

ABSTRACT
Immune checkpoint blockade targeting programmed cell death protein 1 (PD-1) is emerging as an important treatment strategy in a growing list of cancers, yet its clinical benefits are limited to a subset of patients. Further investigation of tumor-intrinsic predictors of response and how extrinsic factors, such as iatrogenic immunosuppression caused by conventional therapies, impact the efficacy of anti-PD-1 therapy are paramount. Given the widespread use of corticosteroids in cancer management and their immunosuppressive nature, this study sought to determine how corticosteroids influence anti-PD-1 responses and whether their effects were dependent on tumor location within the periphery versus central nervous system (CNS), which may have a more limiting immune environment. In well-established anti-PD-1-responsive murine tumor models, corticosteroid therapy resulted in systemic immune effects, including severe and persistent reductions in peripheral CD4+ and CD8 + T cells. Corticosteroid treatment was found to diminish the efficacy of anti-PD-1 therapy in mice bearing peripheral tumors with responses correlating with peripheral CD8/Treg ratio changes. In contrast, in mice bearing intracranial tumors, corticosteroids did not abrogate the benefits conferred by anti-PD-1 therapy. Despite systemic immune changes, anti-PD-1-mediated antitumor immune responses remained intact during corticosteroid treatment in mice bearing intracranial tumors. These findings suggest that anti-PD-1 responses may be differentially impacted by concomitant corticosteroid use depending on tumor location within or outside the CNS. As an immune-specialized site, the CNS may potentially play a protective role against the immunosuppressive effects of corticosteroids, thus sustaining antitumor immune responses mediated by PD-1 blockade.
Keywords: PD-1, immunotherapy, corticosteroid, dexamethasone, colon adenocarcinoma, brain tumor, glioma, central nervous system
Introduction
Blockade of the key inhibitory immune checkpoint, programmed cell death protein 1 (PD-1), aims to reinvigorate antitumor immune responses and has yielded significantly improved outcomes in a variety of advanced hematologic and solid malignancies.1–10 As the list of approvals and clinical trials investigating its efficacy in other tumor types continues to grow, it is paramount to determine factors, both tumor-intrinsic and extrinsic, that influence responses to anti-PD-1 therapy.11 An important concern to address is how iatrogenic immunosuppression caused by conventional therapies such as chemotherapy, fractionated radiotherapy, and corticosteroids may potentially impact the efficacy of anti-PD-1 as well as other immunotherapy strategies.
Corticosteroids are immunomodulatory agents that exert their actions by binding to intracellular receptors, which go on to regulate gene expression and signaling pathways through both genomic and non-genomic mechanisms.12,13 Clinically, corticosteroids are routinely used in the management of cancer for fatigue, night sweats, appetite stimulation, antiemesis, and the reduction of side effects associated with certain chemotherapies (platinum-based agents and taxanes) and immune checkpoint inhibitors.14,15 Corticosteroids are especially important in the management of tumors residing in the central nervous system (CNS), since they can effectively palliate neurological deficits and reduce complications caused by cerebral edema through restoring the integrity of the blood-brain barrier (BBB).16 Moreover, cerebral edema is often potentiated by the additive pro-inflammatory effects of brain radiation plus immunotherapy, even when not given concurrently, thus necessitating the use of corticosteroids post-treatment. Despite these clinical uses, corticosteroids are potent immunosuppressive agents that act on both innate and adaptive immunity. Well-established effects include the induction of T cell apoptosis and the maturation impairment of dendritic cells (DCs).12 For these reasons, concomitant corticosteroid use is thought to be counterproductive in the setting of cancer immunotherapy. Although the issue has not been well investigated, concern about the immunosuppressive effects of corticosteroids has precluded patients from receiving anti-PD-1 therapy, even when otherwise clinically indicated, and has limited eligibility for clinical trials.17
The purpose of this study was to determine whether anti-PD-1 responses were influenced by co-therapy with dexamethasone, which is the most potent synthetic corticosteroid. Recognizing that the immune environment within the CNS is unique and that patients with CNS tumors frequently benefit from corticosteroid medications, the efficacy of anti-PD-1 therapy in the setting of corticosteroids was examined for tumor histologies located within or outside the CNS. Whereas dexamethasone treatment disrupted the benefits of PD-1 blockade in mice bearing peripheral colon adenocarcinoma tumors, antitumor immunity and responses mediated by anti-PD-1 therapy remained intact for those bearing intracranial glioma tumors.
Materials and Methods
Mice, cell lines, therapeutic antibodies, and other reagents
Female wild-type C57BL/6J mice (The Jackson Laboratory), 6–8 weeks old, were used for all described experiments and maintained under pathogen-free conditions at the Sidney Kimmel Comprehensive Cancer Center Animal Facilities. All animal handling and procedures were conducted in accordance with approved protocols by the Institutional Animal Care and Use Committee (IACUC) at Johns Hopkins University. MC38 (kindly provided by Dr. Charles Drake) and luciferase-expressing GL261 (GL261-Luc, purchased from Caliper Life Sciences) cells are syngeneic murine-derived colon adenocarcinoma and glioma cell lines, respectively, that are sensitive to PD-1 blockade in vivo.18–21 Both cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Gemini Bio-Products) and 1% penicillin-streptomycin (Quality Biological) at 37°C in a humidified incubator maintained at 5% CO2 and 5% O2. Additionally, GL261-Luc cells were kept under selection conditions with culture media containing 100 ug/mL G418 sulfate antibiotic (Corning). Both cell lines underwent routine mycoplasma infectivity testing using MycoAlert Mycoplasma Detection Kit (Lonza).
For mouse therapy, hamster anti-murine PD-1 monoclonal antibodies were purified from the G4 hybridoma cell line, and three doses were administered every other day at 10 mg/kg (approximately 200 ug/dose) via intraperitoneal injection.22 Dexamethasone 21-phosphate (Sigma-Aldrich) reconstituted in PBS was also administered at a high dose of 10 mg/kg (approximately 200 ug/dose) via intraperitoneal injection. For experiments, three dexamethasone regimens were investigated – (1) short-course (5 days), concurrent with anti-PD-1 therapy (Dex-1); (2) short-course (5 days), following anti-PD-1 therapy (Dex-2); and (3) long-course (4 weeks), continuous therapy with taper starting on date of randomization (Dex-Cont). Untreated (Control) mice were used as a reference group for single and combination treatment groups.
In vivo flank tumor model
Flank tumors were established by subcutaneously injecting 500,000 MC38 cells in a volume of 200 uL PBS in the shaved right flanks of C57BL/6J mice (day 0). Once all tumors were palpable and measurable, mice were randomized to ensure tumor volumes were equal amongst the groups (day 8). Perpendicular tumor diameters, length (mm, longer dimension) and width (mm, shorter dimension), were then measured every other day using calibrated calipers. Tumor volumes were calculated using the following formula: 0.5 x length x width2. Mice were euthanized when their tumor volumes exceeded 2000 mm3 or when they displayed signs of functional decline (hunched posture, poor grooming, lethargy, extreme weight loss, feeding difficulties, or inability to ambulate).
In vivo intracranial tumor model
To establish orthotopic gliomas, 130,000 GL261-Luc cells in a 1-uL volume of PBS were stereotactically injected into the left striatum of C57BL/6J mice (day 0), as previously described.19 Briefly, mice were properly anesthetized prior to procedure with a ketamine (5 mg/mL, Henry Schein)/xylazine (0.5 mg/mL, Akorn) solution in 0.9% saline. A small midline incision was made, and a burr hole was then placed on the left-side 1 mm anterolateral to bregma. Mice were positioned in a stereotactic frame, and the GL261-Luc cell suspension was injected approximately 3 mm below the cortical surface using a Hamilton micro-syringe (Sigma-Aldrich). Incisions were closed using staples. On day 7, mice underwent bioluminescence imaging using an In Vivo Imaging System (IVIS, Caliper Life Sciences) to confirm tumor establishment. Mice were also randomly assigned that day to ensure tumor burden (average radiance) was equal amongst the groups. Glioma-bearing mice were followed for survival for 90 days or euthanized for flow cytometry experiments for immunophenotyping. For survival experiments, mice were euthanized if they displayed neurological deficits or signs of functional decline (described above). For tumor rechallenge experiments, long-term responders (survival > 90 days) and naïve control mice were implanted in a similar fashion with 260,000 GL261-Luc cells within the contralateral cerebral hemisphere.
Blood and organ collection and processing
Blood (approximately 175 uL) was collected from the retro-orbital venous plexus of each mouse using a heparinized capillary tube (Thermo Fisher Scientific). Red blood cells were removed using 200 uL of ammonium-chloride-potassium (ACK) lysing buffer (Quality Biological) followed by thorough washes with PBS. Cervical tumor-draining lymph nodes (TDLN) were carefully dissected from the surrounding tissue on post-tumor implantation day 19. TDLN were mechanically dissociated in Roswell Park Memorial Institute (RPMI) 1640 Medium (Thermo Fisher Scientific) supplemented with 10% FBS and 1% penicillin-streptomycin followed by PBS washes.
Flow cytometry
For flow cytometric analyzes, recovered immune cells from processed blood and organs were first stained for surface markers using the following anti-mouse antibodies: CD3 BV421 (clone 17A2, BioLegend), CD4 FITC (clone RM4-4, eBioscience), CD8a BV605 (clone 53–6.7, BioLegend), and/or CD44 AF700 (clone IM7, BioLegend). Live/dead cell discrimination was done using LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific). Cells were then fixed followed by staining with anti-mouse FOXP3 AF700 or APC (clone FJK-16s, eBioscience). For blood samples, 25 uL of AccuCheck Counting Beads (Thermo Fisher Scientific) were added to determine absolute cell counts. Samples were acquired on an LSR II Flow Cytometer (Becton Dickinson). Flow cytometry data were analyzed using FlowJo software (FlowJo, LLC). Lymphocytes were gated on using side vs. forward scatter plots followed by doublet and live/dead discrimination. CD8+ and CD4+ T cells were then gated on live CD3+ T cells. Regulatory T cells (Treg) and effector CD8+ T cells (CD8eff) were defined as CD4+ FOXP3+ and CD8+ CD44hi cells, respectively. Fluorescence Minus One (FMO) controls were used to guide FOXP3 and CD44 gating strategies. Absolute cell counts were calculated using the counting beads according to the manufacturer’s protocol.
Statistical analysis
Throughout this manuscript, group statistics are represented as mean ± standard error of mean (SEM). Unless stated otherwise, Student’s t-test, which assumes equal variances, was used to compare means between two groups. In some circumstances, Welch’s t-test was utilized if the assumption of equal variances was invalid. Means across multiple groups were compared using one-way analysis of variance (ANOVA) with Tukey’s or Dunnett’s multiple comparisons test (usually untreated control mice as reference group). When appropriate, individual tumor growth curves were fitted with exponential growth equations, and tumor doubling times were calculated by dividing the natural logarithm of 2 by the exponential growth constant. If a mouse’s tumor growth could not be properly fitted (R2 < 0.95) with an exponential growth equation, its tumor growth patterns were followed for a total of 50 days. If the mouse’s tumor failed to double in size within that period, the doubling time was set to 50 days. For survival experiments, Kaplan-Meier curves were constructed and analyzed using the log-rank test. Linear and Cox regression analyzes were used to determine predictors of tumor growth and survival, respectively. Two-tailed P-values less than a type I error rate of 0.05 were considered statistically significant. All statistical analyzes were carried out using either Prism 7 (GraphPad) or SAS version 9.2 (SAS Institute) software.
Results
Anti-PD-1 therapy inhibits tumor growth and mediates tumor regressions in mice bearing colon adenocarcinoma MC38 flank tumors while dexamethasone treatment abrogates these effects
First, this study aimed to determine whether the timing and duration of dexamethasone would impact anti-PD-1 therapy in an anti-PD-1-responsive peripheral flank tumor model (N = 64) (Figure 1A). Untreated mice and those receiving only dexamethasone treatment (Dex-1, Dex-2, and Dex-Cont) had uninhibited, exponential tumor growth patterns (Figure 1B). Consequently, no mice (0/8, 0.0%) were found to be tumor-free on day 50 within these groups. As expected, anti-PD-1 monotherapy inhibited tumor growth and mediated tumor regressions in this flank tumor model. Four of the 8 mice (50.0%) given anti-PD-1 therapy alone had complete responses and were without tumors long-term. However, this level of complete response rate was not observed in the combination anti-PD-1 plus dexamethasone therapy groups. In mice that received short-course dexamethasone either before or after anti-PD-1 therapy (anti-PD-1 + Dex-1 and anti-PD-1 + Dex-2, respectively), only 1 of the 8 mice (12.5%) were tumor-free on day 50. Long-term responses were completely abrogated in the anti-PD-1 + Dex-Cont group (0/8, 0.0% tumor-free on day 50). Interestingly, 2 of the 8 (25.0%) mice within the anti-PD-1 + Dex-2 group initially experienced a partial response (≥ 50% decrease in tumor volume from baseline but still with measurable tumor) with therapy but ultimately had progressive disease. These observed differences in tumor growth kinetics between the groups were confirmed by comparing tumor doubling times (Figure 1C). Compared to untreated controls, mice that received anti-PD-1 therapy alone experienced significantly longer tumor doubling times thus delaying tumor growth (P = 0.0023). On the contrary, doubling times for the dexamethasone alone and anti-PD-1 combination treatment groups were not significantly different compared to control mice.
Figure 1.
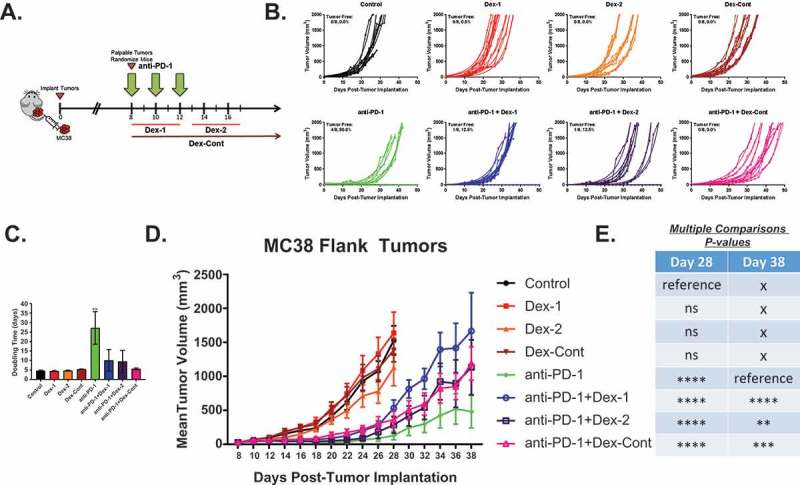
Dexamethasone impairs colon adenocarcinoma MC38 flank tumor growth patterns in mice administered anti-PD-1 therapy. (A) Schematic diagram of experimental design and treatment paradigm: Female C57BL/6J mice (N = 64) were subcutaneously inoculated with 500,000 colon adenocarcinoma MC38 cells in the right flank (day 0) and monitored for tumor growth. Once tumors were palpable, mice were randomized (8 mice/group) to ensure tumor burden was equal amongst the groups (day 8). Mice were administered anti-PD-1 antibody (200 ug/dose, 10 mg/kg, intraperitoneal injection) on days 8, 10, and 12; concurrent dexamethasone (200 ug/dose, 10 mg/kg, intraperitoneal injection) on days 8 through 12 (Dex-1); sequential dexamethasone on days 13 through 17 (Dex-2); and/or continuous dexamethasone from days 8 (randomization date) through 36 (4 weeks) with taper (Dex-Cont). (B) Individual tumor volume growth curves by treatment group: Mice that did not receive anti-PD-1 therapy had continued exponential tumor growth, resulting in no tumor-free mice (0.0%). Anti-PD-1 monotherapy resulted in 50.0% tumor-free mice. However, combination anti-PD-1 and dexamethasone therapy appeared to disrupt this trend (% tumor-free: anti-PD-1 + Dex-1, 12.5%; anti-PD-1 + Dex-2, 12.5%; anti-PD-1 + Dex-Cont, 0.0%). (C) Anti-PD-1 monotherapy resulted in a significantly prolonged tumor doubling time compared to untreated control mice. This effect was abrogated by dexamethasone therapy. (D) and (E) Tumor volume growth patterns between the groups: Anti-PD-1 therapy, regardless of dexamethasone treatment, delayed the early growth of tumors compared to untreated control mice on day 28. However, this effect was abrogated with dexamethasone treatment long-term on day 38. Means were compared using one-way ANOVA and Dunnett’s multiple comparisons test (untreated control and anti-PD-1 alone arms were the designated reference groups on days 28 and 38, respectively). ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. If no asterisks are shown in (C), statistical analyzes yielded non-significant P-values.
At day 28 (approximately 2 weeks following completion of anti-PD-1 therapy), anti-PD-1 therapy, regardless of dexamethasone treatment, inhibited tumor growth compared to untreated mice (all P < 0.0001, Figure 1D and 1E). However, this effect was short-lived as the anti-PD-1 + Dex-1 (P < 0.0001), anti-PD-1 + Dex-2 (P = 0.0017), and anti-PD-1 + Dex-Cont (P = 0.0003) groups were all found to have significantly larger tumor volumes compared to mice receiving anti-PD-1 therapy alone on day 38. Ultimately, dexamethasone impaired the long-term benefits conferred by anti-PD-1 therapy in this flank tumor model, as shown by comparing individual tumor growth curves and doubling times. Nonetheless, mice that received combination anti-PD-1 and dexamethasone therapy did experience some temporary benefits from anti-PD-1 therapy suggesting that PD-1 blockade in the setting of dexamethasone could produce clinical, albeit non-durable, benefits in peripheral tumors.
Dexamethasone therapy significantly alters T cell compartments in peripheral blood
Corticosteroids dramatically affect peripheral T cell survival and homeostasis in vivo.23–25 These potential dexamethasone-induced effects were investigated within the MC38 flank tumor model by measuring absolute cell counts of various T cell subsets in peripheral blood. To accomplish this, peripheral blood from the mice (N = 64) of the previously discussed tumor growth experiment were analyzed at multiple time points including days 8 (pre-treatment), 15 (shortly after anti-PD-1 therapy completion), and 27 (long time after anti-PD-1 therapy) (Figure 2A). In addition to determining dexamethasone’s systemic immune effects, potential peripheral T cell changes associated with anti-PD-1 responses in the flank tumor model were also evaluated.
Figure 2.
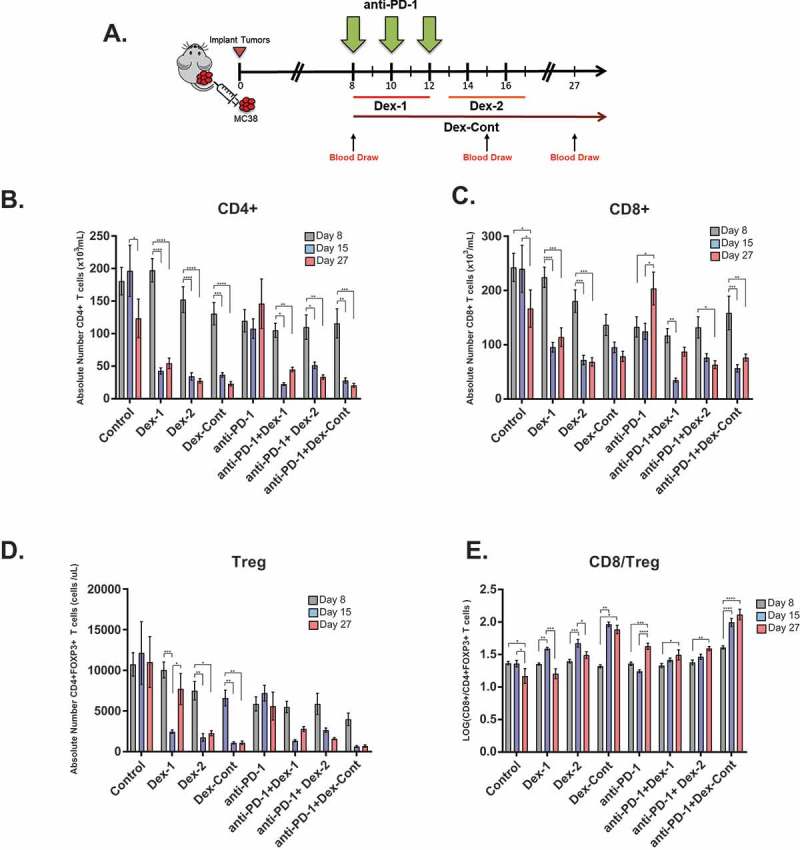
Dexamethasone alone or combined with anti-PD-1 therapy significantly alters peripheral T cell compartments in mice bearing colon adenocarcinoma MC38 flank tumors. (A) Timeline of blood draws relative to treatment schedule in MC38 flank tumor experiment (N = 64, 8 mice/group): Blood was collected from the retro-orbital venous plexus of each mouse on days 8 (pre-treatment), 15 (shortly after anti-PD-1 therapy completion), and 27 (long time after anti-PD-1 therapy). Depicted are absolute numbers of (B) CD4+, (C) CD8+, (D) CD4+ FOXP3+ regulatory T cells (Treg), and (E) CD8/Treg ratios in peripheral blood by treatment group. Means were compared within each group using one-way ANOVA and Tukey’s multiple comparisons test. Dexamethasone treatment, either alone or in combination with anti-PD-1 therapy, resulted in persistent reductions within CD4+, CD8+, and Treg compartments. Anti-PD-1 therapy improved peripheral CD8/Treg ratios. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. If no asterisks are shown, statistical analyzes yielded non-significant P-values.
All dexamethasone regimens, whether given alone or in combination with anti-PD-1 therapy, resulted in significant reductions of circulating CD4+ T cells (Figure 2B). These levels were persistently low as witnessed on day 27. Absolute numbers of circulating CD8+ T cells also displayed similar significant trends with dexamethasone treatment (Figure 2C). In untreated control mice, there were significant decreases in both peripheral CD4+ and CD8+ T cell compartments from day 15 to 27. Anti-PD-1 monotherapy had no apparent effects on the number of circulating CD4+ T cells, but it did result in significantly higher levels of CD8+ T cells on day 27.
Previous studies have shown that dexamethasone can cause dose-dependent decreases in the number of immunosuppressive CD4+FOXP3+ Treg cells within peripheral blood of mice.26 Consistent with these studies, the Dex-1, Dex-2, and Dex-Cont groups had significant reductions in circulating Treg cell levels from baseline (Figure 2D). Absolute Treg cell counts were also found to be lower in mice receiving combination anti-PD-1 and dexamethasone therapy, but these trends were not statistically significant. Lack of treatment and anti-PD-1 monotherapy did not appear to alter circulating Treg cell levels in any significant way.
As successful antitumor immunity requires an improved balance between effector and suppressor immune cells, peripheral CD8/Treg ratio changes in the MC38 flank tumor model were analyzed. Untreated mice had significant declines in CD8/Treg ratios on day 27 compared to either day 8 or 15 (Figure 2E). Mice treated with dexamethasone alone exhibited an initial elevation in their CD8/Treg ratios from day 8 to 15, which were likely due to the dramatic reductions within the Treg compartment relative to the CD8 + T cell compartment (Figure 2D). CD8/Treg ratios failed to remain elevated on day 27 with short-course dexamethasone use (Dex-1 and Dex-2 groups); however, they remained elevated in mice receiving the Dex-Cont regimen. Conversely, anti-PD-1 therapy, regardless of dexamethasone treatment, significantly increased peripheral CD8/Treg ratios on day 27 compared to baseline.
Peripheral CD8/Treg ratio changes associate with anti-PD-1 responses in mice bearing colon adenocarcinoma MC38 flank tumors
Anti-PD-1 monotherapy inhibited tumor growth patterns and mediated durable tumor regressions in the MC38 flank tumor model, which were diminished by combination dexamethasone treatment. Using mouse-matched peripheral blood data, this study sought to determine peripheral T cell changes associated with these anti-PD-1 responses. Compared to untreated controls, mice receiving anti-PD-1 monotherapy had significantly higher changes in their CD8/Treg ratios shortly following completion of anti-PD-1 therapy on days 15 to 27 (+31.4% ± 4.8% vs. −13.4% ± 9.3%, P < 0.0001, Supplementary Figure 1A). Similar to controls, mice receiving only dexamethasone treatment had negative CD8/Treg ratio changes (Dex-1: −22.8% ± 5.6%; Dex-2: −10.1% ± 13.0%; Dex-Cont: −3.1% ± 4.7%, all P > 0.05). Mice that received combination anti-PD-1 and dexamethasone therapy had positive CD8/Treg ratio changes, but these were not as robust compared to anti-PD-1 monotherapy (anti-PD-1 + Dex-1: +6.1% ± 6.5%, anti-PD-1 + Dex-2: +9.2% ± 3.1%, and anti-PD-1 + Dex-Cont: +6.7% ± 5.6%). These changes were not significantly different compared to untreated controls. Furthermore, there was a negative correlation between CD8/Treg ratio and tumor volume changes amongst the groups (slope = −0.35, R2 = 0.82, P = 0.0019, Supplementary Figure 1B).
Given these group findings, CD8/Treg ratio changes and tumor growth patterns were evaluated on an individual basis to determine if there was a correlation. On analysis, CD8/Treg ratio changes were found to be inversely related to tumor volume changes (slope = −1.20, R2 = 0.23, P < 0.0001, Figure 3A). Of note, six mice were excluded from this analysis because they reached euthanization endpoints and lacked data (Control: n = 3, Dex-1: n = 1, Dex-2: n = 1, Dex-Cont: n = 1). Tumor regressions (n = 8) were observed among mice receiving anti-PD-1 therapy. Complete responses were seen in 6 of 8 mice (anti-PD-1: n = 4, anti-PD-1 + Dex-1: n = 1, anti-PD-1 + Dex-2: n = 1), which had corresponding positive CD8/Treg ratio changes (purple open circles). Two mice within the anti-PD-1 + Dex-2 group had partial responses that were then followed by progressive disease (gray open circles, mice labeled #1–2). One of these mice had a positive CD8/Treg ratio change (mouse #2), but the other had a negative change (mouse #1). As a reference, exponential tumor growth in untreated mice coincided with decreasing, negative CD8/Treg ratio changes from baseline over time (−1.65% ± 5.14% to −14.78% ± 8.58%, Figure 3B, top left panel). In complete responders, tumor growth at earlier time points of the experiment coincided with an overall negative CD8/Treg ratio change from baseline (−5.42% ± 3.52%, Figure 3B, bottom panel). Interestingly, as the tumors began to shrink, peripheral CD8/Treg ratio changes transitioned from net negative to positive and became significantly elevated compared to baseline (+20.24% ± 3.02%, P = 0.0002). On the contrary, CD8/Treg ratio changes remained unchanged in those with partial responses followed by progressive disease (+2.66% ± 7.52% to +2.32% ± 3.65%, Figure 3B, top right panel). Supplementary Figure 2 shows the different patterns of peripheral CD8/Treg ratio changes and tumor responses for the individual mice with regressions.
Figure 3.
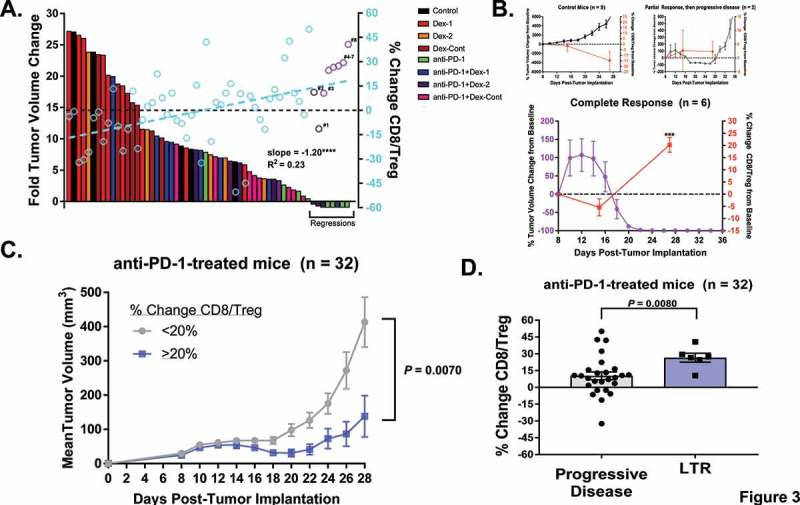
Peripheral CD8/Treg ratio changes correlate with tumor regressions and long-term responses in mice bearing colon adenocarcinoma MC38 flank tumors given anti-PD-1 therapy with or without dexamethasone. (A) Waterfall plot (n = 58) of fold tumor volume changes from day 12 (immediately after last anti-PD-1 dose) to day 28 (approximately 2 weeks following anti-PD-1 treatment) overlaid with scatter plot of peripheral CD8/Treg ratio changes from day 15 to 27 (open circles): As visualized, mice treated with anti-PD-1 therapy alone represented the majority of mice with tumor regressions (n = 8). Two mice within the anti-PD-1 + Dex-2 group (gray open circles, labeled #1–2) were found to have partial responses, which were then followed by progressive disease. Six mice (purple open circles, labeled #3–8, anti-PD-1: n = 4, anti-PD-1 + Dex-1: n = 1, anti-PD-1 + Dex-2: n = 1) had complete responses, which were durable. Additionally, tumor volume changes were inversely related to CD8/Treg ratio changes (slope = −1.20, R2 = 0.23, P < 0.0001). (B) Dynamics between tumor volume and peripheral CD8/Treg ratio changes from baseline in untreated controls (top left panel), partial responders followed by progressive disease (top right panel), and complete responders (bottom panel): As visualized, tumor growth typically coincided with drops in CD8/Treg ratios in control mice. However, complete responders had a significant increase in their CD8/Treg ratios, which correlated with tumor regressions. The partial responders who ultimately had PD failed to display this trend. (C) In all mice administered anti-PD-1 therapy (n = 32), regardless of dexamethasone treatment, those with ≥ 20% change in peripheral CD8/Treg ratios had significantly slower tumor growths compared to those with < 20% changes. (D) Moreover, in all mice treated with anti-PD-1 therapy (n = 32), long-term responders (LTR) had higher peripheral CD8/Treg ratio changes compared to those with progressive disease (P = 0.0080). Means for (C) and (D) were compared using Welch’s t-test assuming unequal variances. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. If no asterisks are shown, statistical analyzes yielded non-significant P-values.
Lastly, all mice treated with anti-PD-1 therapy (n = 32) were analyzed to determine whether peripheral CD8/Treg ratio changes could predict responses, regardless of dexamethasone treatment. In anti-PD-1-treated mice, those with ≥ 20% increases in their CD8/Treg ratios had slower tumor growth patterns compared to those with < 20% increases (tumor volumes on day 28: 138 mm3 ± 60 mm3 vs. 413 mm3 ± 73 mm3, P = 0.0070, comparison done using Welch’s t-test, Figure 3C). Additionally, long-term responders had significantly higher CD8/Treg ratio changes compared to non-responders with progressive disease (26.51% ± 3.95% vs. 10.31% ± 3.44%, P = 0.008, comparison done using Welch’s t-test, Figure 3D). Taken together, these results suggest that monitoring peripheral CD8/Treg ratio changes could predict anti-PD-1 responses in mice bearing colon adenocarcinoma flank tumors.
Dexamethasone does not abrogate the survival benefit conferred by anti-PD-1 therapy in intracranial glioma-bearing mice
Aforementioned findings reveal dexamethasone treatment disrupts the benefits conferred by anti-PD-1 therapy in a flank tumor model. The next analysis was to determine whether dexamethasone’s influence on anti-PD-1 therapy varied depending on tumor location within the CNS. To investigate this question, a well-established anti-PD-1-responsive, intracranial GL261-Luc tumor model (N = 80) was utilized (Figure 4A).19–21 Single therapy with Dex-1, Dex-2, or Dex-Cont did not appear to improve survival compared to control mice (Figure 4B through 4D). In accordance with previous studies, anti-PD-1 monotherapy significantly prolonged survival compared to untreated controls in the intracranial tumor model (P = 0.020).19–21 Combination anti-PD-1 and dexamethasone therapy prolonged survival compared to untreated mice (anti-PD-1 + Dex-1: P = 0.0047, anti-PD-1 + Dex-2: P = 0.020, anti-PD-1 + Dex-Cont: P = 0.0061). There were no significant differences in survival between the anti-PD-1 monotherapy and combination treatment groups. Additionally, there were no significant differences in tumor burdens as measured by bioluminescence intensities using an IVIS between the anti-PD-1 monotherapy and combination treatment groups at multiple time points (Supplementary Figure 3).
Figure 4.
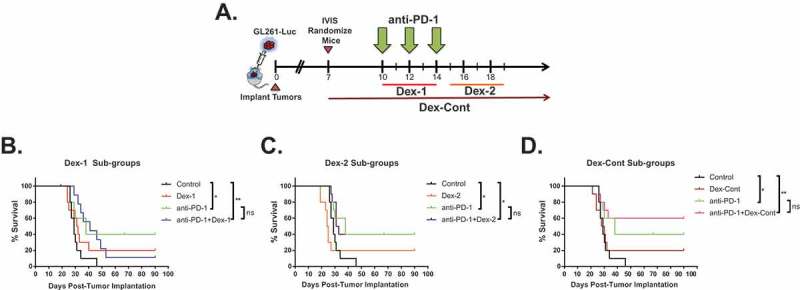
Dexamethasone does not abrogate the survival benefit conferred by anti-PD-1 therapy in intracranial glioma-bearing mice. (A) Schematic diagram of experimental design and treatment paradigm: Female C57BL/6J mice (n = 80) were intracranially implanted with 130,000 luciferase-expressing glioma (GL261-Luc) cells within the left striatum and monitored for tumor growth by bioluminescence imaging using an In Vivo Imaging System (IVIS). On day 7, mice were randomized (10 mice/group) to ensure tumor burden was equal amongst the groups. Mice were administered anti-PD-1 antibody (200 ug/dose, 10 mg/kg intraperitoneal injection) on days 10, 12, and 14; concurrent dexamethasone (200 ug/dose, 10 mg/kg, intraperitoneal injection) on days 10 through 14 (Dex-1); sequential dexamethasone on days 15 through 19 (Dex-2); and/or continuous dexamethasone from days 7 (randomization date) through 35 (4 weeks) with taper (Dex-Cont). (B) Dex-1, (C) Dex-2, and (D) Dex-Cont sub-group analyzes demonstrate combination therapy with dexamethasone does not significantly impair the survival advantage conferred by PD-1 blockade in mice harboring intracranial GL261-Luc tumors. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Alterations in systemic T cell compartments displays dexamethasone’s efficacy in intracranial glioma-bearing mice
In comparison to the flank tumor model, the benefits conferred by anti-PD-1 therapy were not significantly impaired by combination dexamethasone treatment in mice bearing intracranial tumors. Given these findings, peripheral blood on experiment days 19 (shortly after anti-PD-1 therapy completion) and 27 (long time after anti-PD-1 therapy completion) were analyzed to determine whether dexamethasone treatment produced its stereotypical systemic effects in the intracranial tumor model (N = 80) (Figure 5A). Consistent with the flank tumor model, all dexamethasone regimens, whether given alone or in combination with anti-PD-1 therapy, resulted in significant reductions in peripheral CD4+, CD8+, and Treg cell compartments on day 19 compared to untreated controls (Figure 5B through 5D). Peripheral CD4+, CD8+, and Treg cell counts remained persistently low on day 27 but were not significantly different compared to untreated mice, who exhibited decreased T cell counts at day 27. In mice given anti-PD-1 therapy alone, circulating CD4+ and CD8+ T cell levels were maintained throughout the experiment and were significantly higher compared to untreated mice on day 27 (Figure 5B and 5C). There were no significant differences in circulating Treg cell counts on days 19 and 27 between untreated controls and mice receiving anti-PD-1 monotherapy (Figure 5D). Peripheral CD8/Treg ratios were significantly elevated on days 15 and 27 in mice administered the Dex-Cont regimen compared to control mice (Figure 5E). Similar to the flank tumor model, these findings were likely due to dramatic reductions within the Treg compartment relative to CD8+ T cell compartment in mice that received the Dex-Cont regimen.
Figure 5.
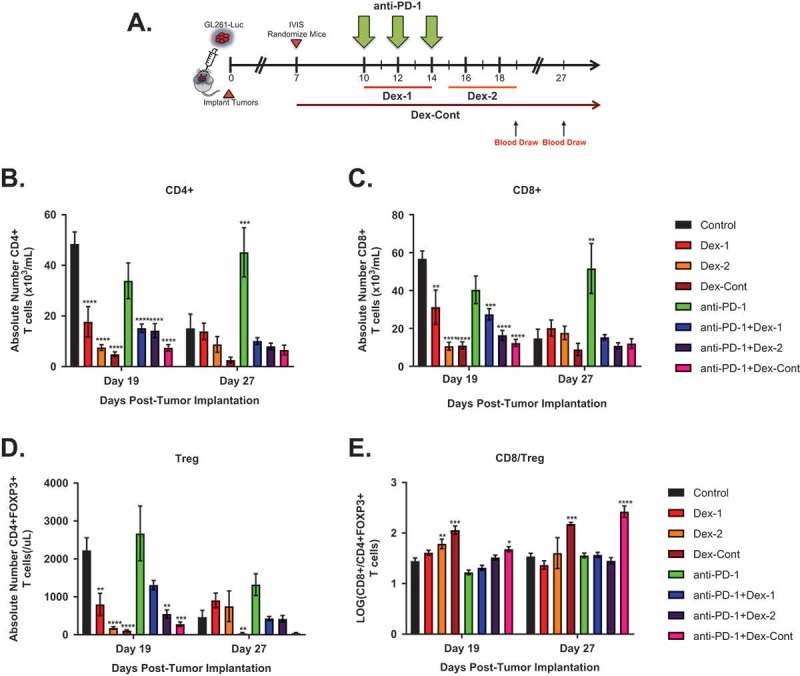
Dexamethasone alone or combined with anti-PD-1 therapy significantly alters peripheral T cell compartments in mice bearing intracranial glioma tumors. (A) Timeline of blood draws relative to treatment schedule in the intracranial GL261-Luc experiment (N = 80, 10 mice/group): Blood was collected from the retro-orbital venous plexus of each mouse on days 19 and 27. Depicted are absolute numbers of (B) CD4+, (C) CD8+, (D) CD4+ FOXP3+ regulatory T cells (Treg), and (E) CD8/Treg ratios in peripheral blood by treatment group. Dexamethasone treatment, either alone or in combination with anti-PD-1 therapy, resulted in persistent reductions within CD4+, CD8+, and Treg compartments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. If no asterisks are shown, statistical analyzes yielded non-significant P-values.
Overall, there were similar trends in peripheral T cell compartments between the flank and intracranial tumor models. However, unlike the flank tumor model where peripheral CD8/Treg ratio changes correlated with anti-PD-1 responses, peripheral T cell changes were not found to associate with survival in mice bearing intracranial glioma tumors (Supplementary Figure 4A). Notably, in mice given anti-PD-1 therapy, peripheral CD4+ (HR = 0.95, 95% CI = 0.59–1.52, P > 0.05), CD8+ (HR = 0.98, 95% CI = 0.58–1.68, P > 0.05), Treg (HR = 1.24, 95% CI = 0.95–1.62, P > 0.05), and CD8/Treg ratio (HR = 1.09, 95% CI = 0.54–2.20, P > 0.05) changes were not significantly associated with survival (Supplementary Figure 4B). These findings suggest that survival in the intracranial tumor model may be independent of the systemic CD4+, CD8+, Treg, or CD8/Treg ratio alterations caused by dexamethasone therapy.
Dexamethasone does not disrupt anti-PD-1-mediated nodal antitumor immune responses in intracranial glioma-bearing mice despite systemic effects
For a comparison to the peripheral blood data, cervical tumor-draining lymph nodes (TDLN) of glioma-bearing mice (5 mice/group) were next analyzed to determine whether anti-PD-1-mediated antitumor immune responses remained intact during combination dexamethasone therapy. TDLN were carefully dissected and processed for flow cytometry analysis following completion of the Dex-2 regimen on day 19. As an observation, TDLN were atrophic in mice receiving dexamethasone alone but remained bulky in those receiving combination anti-PD-1 and dexamethasone therapy. These observations are illustrated when examining flow cytometry scatter plots of CD4+ vs. CD8 + T cells (Figure 6). Unlike the peripheral blood results on day 19, CD4+ and CD8+ T cell levels in TDLN of mice receiving combination anti-PD-1 and dexamethasone therapy remained relatively unchanged compared to untreated mice. Relative numbers of activated CD8eff T cells were significantly higher in mice treated with anti-PD-1 therapy despite dexamethasone treatment (anti-PD-1 vs. Control: P = 0.026, anti-PD-1 + Dex-1 vs. Dex-1: P = 0.014, anti-PD-1 + Dex-2 vs. Dex-2: P < 0.0001, anti-PD-1 + Dex-Cont vs. Dex-Cont: P = 0.0056), whereas Treg cell levels remained unchanged (Supplementary Figure 5). This resulted in improved CD8eff/Treg ratios with anti-PD-1 therapy despite dexamethasone treatment (anti-PD-1 vs. Control: P = 0.014, anti-PD-1 + Dex-1 vs. Dex-1: trending towards significance P = 0.067, anti-PD-1 + Dex-2 vs. Dex-2: P = 0.0016, anti-PD-1 + Dex-Cont vs. Dex-Cont: P = 0.017). Taken together, these data suggest antitumor immune responses mediated by anti-PD-1 therapy may remain intact during dexamethasone treatment in the intracranial tumor model.
Figure 6.
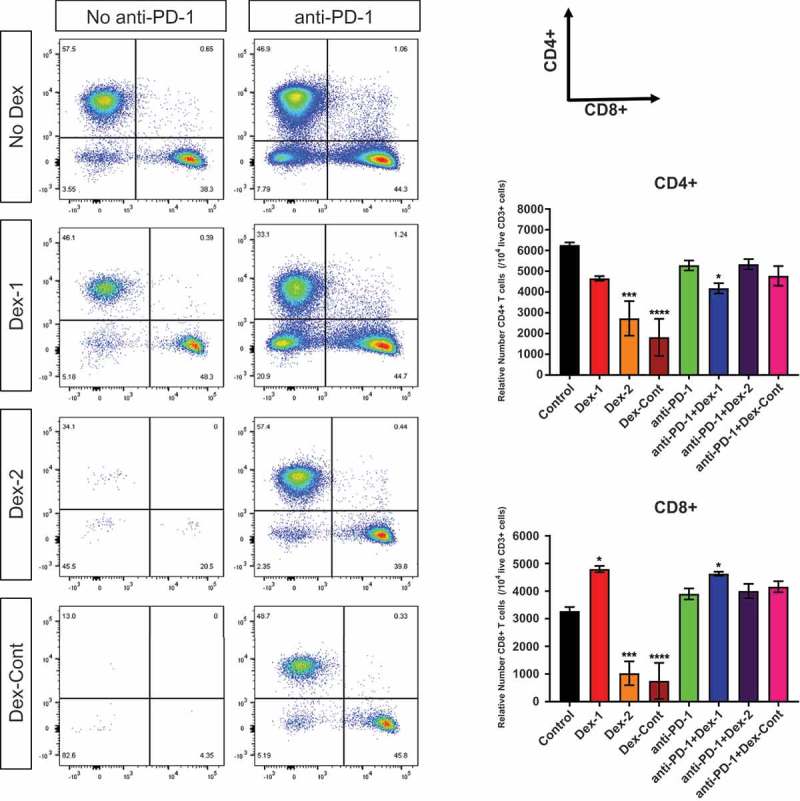
Anti-PD-1-mediated antitumor immune responses are maintained during combination dexamethasone therapy, as witnessed in tumor-draining lymph nodes (TDLN) of glioma-bearing mice. Representative flow cytometry scatter plots of CD4+ vs. CD8+ surface markers (gated on live CD3+ T cells) and their aggregate analyzes derived from TDLN (harvested on day 19, 5 mice/group): Compared to untreated control mice, dexamethasone monotherapy substantially reduced CD4+ and CD8+ T cell numbers in TDLN. However, these levels were maintained in mice receiving combination anti-PD-1 and dexamethasone therapy. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. If no asterisks are shown, statistical analyzes yielded non-significant P-values.
Combination anti-PD-1 and dexamethasone therapy results in long-term tumor-specific immunologic memory
Lastly, long-term responders to anti-PD-1 therapy were tumor rechallenged to determine whether tumor-specific immunologic memory was compromised in the setting of combination dexamethasone therapy. On day 7 post-tumor rechallenge, all naïve control mice had tumor formation as confirmed by bioluminescence imaging (Figure 7A). Consequently, these mice died due to their large tumor burdens (Figure 7B). On the contrary, long-term responders treated with anti-PD-1 monotherapy were protected against tumor formation and had prolonged survival, suggesting intact immunologic memory. These effects were preserved in long-term responders treated with combination anti-PD-1 and dexamethasone therapy. Interestingly, 1 of the 6 mice (17%) in the anti-PD-1 + Dex-Cont group established tumor on day 21 and died on day 37.
Figure 7.
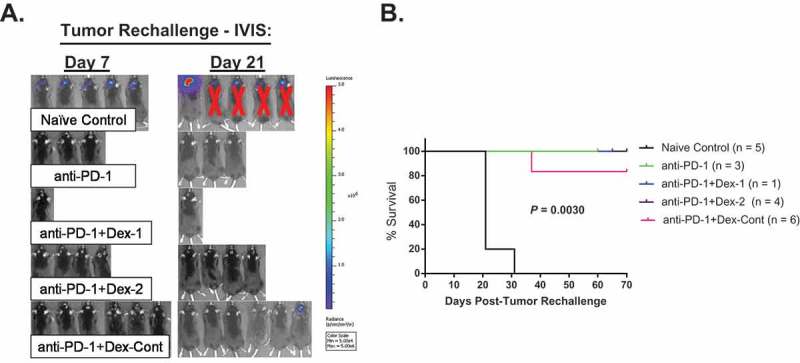
Tumor-specific immunologic memory is grossly preserved in long-term responders treated with combination anti-PD-1 and dexamethasone therapy. Briefly, long-term survivors along with naïve control mice (n = 5) underwent tumor rechallenge by implanting 260,000 GL261-Luc cells within the contralateral cerebral hemisphere (day 0). Tumor burden was monitored via bioluminescence using an In Vivo Imaging System (IVIS). (A) IVIS imaging results on days 7 and 21 following tumor rechallenge: On day 7, all naïve control mice established tumors, while none of the long-term survivors had tumors. However, 1 of 6 mice (17%) in the anti-PD-1 + Dex-Cont group was found to have a tumor on day 21. (C) Survival analyzes of tumor rechallenge experiment: Except for the naïve controls and the one anti-PD-1 + Dex-Cont mouse with tumor on day 21, all other mice had prolonged survival following tumor rechallenge, suggesting intact immunologic memory.
Discussion
Monoclonal antibodies targeting PD-1 have produced durable tumor regressions and survival benefits in an expanding list of solid and hematologic malignancies.1–10 Given the ubiquitous use of corticosteroids in the medical management of cancer patients and their immunosuppressive effects, this study primarily sought to determine whether responses mediated by anti-PD-1 therapy are influenced by corticosteroids in a preclinical setting. Utilizing well-established in vivo tumor models that are sensitive to PD-1 blockade, this study examined whether dexamethasone’s impact was dependent on its timing, duration, and tumor location within or outside the CNS. Overall, short- or long-course dexamethasone treatment resulted in persistent systemic lymphodepletion and ultimately impaired anti-PD-1 responses in mice bearing flank tumors. Contrary to these findings, the survival benefit conferred by anti-PD-1 therapy was not significantly impacted by dexamethasone in the intracranial tumor model. Taken together, these findings suggest that the anatomical location of tumors may influence anti-PD-1 responses in the setting of corticosteroid administration.
Classically regarded as immunosuppressants, corticosteroids modulate both the innate and adaptive arms of the immune system.12 Corticosteroids can prevent adequate T cell responses by inhibiting the antigen presenting capabilities of immature DCs through the downregulation of major histocompatibility complex (MHC) class II and co-stimulatory molecules, including CD80 (B7.1) and CD86 (B7.2).27 Supra-physiologic doses of corticosteroids can induce T cell apoptosis, preferentially in CD4+ compared to CD8+ T cells due to differential Bcl-2 expression profiles.23–25 Additionally, corticosteroids enhance humoral T helper (TH) 2 immune responses over cellular responses (TH1), which are mainly thought to promote antitumor immunity.12,28 For these reasons, corticosteroids are considered antithetical to the immune-stimulatory effects of cancer immunotherapy. Alternatively, it is important to note that the effects of corticosteroids on innate and adaptive immunity are context-dependent. For instance, mature DCs are resistant to the suppressive actions of corticosteroids.29 Corticosteroids are also known to inhibit T cell receptor (TCR)-mediated activation-induced cell death (AICD), which serves to attenuate T cell responses and promote peripheral T cell deletion.30,31 In these contexts, corticosteroids may positively influence and help sustain T cell responses to overcome foreign antigens, pathogens, and tumors.
Consistent with previously mentioned effects, dexamethasone therapy resulted in significant reductions in peripheral CD4+ and CD8+ T cells in both tumor models, thus confirming the expected in vivo effects of the dexamethasone regimens. In mice receiving short-course dexamethasone (Dex-1 or Dex-2), these peripheral reductions were persistent as cell counts failed to recover on day 27. Additionally, dexamethasone treatment also reduced the number of circulating Treg cells. The sensitivity of Treg cells to dexamethasone is controversial with multiple studies showing conflicting results.26,32–36 Given this study’s findings, dexamethasone treatment could potentially have a positive influence on shaping antitumor immunity by reducing circulating Treg cell numbers and their subsequent tumor infiltration.37 However, this potential positive influence must be counterbalanced with dexamethasone’s negative effects on conventional CD4+ and CD8+ T cells.
In cancer, PD-1 mainly exerts its immune-inhibitory effects locally within the tumor microenvironment rather than systemically.38 Hence, the mechanism of action of anti-PD-1 therapy is to reverse the exhaustion of pre-existing tumor-residing T cells, thus improving their effector functions to carry out tumor regressions.39–41 Depending on the context and their sensitivity, if tumor-infiltrating T cells undergo adequate exposure to corticosteroids, then the benefits conferred by anti-PD-1 therapy may be diminished or completely lost. Endogenous and exogenous corticosteroids have been demonstrated to reduce antitumor immunity and counteract cancer immunotherapy in a murine autochthonous pancreatic ductal carcinoma model.42 In agreement, dexamethasone therapy disrupted the efficacy of PD-1 blockade in the flank tumor model. Anti-PD-1 therapy initially inhibited tumor growth, but this benefit was ultimately lost long-term in mice receiving co-therapy with dexamethasone. Interestingly, there were also trends in complete and non-durable partial responses regarding the timing and duration of dexamethasone therapy. Whereas anti-PD-1 monotherapy achieved a complete response rate of 50.0%, this down-trended to 12.5% with short-course dexamethasone (anti-PD-1 + Dex-1 and anti-PD-1 + Dex-2) and was completely lost (0.0%) with continuous administration (anti-PD-1 + Dex-Cont). Partial responses (25.0%) were only observed in mice receiving dexamethasone following the completion of anti-PD-1 therapy (anti-PD-1 + Dex-2). These partial responses were not durable though, suggesting the initial benefits of anti-PD-1 therapy could be overturned with subsequent dexamethasone treatment. Taken together, these findings suggest that co-therapy with corticosteroids does not necessarily preclude objective responses seen with anti-PD-1 therapy in peripheral tumors. However, these objective responses may less likely occur and may be short-lived.
Although historic views regarding the CNS as an “immune-privileged” site are being challenged, the CNS is nonetheless subject to interrogation by the immune system in a manner unique to other anatomic sites.43,44 Unlike peripheral tissues, the CNS is protected by the BBB,45,46 possesses a unique non-classical lymphatic drainage system,47 and contains distinct tissue-resident cells that T cells inevitably interact with including neurons, astrocytes, and microglia.48 Since the CNS has limited abilities to regenerate or accommodate large volume changes in a skull-confined space (cellular infiltrates and/or edema), these specialized cellular and anatomic structures serve to protect the host from the fatal consequences of excessive neuro-inflammation. These inherent differences between peripheral tissues and CNS may provide a possible explanation for the discrepancy seen in the flank and intracranial tumor models. Whereas peripheral CD8/Treg ratio changes associated with flank tumor responses, this study failed to uncover peripheral T cell changes that predicted intracranial responses to anti-PD-1 therapy with or without dexamethasone therapy. These findings suggest intracranial responses may be independent of the systemic effects caused by anti-PD-1 and dexamethasone therapy. Despite systemic lymphodepletion, antitumor immune responses were maintained regionally in glioma-bearing mice receiving combination anti-PD-1 and dexamethasone therapy, and the survival benefit conferred by anti-PD-1 therapy was not diminished. Caution should be taken when interpreting these results as tumor histology was not held constant between the implantation sites. However, since anti-PD-1 therapy targets T cells rather than tumor cells, tumor histology becomes a less concerning factor given both models respond to PD-1 blockade alone within their intended implantation sites.
From a teleologic standpoint, corticosteroids are critical to control neuro-inflammatory responses, where they must counterbalance the selective destruction of peripheral immunologically ignorant T cells that can propagate neuro-pathology over CNS-resident, pathogen-specific T cells that are needed to effectively clear neuro-pathogens.49,50 Thus, in comparison to the flank tumor model, dexamethasone may not be reaching adequate levels to negatively impact CNS tumor-resident T cells that mediate the benefits of anti-PD-1 therapy. In support of these claims, dexamethasone is known to restore the integrity of the BBB and has limited cerebrospinal fluid penetration.51,52 Additionally, in studies of experimental autoimmune encephalomyelitis (EAE), a well-established rodent model of multiple sclerosis (MS), dexamethasone treatment induced apoptosis and inhibited the CNS migration of peripheral, bystander T cells with limited impact on CNS-resident, antigen-specific T cells.35,53 Taken together, these data suggest there may be variability of corticosteroid’s influence on CNS-resident vs. peripheral T cells, and the current study’s findings demonstrate that anti-PD-1-mediated immune responses against intracranial tumors may potentially go undeterred in the setting of corticosteroids. Corticosteroid use did not correlate with the density of infiltrating T cells in a large cohort of patients with brain metastases.54 In a recent case report, a patient with disseminated non-small cell lung cancer was able to experience partial responses with anti-PD-1 therapy in symptomatic brain metastases, for which he was receiving concomitant high-dose corticosteroids.55 As the current study’s findings may be anti-PD-1-specific, caution must be taken when extrapolating these results to other forms of immunotherapy, especially those whose mechanism of action rely on the circulation and subsequent tumor infiltration by tumor-reactive T cells (e.g., adoptive T cell transfer, tumor vaccines, oncolytic virotherapy, etc.). This study’s results indirectly support this claim as dexamethasone treatment caused severe reductions in circulating T cells.
Contrary to these findings, previous studies have shown that dexamethasone abrogates the survival benefit conferred by non-anti-PD-1 immunotherapy regimens in orthotopic glioma models.56,57 Although different immune-stimulatory agents were investigated, the discrepancies between these results may be due to the timing of dexamethasone treatment relative to tumor implantation. Whereas treatment was postponed until tumor establishment, other studies gave dexamethasone either before or immediately following tumor implantation. Theoretically, early dexamethasone administration would reduce the peripheral pool and early CNS infiltration of tumor-reactive T cells, therefore not affording the opportunity for an early adaptive immune response that may be enhanced by anti-PD-1 therapy. As a more clinically relevant scenario, dexamethasone was given later in the disease course as patients suffering from CNS malignancies are more likely to be prescribed corticosteroids after tumor establishment. In the intracranial tumor model, early dexamethasone administration may be counterproductive to anti-PD-1 therapy. In the early phases of tumor development, early dexamethasone treatment would potentially deprive peripheral tumor-reactive T cells’ the opportunities to infiltrate and become exhausted tumor-resident T cells, thus providing no substrate for anti-PD-1 therapy.
It will be equally important to assess how other systemic agents associated with lymphodepletion, such as chemotherapy, unintentionally interact with anti-PD-1 therapy or other immunotherapy strategies. In another study utilizing the same intracranial tumor model, systemic chemotherapy, despite causing severe lymphodepletion, did not interfere with the survival benefits when combined with anti-PD-1 therapy.20 Interestingly, local chemotherapy in the form of biodegradable impregnated wafers was found to be synergistic with PD-1 blockade while avoiding the systemic toxicities associated with chemotherapy. As a negative consequence, long-term responders that were previously treated with anti-PD-1 and systemic chemotherapy (but not local) had impaired tumor-specific immunologic memory and were unable to reject tumor formation upon rechallenge. In this study, immunologic memory against CNS tumors was grossly preserved in mice treated with combination anti-PD-1 and dexamethasone therapy.
In conclusion, these findings suggest that corticosteroids may have differential impacts on the efficacy of anti-PD-1 therapy depending on the anatomical location of tumors within or outside the CNS. Whereas corticosteroids attenuated anti-PD-1-mediated antitumor immune responses against tumors in the periphery, the benefits conferred by PD-1 blockade remained intact when targeting CNS tumors. This evidence suggests that strategies and selection criteria may be developed to guide appropriate use of corticosteroid therapy without limiting the effectiveness of anti-PD-1 therapy, especially for patients with tumors residing within the CNS. Clinical trials are warranted to further explore these questions.
SUPPLEMENTAL DATA
Supplemental data for this article can be accessed at here
Disclosure of Potential Conflicts of Interest
ML has received research support from Arbor Pharmaceuticals, Aegenus, Altor, Bristol-Myers Squibb, Immunocellular, Celldex, and Accuray. ML is also a consultant for Aegenus, Bristol-Myers Squibb, Oncorus, Boston Biomedical, Stryker, and Baxter.
LRK has received research support from and is on the advisory board of Novocure. LRK has received research support and honoraria from Accuray.
DMP has received research support from Bristol-Myers Squibb and the Melanoma Research Alliance. DMP has also received personal fees from Five Prime Therapeutics, Aduro, Compugen, GlaxoSmithKline, Medimmune/AstraZeneca, Merck, Potenza Therapeutics, Sanofi, Tizona, DNatrix, Amgen, Rock Springs Capital, Immunomic Therapeutics, Janssen, Astellas, WindMill Therapeutics, and Bayer. DMP has pending patents including biomarkers useful for determining response to PD-1 blockade, cancer therapy via combination of epigenetic modulation and immune modulation, method of preventing organ transplant rejections using agonists to PD-1 checkpoint pathway, cancer immunotherapy, compositions and methods for targeting activin signaling to treat cancer, combinatorial therapy of cancer and infectious diseases with anti-B7-H1 antibodies, combination of immunotherapy with local chemotherapy for the treatment of malignancies, inhibition of YAP for breaking tumor immune tolerance, compositions and methods for cancer immunotherapy (licensed to Aduro Biotech), and T cell regulation (licensed to Bristol- Myers Squibb).
HB has received research support from Arbor Pharmaceuticals, Bristol-Myers Squibb, and Accurexa. HB is a consultant for AsclepiX Therapeutics, Celsion-EGEN, Perosphere Inc., StemGen, Accelerating Combination Therapies, Camden Partners, LikeMinds Inc., and Acuity Bio Corp.
KJR has received research support from Elekta AB and honoraria for education activities from AstraZeneca. KJR has received research support and honoraria from Accuray.
References
- 1.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 2.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 3.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 8.Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellmunt J, De Wit R, Vaughn DJ, Fradet Y, Lee J-L, Fong L, Vogelzang NJ, Climent MA, Petrylak DP, Choueiri TK, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015–1026. doi: 10.1056/NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topalian SL, Taube JM, Anders RA, Pardoll DM.. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franchimont D. Overview of the actions of glucocorticoids on the immune response: a good model to characterize new pathways of immunosuppression for new treatment strategies. Ann N Y Acad Sci. 2004;1024:124–137. doi: 10.1196/annals.1321.009. [DOI] [PubMed] [Google Scholar]
- 13.Flammer JR, Rogatsky I. Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol. 2011;25(7):1075–1086. doi: 10.1210/me.2011-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shih A, Jackson KC 2nd. Role of corticosteroids in palliative care. J Pain Palliat Care Pharmacother. 2007;21(4):69–76. [PubMed] [Google Scholar]
- 15.Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2(10):1346–1353. doi: 10.1001/jamaoncol.2016.1051. [DOI] [PubMed] [Google Scholar]
- 16.Drappatz J, Schiff D, Kesari S, Norden AD, Wen PY. Medical management of brain tumor patients. Neurol Clin. 2007;25(4):1035–71, ix. doi: 10.1016/j.ncl.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 17.Connell CM, Raby S, Beh I, Flint TR, Williams EH, Fearon DT, Jodrell DI, Janowitz T. Cancer immunotherapy trial registrations increase exponentially but chronic immunosuppressive glucocorticoid therapy may compromise outcomes. Ann Oncol. 2017;28(7):1678–1679. doi: 10.1093/annonc/mdx181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathios D, Kim JE, Mangraviti A, Dewhirst M, Fan TM, Gustafson DL, Helman LJ, Kastan MB, Knapp DW, Levin WJ, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. 2016;8(370):370ra180. doi: 10.1126/scitranslmed.aaf0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, Liu A, Sankey EW, Tam A, Xu H, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124–136. doi: 10.1158/1078-0432.CCR-15-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65(3):1089–1096. [PubMed] [Google Scholar]
- 23.Migita K, Eguchi K, Kawabe Y, Nakamura T, Shirabe S, Tsukada T, Ichinose Y, Nakamura H, Nagataki S. Apoptosis induction in human peripheral blood T lymphocytes by high-dose steroid therapy. Transplantation. 1997;63(4):583–587. [DOI] [PubMed] [Google Scholar]
- 24.Leussink VI, Jung S, Merschdorf U, Toyka KV, Gold R. High-dose methylprednisolone therapy in multiple sclerosis induces apoptosis in peripheral blood leukocytes. Arch Neurol. 2001;58(1):91–97. [DOI] [PubMed] [Google Scholar]
- 25.Cook AM, McDonnell AM, Lake RA, Nowak AK. Dexamethasone co-medication in cancer patients undergoing chemotherapy causes substantial immunomodulatory effects with implications for chemo-immunotherapy strategies. Oncoimmunology. 2015;5(3):e1066062. doi: 10.1080/2162402X.2015.1066062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sbiera S, Dexneit T, Reichardt SD, Michel KD, Van Den Brandt J, Schmull S, Kraus L, Beyer M, Mlynski R, Wortmann S, et al. Influence of short-term glucocorticoid therapy on regulatory T cells in vivo. PLoS One. 2011;6(9):e24345. doi: 10.1371/journal.pone.0024345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanderheyde N, Verhasselt V, Goldman M, Willems F. Inhibition of human dendritic cell functions by methylprednisolone. Transplantation. 1999;67(10):1342–1347. [DOI] [PubMed] [Google Scholar]
- 28.Blotta MH, DeKruyff RH, Umetsu DT. Corticosteroids inhibit IL-12 production in human monocytes and enhance their capacity to induce IL-4 synthesis in CD4+ lymphocytes. J Immunol. 1997;158(12):5589–5595. [PubMed] [Google Scholar]
- 29.Matyszak MK, Citterio S, Rescigno M, Ricciardi-Castagnoli P. Differential effects of corticosteroids during different stages of dendritic cell maturation. Eur J Immunol. 2000;30(4):1233–1242. doi:. [DOI] [PubMed] [Google Scholar]
- 30.Zacharchuk CM, Mercep M, Chakraborti PK, Simons SS Jr, Ashwell JD. Programmed T lymphocyte death. cell activation- and steroid-induced pathways are mutually antagonistic. J Immunol. 1990;145(12):4037–4045. [PubMed] [Google Scholar]
- 31.D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, Cannarile L, Migliorati G, Riccardi C. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997;7(6):803–812. [DOI] [PubMed] [Google Scholar]
- 32.Karagiannidis C, Akdis M, Holopainen P, Woolley NJ, Hense G, Rückert B, Mantel P-Y, Menz G, Akdis CA, Blaser K, et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol. 2004;114(6):1425–1433. doi: 10.1016/j.jaci.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Oppenheim JJ, Winkler-Pickett RT, Ortaldo JR, Howard OM. Glucocorticoid amplifies IL-2-dependent expansion of functional FoxP3(+)CD4(+)CD25(+) T regulatory cells in vivo and enhances their capacity to suppress EAE. Eur J Immunol. 2006;36(8):2139–2149. doi: 10.1002/eji.200635873. [DOI] [PubMed] [Google Scholar]
- 34.Stock P, Akbari O, DeKruyff RH, Umetsu DT. Respiratory tolerance is inhibited by the administration of corticosteroids. J Immunol. 2005;175(11):7380–7387. [DOI] [PubMed] [Google Scholar]
- 35.Wust S, Van Den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, Lühder F, Reichardt HM. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol. 2008;180(12):8434–8443. [DOI] [PubMed] [Google Scholar]
- 36.Xiang L, Marshall GD Jr. Immunomodulatory effects of dexamethasone on gene expression of cytokine and stress hormone receptors in peripheral blood mononuclear cells. Int Immunopharmacol. 2013;17(3):556–560. doi: 10.1016/j.intimp.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 37.Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. 2017;20(3):558–571. doi: 10.1016/j.celrep.2017.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kansy BA, Concha-Benavente F, Srivastava RM, Jie H-B, Shayan G, Lei Y, Moskovitz J, Moy J, Li J, Brandau S, et al. PD-1 status in CD8+ T cells associates with survival and anti-PD-1 therapeutic outcomes in head and neck cancer. Cancer Res. 2017. doi: 10.1158/0008-5472.CAN-16-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-A-AS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170(6):1120–1133.e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, Jodrell DI, Fearon DT. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 2016;24(5):672–684. doi: 10.1016/j.cmet.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18(2):123–131. doi: 10.1038/ni.3666. [DOI] [PubMed] [Google Scholar]
- 44.Jackson CM, Kochel CM, Nirschl CJ, Durham NM, Ruzevick J, Alme A, Francica BJ, Elias J, Daniels A, Dubensky TW, et al. Systemic tolerance mediated by melanoma brain tumors is reversible by radiotherapy and vaccination. Clin Cancer Res. 2016;22(5):1161–1172. doi: 10.1158/1078-0432.CCR-15-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. 2011;1812(2):220–230. doi: 10.1016/j.bbadis.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cserr HF, Knopf PM. Cervical lymphatics, the blood-brain barrier and the immunoreactivity of the brain: a new view. Immunol Today. 1992;13(12):507–512. doi: 10.1016/0167-5699(92)90027-5. [DOI] [PubMed] [Google Scholar]
- 47.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122(4):1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGavern DB. The role of bystander T cells in CNS pathology and pathogen clearance. Crit Rev Immunol. 2005;25(4):289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tischner D, Reichardt HM. Glucocorticoids in the control of neuroinflammation. Mol Cell Endocrinol. 2007;275(1–2):62–70. doi: 10.1016/j.mce.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 51.Balis FM, Lester CM, Chrousos GP, Heideman RL, Poplack DG. Differences in cerebrospinal fluid penetration of corticosteroids: possible relationship to the prevention of meningeal leukemia. J Clin Oncol. 1987;5(2):202–207. doi: 10.1200/JCO.1987.5.2.202. [DOI] [PubMed] [Google Scholar]
- 52.Salvador E, Shityakov S, Forster C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res. 2014;355(3):597–605. doi: 10.1007/s00441-013-1762-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCombe PA, Nickson I, Tabi Z, Pender MP. Corticosteroid treatment of experimental autoimmune encephalomyelitis in the lewis rat results in loss of V beta 8.2+ and myelin basic protein-reactive cells from the spinal cord, with increased total T-cell apoptosis but reduced apoptosis of V beta 8.2+ cells. J Neuroimmunol. 1996;70(2):93–101. [DOI] [PubMed] [Google Scholar]
- 54.Berghoff AS, Fuchs E, Ricken G, Mlecnik B, Bindea G, Spanberger T, Hackl M, Widhalm G, Dieckmann K, Prayer D, et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology. 2015;5(1):e1057388. doi: 10.1080/2162402X.2015.1057388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pluchart H, Pinsolle J, Cohen J, Ferretti GR, Bedouch P, Giaj Levra M, Toffart A-C, Moro-Sibilot D. Partial response of pulmonary adenocarcinoma with symptomatic brain metastasis to nivolumab plus high-dose oral corticosteroid: A case report. J Med Case Rep. 2017;11(1):183. doi: 10.1186/s13256-017-1334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kleijn A, Kloezeman J, Treffers-Westerlaken E, Fulci G, Leenstra S, Dirven C, Debets R, Lamfers M, Castro MG. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PLoS One. 2014;9(5):e97495. doi: 10.1371/journal.pone.0097495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, Dai C, Ozawa T, Chang M, Chan TA, et al. Corticosteroids compromise survival in glioblastoma. Brain. 2016;139(Pt 5):1458–1471. doi: 10.1093/brain/aww046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.