibodies were detected by a whole cell ELISA and antigen specific ELISA.13,22 (supplemental information).
Statistical analyses
Wilcoxon signed-rank test was used to compare baselines with each time point from pre-study for cytotoxicity and IFN-γ Elispots against SK-BR-3 cells. Spearman correlation and log-rank tests were used to test the correlation between ATC cytotoxicity and TTP. GraphPad Prism version 6 for Windows (GraphPad Software, San Diego, CA) was used for statistical analysis.
Funding Statement
This study was primarily supported by funding from the Susan G. Komen for the cure (Grant #BCTR0707125) and in part by DHHS R01 CA 092344, R01 CA 140314, and P30CA022453 (Microscopy, Imaging, and Cytometry Resources Core), a startup funds from the University of Virginia Cancer Center.
Acknowledgments
Special thanks to the nurse clinical coordinators: Wendy Young, Lori Hall, Janet McIntyre, Patricia Steele and Kristie Fields who provided clinical and emotional support to the women who participated in this clinical trial. We thank Melissa Dufresne for performing the infusions and scoring toxicities. The immunotherapy team acknowledges the special efforts of all of the members of the Immunotherapy Program at Roger Williams Hospital and the BMT/Immunotherapy Program at Karmanos Cancer Institute who have provided support and infrastructure for the compassionate care of the women with MBC. The Microscopy, Imaging and Cytometry Resources Core is supported, in part, by NIH Center grant P30CA22453 to The Karmanos Cancer Institute, Wayne State University and the Perinatology Research Branch of the National Institutes of Child Health and Development, Wayne State University.
Contributions
AT and LGL wrote the original draft of the manuscript, edited and reviewed by AT, LGL, JPU and VR. AT supervised and analyzed the immune monitoring data. LGL, RR, AT, JPU and VR were involved in the design, writing, regulatory, clinical management of the protocol, cell infusions. AT, LGL and SVK were involved in producing the cGMP product, immune evaluation assays, data collection and management.
Originality Disclosure
The data presented in this manuscript are original and have not been published elsewhere except in the form of abstracts and poster presentations at symposium and meetings.
Conflict of Interest
LGL is co-founder of Transtarget Inc.; AT is co-founder of Nova Immune Platform Inc.; RR, SVK, JPU and VR have no conflicts of interest.
Supplementary Material
The Supplemental data can be accessed here
References
- 1.Sen M, Wankowski DM, Garlie NK, Siebenlist RE, Van Epps D, LeFever AV, Lum LG. . Use of anti-CD3 x anti-HER2/neu bispecific antibody for redirecting cytotoxicity of activated T cells toward HER2/neu+ tumors. J Hematother Stem Cell Res. 2001;10(2):247–260. doi: 10.1089/15258160151134944. [DOI] [PubMed] [Google Scholar]
- 2.Lum LG, Thakur A, Al-Kadhimi Z, Colvin GA, Cummings FJ, Legare RD, Dizon DS, Kouttab N, Maizel A, Colaiace W , et al. Targeted T-cell therapy in stage iv breast cancer: A phase I clinical trial. Clin Cancer Res. 2015;21(10):2305–2314. doi: 10.1158/1078-0432.CCR-14-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lum LG. Immunotherapy with activated T cells after high dose chemotherapy and PBSCT for breast cancer. In: Dicke KA, Keating A, editors: Proc of the 10th International Symposium on Autologous Blood and Marrow Transplantation, Charlottesville, VA: Carden Jennings; 2000, pp 95–105. [Google Scholar]
- 4.Berry DA, Ueno NT, Johnson MM, Lei X, Caputo J, Rodenhuis S, Peters WP, Leonard RC, Barlow WE, Tallman MS , et al. High-dose chemotherapy with autologous stem-cell support as adjuvant therapy in breast cancer: overview of 15 randomized trials. J Clin Oncol. 2011;29(24):3214–3223. doi: 10.1200/JCO.2010.32.5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson-Abelson MR, Purohit VS, Pang WM, Iyer V, Odunsi K, Demmy TL, Yokota SJ, Loyall JL, Kelleher RJ, Jr., Balu-Iyer S , et al. IL-12 delivered intratumorally by multilamellar liposomes reactivates memory T cells in human tumor microenvironments. Clin Immunol. 2009;132(1):71–82. doi: 10.1016/j.clim.2009.03.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curti BD, Longo DL, Ochoa AC, Conlon KC, Smith JW II, Alvord WG, Creekmore SP, Fenton RG Gause BL, Holmlund J, et al. Treatment of cancer patients with ex vivo anti-CD3-activated killer cells and interleukin-2. J Clin Oncol. 1993;11:652–660. doi: 10.1200/JCO.1993.11.4.652. [DOI] [PubMed] [Google Scholar]
- 7.Curti BD, Ochoa AC, Powers GC, Kopp WC, Alvord WG, Janik JE, Gause BL, Dunn B, Kopreski MS, Fenton R , et al. Phase I trial of anti-CD3-stimulated CD4+ T cells, infusional interleukin-2, and cyclophosphamide in patients with advanced cancer. J Clin Oncol. 1998;16:2752–2760. doi: 10.1200/JCO.1998.16.8.2752. [DOI] [PubMed] [Google Scholar]
- 8.Grabert RC, Cousens LP, Smith JA, Olson S, Gall J, Young WB, Davol PA, Lum LG.. Human T cells armed with Her2/neu bispecific antibodies divide, are cytotoxic, and secrete cytokines with repeated stimulation. Clin Cancer Res. 2006;12(2):569–576. doi: 10.1158/1078-0432.CCR-05-2005. [DOI] [PubMed] [Google Scholar]
- 9.Lum LG, Culbertson NJ.. The induction and suppression of in vitro IgG anti-tetanus toxoid antibody synthesis by human lymphocytes stimulated with tetanus toxoid in the absence of in vivo booster immunizations. J Immunol. 1985;135:185–191. [PubMed] [Google Scholar]
- 10.Stadtmauer EA, Vogl DT, Luning PE, Boyer J, Aqui NA, Rapoport AP, McDonald KR, Hou X, Murphy H, Bhagat R , et al. Transfer of influenza vaccine-primed costimulated autologous T cells after stem cell transplantation for multiple myeloma leads to reconstitution of influenza immunity: results of a randomized clinical trial. Blood. 2011;117(1):63–71. doi: 10.1182/blood-2010-07-296822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Fang HB, Cai L, Janofsky S, Chew A, Storek J, Akpek G , et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood. 2011;117(3):788–797. doi: 10.1182/blood-2010-08-299396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, Chrisley L, Veloso E, Zheng Z, Westphal S, Mair R , et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005;11(11):1230–1237. [DOI] [PubMed] [Google Scholar]
- 13.Thakur A, Littrup P, Paul EN, Adam B, Heilbrun LK, Lum LG. Induction of specific cellular and humoral responses against renal cell carcinoma after combination therapy with cryoablation and granulocyte-macrophage colony stimulating factor: a pilot study. J Immunotherapy. 2011;34(5):457. doi: 10.1097/CJI.0b013e31821dcba5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lum LG. Review: the kinetics of immunologic recovery after human marrow transplantation. Blood. 1987;69:369–380. [PubMed] [Google Scholar]
- 15.Witherspoon RP, Lum LG, Storb R, Thomas ED. In vitro regulation of immunoglobulin synthesis after human marrow transplantation. II. Deficient T and non-T lymphocyte function within 3–4 months of allogeneic, syngeneic, or autologous marrow grafting for hematologic malignancy. Blood. 1982;59:844–850. [PubMed] [Google Scholar]
- 16.Wahren B, Gahrton G, Linde A, Ljungman P, Lonnqvist B, Ringden O, Sundqvist V-A. . Transfer and persistence of viral antibody-producing cells in bone marrow transplantation. J Infect Dis. 1984;150:358–365. doi: 10.1093/infdis/150.3.358. [DOI] [PubMed] [Google Scholar]
- 17.Matsue K, Lum LG, Witherspoon RP, Storb R. Proliferative and differentiative responses of B cells from human marrow graft recipients to T cell-derived factors. Blood. 1987;69:308–315. [PubMed] [Google Scholar]
- 18.Lum LG, Seigneuret MC, Storb R. The transfer of antigen-specific humoral immunity from marrow donors to marrow recipients. J Clin Immunol. 1986;6:389–396. [DOI] [PubMed] [Google Scholar]
- 19.Stevens PH, Saxon A. Immunoregulation in humans. Control of anti-tetanus toxoid antibody production after booster immunization. J Clin Invest. 1978;62:1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saxon A, Mitsuyasu R, Stevens R, Champlin RE, Kimata H, Gale RP. Designed transfer of specific immune responses with bone marrow transplantation. J Clin Invest. 1986;78:959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gall JM, Davol PA, Grabert RC, Deaver M, Lum LG. T cells armed with anti-CD3 x anti-CD20 bispecific antibody enhance killing of CD20+ malignant B-cells and bypass complement-mediated Rituximab-resistance in vitro. Exp Hematol. 2005;33(4):452–459. doi: [DOI] [PubMed] [Google Scholar]
- 22.Thakur A, Norkina O, Lum LG. In vitro synthesis of primary specific anti-breast cancer antibodies by normal human peripheral blood mononuclear cells. Cancer Immun Immunotherapy. 2011;60(12):1707–1720. doi: 10.1007/s00262-011-1056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.