

The GTPase atlastin mediates homotypic ER fusion through trans-crossover dimerization, but how dimerization is coupled to the GTPase cycle has remained unclear. Winsor et al. show that GTP binding causes crossover dimerization for fusion, whereas GTP hydrolysis promotes disassembly of the crossover dimer for subunit recycling.
Abstract
Membrane fusion of the ER is catalyzed when atlastin GTPases anchored in opposing membranes dimerize and undergo a crossed over conformational rearrangement that draws the bilayers together. Previous studies have suggested that GTP hydrolysis triggers crossover dimerization, thus directly driving fusion. In this study, we make the surprising observations that WT atlastin undergoes crossover dimerization before hydrolyzing GTP and that nucleotide hydrolysis and Pi release coincide more closely with dimer disassembly. These findings suggest that GTP binding, rather than its hydrolysis, triggers crossover dimerization for fusion. In support, a new hydrolysis-deficient atlastin variant undergoes rapid GTP-dependent crossover dimerization and catalyzes fusion at an initial rate similar to WT atlastin. However, the variant cannot sustain fusion activity over time, implying a defect in subunit recycling. We suggest that GTP binding induces an atlastin conformational change that favors crossover dimerization for fusion and that the input of energy from nucleotide hydrolysis promotes complex disassembly for subunit recycling.
Introduction
The substantial energy barriers that prevent spontaneous lipid bilayer fusion allow for the formation and maintenance of distinct subcellular compartments in eukaryotic cells. However, exchange of content between compartments must also regularly occur, so cells maintain specialized fusion catalysts that overcome these barriers (Tamm et al., 2003; Cohen and Melikyan, 2004; Frolov and Zimmerberg, 2010; Kozlov et al., 2010). The two best-understood fusion catalysts are the viral fusion proteins, which mediate membrane fusion during viral entry into host cells (Weissenhorn et al., 1999; Skehel and Wiley, 2000; Eckert and Kim, 2001), and the SNARE proteins, responsible for fusion events during vesicle trafficking (Chen and Scheller, 2001; Jahn and Scheller, 2006; Südhof and Rothman, 2009). Wide-ranging studies over the past several decades have shown that while the two types of fusion catalysts are evolutionarily unrelated and structurally distinct, once the proteins come to span opposing membranes, they share a common strategy for fusion catalysis. Each undergoes a series of energetically favorable conformational changes that draw the membranes together, converting a prefusion membrane-spanning complex into a stable postfusion complex whose binding energy overcomes the barriers to spontaneous fusion (Söllner, 2004).
The last several years have seen the emergence of a new type of fusion catalyst that differs from previously studied examples by virtue of possessing intrinsic GTP hydrolytic activity (Hoppins and Nunnari, 2009; McNew et al., 2013). Of these, atlastin and atlastin-like proteins are sufficient to trigger fusion of synthetic liposomes and are required for the branched morphology of the ER in organisms ranging from yeast to humans (Hu et al., 2009; Orso et al., 2009; Chen et al., 2011; Anwar et al., 2012; Zhang et al., 2013). In addition to an N-terminal dynamin-related GTPase (G) domain (Praefcke and McMahon, 2004) that couples GTP hydrolysis to fusion activity (Orso et al., 2009), atlastins possess a fully folded three-helix bundle (3HB) connected to the G domain via a short linker (Bian et al., 2011; Byrnes and Sondermann, 2011), two closely spaced trans-membrane helices, and a C-terminal tail with an amphipathic helix whose insertion into the bilayer helps promote fusion (Moss et al., 2011; Liu et al., 2012; Faust et al., 2015).
Despite little resemblance to previously studied fusion catalysts, the earliest crystal structures of the truncated N-terminal cytoplasmic domain of human atlastin1 (hATL1) suggested a way by which atlastins might also span opposing membranes and undergo a series of favorable conformational changes to drive fusion (Bian et al., 2011; Byrnes and Sondermann, 2011; Byrnes et al., 2013). In one conformation (form 2), the G domain heads of two atlastin monomers were seen to interact in a head-to-head (H/H) fashion, with each 3HB packed against its respective G domain head and pointed away from the dimer interface (Bian et al., 2011; Byrnes and Sondermann, 2011). With its relatively low surface area of binding (756 Å2), this putative extended dimer conformation could represent an initial encounter complex between atlastins in opposing membranes; therefore, it is commonly referred to as the prefusion dimer. In another more tightly packed dimer conformation (form 1), the G domains were observed to interact more closely with one another, and furthermore, the 3HBs were dislodged from their respective heads and rotated toward the dimer interface, forming a crossed over dimer conformation with the 3HBs now in close parallel alignment and a substantially increased overall binding interface of 2,797 Å2 (Bian et al., 2011; Byrnes and Sondermann, 2011). In yet a third conformation (form 3), an even more tightly packed crossover dimer was observed with a buried surface area of 3,852 Å2 (Byrnes et al., 2013). The parallel alignment of the 3HBs in either the loose (2,797 Å2) or tight (3,852 Å2), but especially in the tight, crossover dimer made it difficult to envision how the crossover conformation could be adopted by full-length atlastins in separate membranes. Consequently, the crossover dimer state is typically referred to as the postfusion state, by analogy to the stable postfusion complexes of viral and SNARE fusion proteins (Sutton et al., 1998; Weber et al., 1998; Weissenhorn et al., 1999). In support of a model in which fusion is driven by crossover dimer formation between atlastins in opposing membranes (Bian et al., 2011; Daumke and Praefcke, 2011), we recently showed that the relative binding energy of the crossover dimer (most likely form 3) correlates strongly with fusion capacity (Winsor et al., 2017).
The role of GTP hydrolysis in the atlastin fusion mechanism has remained far more enigmatic. While two different laboratories observed the same initial pre- and postfusion dimer conformations, one group obtained both pre- (form 2) and postfusion (form 1) dimers using the nonhydrolysable analogue GTPγS (Byrnes and Sondermann, 2011), while the other group obtained the prefusion dimer using GDP ⋅ phosphate (Pi) and the postfusion dimer using GDP (Bian et al., 2011). Adding to the confusion, only GDP was detected in each of the four structures (Bian et al., 2011; Byrnes and Sondermann, 2011). Subsequently, the more tightly packed postfusion (form 3) dimer was observed with either the nonhydrolysable analogue GMPPNP or the transition state analogue GDP ⋅ AlF4 (Byrnes et al., 2013). Thus, whether crossover dimerization occurs in the GTP-bound state, the GTP hydrolysis transition state, the GDP ⋅ Pi state, or the GDP-bound state has since been debated.
By analogy to other dimerizing GTPases (Gasper et al., 2009), arguably the simplest model would have been one in which the binding of nucleotide triggers a conformational change that favors dimerization and crossover, with the subsequent hydrolysis of GTP and release of inorganic Pi driving dimer disassembly to reset the molecules to their original conformation. In the context of the fusion reaction cycle, GTP binding, by this model, would be sufficient to promote crossover dimerization to drive fusion, whereas the hydrolysis of nucleotide and release of Pi subsequent to fusion would serve to disassemble the stable dimer for subunit recycling. The appeal of this simple model is that it would further extend the parallels with the SNARE paradigm in which SNARE proteins are recycled for reuse after the completion of fusion through the action of the N-ethylmaleimide–sensitive fusion (NSF) protein coupling the hydrolysis of ATP to disassembly of the SNARE postfusion complex (Otto et al., 1997; Zhao and Brunger, 2016).
Despite the appeal of the simple SNARE-like model, mechanistic studies to date on full-length, membrane-anchored atlastin have not supported it. Whereas the simple model predicts at least one round of fusion catalysis in the absence of GTP hydrolysis, replacing GTP with the nonhydrolysable analogue GTPγS renders Drosophila melanogaster atlastin (DATL) completely inert in the fusion assay (Orso et al., 2009). Also, mutation of a key catalytic arginine residue to alanine (R48A in DATL), known to inhibit GTP hydrolysis, has a similar debilitating effect (Pendin et al., 2011). Later studies, again using DATL, have confirmed these findings with the nonhydrolysable analogue GMPPNP, and furthermore, even the initial attachment, or tethering, of opposing membranes to one another, a step presumed to precede fusion, is inhibited by either GMPPNP, GTPγS, or the R48A mutation in DATL (Saini et al., 2014; Liu et al., 2015). One interesting exception has been Sey1p, the yeast atlastin-like protein, which exhibits some, albeit slow and limited, fusion activity with GMPPNP (Yan et al., 2015). Notably, though, Sey1p contains multiple additional 3HBs that endow it with an expansive stalk domain not present in atlastins (Yan et al., 2015). Therefore, the limited fusion by Sey1p observed with GMPPNP has arguably been viewed more as an exception than the rule (Hu and Rapoport, 2016).
Studies to date on the truncated soluble domain of atlastin have also disputed the simple SNARE-like model. The cytoplasmic soluble domain of hATL1 (cyt-hATL1) or DATL (cyt-DATL), truncated to remove the trans-membrane domain and cytoplasmic tail, is induced to undergo dimerization and crossover by either GTP or its nonhydrolysable analogues (Byrnes and Sondermann, 2011; Moss et al., 2011; Byrnes et al., 2013; Liu et al., 2015; Wu et al., 2015; O’Donnell et al., 2017; Winsor et al., 2017). However, GTP triggers crossover dimerization 100× faster than either GMPPNP or GTPγS (Byrnes et al., 2013; O’Donnell et al., 2017; Winsor et al., 2017). Furthermore, the R48A equivalent, hydrolysis-defective truncated soluble domain of hATL1 (R77A), undergoes crossover dimerization very slowly even with GTP (Byrnes et al., 2013). Altogether, these observations have led to the currently accepted view that while atlastin monomers may weakly associate with one another in the GTP-bound state, GTP hydrolysis is strictly required to stabilize the dimer interface for crossover dimerization and fusion (Byrnes et al., 2013; Saini et al., 2014; Liu et al., 2015; Hu and Rapoport, 2016; Winsor et al., 2017). A more recent study has gone further, proposing that the hydrolysis of nucleotide occurs before G domain dimerization and that the role of hydrolysis is to produce a GDP ⋅ Pi–bound, dimerization-competent conformational state (O’Donnell et al., 2017).
Importantly, the idea that the energy of nucleotide hydrolysis is used to drive crossover dimerization for fusion leaves unsettled how the crossover dimer is broken apart after fusion. Given the relative stability of the crossover dimer, its disassembly would also be expected to require substantial energy, but the source of that energy remains ambiguous. In this study, we set out to resolve this issue by using presteady-state and single-turnover approaches. We undertook a comprehensive kinetic analysis of both WT atlastin (DATL) and a new hydrolytic mutant variant D127N DATL, which we identify in this study to reveal the coupling between the GTPase cycle of atlastin and its fusion-associated conformational changes. Much to our surprise, we find dimerization and crossover to consistently precede GTP hydrolysis, not only for the hydrolysis-deficient D127N mutant variant but also for WT DATL as well as for WT hATL1, indicating that the hydrolysis of GTP cannot be the driver of crossover dimerization. In all cases, both the hydrolysis of nucleotide and the release of Pi better coincide with dimer disassembly, indicating for the first time that the energy of GTP hydrolysis is devoted to disassembling the atlastin postfusion complex. These results return us to the simple SNARE-like model in which GTP binding produces crossover dimerization for fusion and the energy of nucleotide hydrolysis is harnessed to break the atlastin crossover dimer apart after fusion to promote subunit recycling. In further support of a model in which nucleotide hydrolysis drives the recycling of subunits for multiple rounds of fusion catalysis, the D127N hydrolysis-deficient DATL variant catalyzes in vitro fusion at a rate similar to the WT early on, but it is unable to sustain fusion activity at later times.
Results
Previous kinetic assays of cyt-hATL1 or cyt-DATL have used excess GTP (Byrnes et al., 2013; Liu et al., 2015; O’Donnell et al., 2017; Winsor et al., 2017), which produces many rounds of dimer assembly and disassembly, obscuring the timing and sequence of later events such as dimer disassembly and Pi release. In this study, we used a substoichiometric GTP concentration to limit the reaction to a single round of GTP hydrolysis and product release. To examine the rate of crossover dimer assembly and disassembly, we used a protein-induced fluorescence enhancement (PIFE) assay (Hwang et al., 2011), which places an environmentally sensitive fluorophore Cy3 on an engineered cysteine residue on the 3HB of cyt-DATL (Winsor et al., 2017). We previously established that the fluorescence of the dye is enhanced as it transitions from being solvent exposed on the monomer to becoming buried at the 3HB dimer interface in the crossover dimer conformation (Fig. 1 A; Winsor et al., 2017). Fluorescence enhancement occurs under assay conditions in which bismaleimideothane, an 8-Å thiol-reactive bifunctional cross-linker, efficiently cross-links the engineered 3HB cysteine of one monomer to that of the other (Saini et al., 2014; Winsor et al., 2017), and this is only expected to occur in the tight (form 3) crossover dimer in which the equivalent residues in the hATL1 crystal structure are 8 Å apart (Byrnes et al., 2013). By contrast, the same residues are 20 Å apart in the loose (form 1) crossover dimer (Bian et al., 2011; Byrnes and Sondermann, 2011) and >100 Å apart in the so-called prefusion (form 2) dimer (Bian et al., 2011; Byrnes and Sondermann, 2011). Of additional note, our previous assays used G343C on the 3HB of cyt-DATL as the labeling site; however, that site also detected an early and transient downward deflection of Cy3 fluorescence likely related to the initial release of the 3HB from the G domain before crossover (Winsor et al., 2017). By moving the labeling site 10 Å further down the 3HB and away from the G domain, we eliminated the early downward deflection while retaining the same sensitivity to crossover (Fig. S1).
Figure 1.

Atlastin (DATL) dimerization and crossover precedes GTP hydrolysis and Pi release. (A–F) Single-turnover kinetics of cyt-DATL. (A) Schematic of Cy3 fluorescence enhancement (PIFE) as Cy3-labeled cyt-DATL (i) undergoes dimer formation and crossover (ii). (B) Stopped-flow PIFE of WT or R48A cyt-DATL after addition of GTP. (C) Schematic of FRET as cyt-DATL monomers labeled with a FRET donor and acceptor (i) undergo H/H dimerization (ii). (D) Comparison of WT H/H FRET with WT crossover PIFE (C/O) after GTP addition under stopped flow. (E) Hydrolysis of GTP (containing a-32P-GTP) by WT cyt-DATL at room temperature. Reactions were acid quenched at the indicated times (one of three replicates shown). (F) Comparison of WT crossover PIFE (C/O), GTP hydrolysis, and Pi release. PIFE (C/O) trace is from B except normalized (initial value = 0; maximum value = 1). GTP hydrolysis was the average of three replicates from E (±SEM). Pi release was measured by including 15 µM MDCC-PBP (final) in the stopped flow and normalized (initial value = 0; maximum value = 1). All reactions contained 15 µM cyt-DATL and 7.5 µM GTP (final concentrations) and were performed at 25°C except where indicated. All traces are the average of three to five individual traces and are representative of two independent protein preparations.
The hydrolysis-defective R48A Cy3-labeled cyt-DATL showed only a negligible PIFE signal at early times after mixing with limiting GTP (Fig. 1 B), consistent with previous results using the equivalent R77A cyt-hATL1 (Byrnes et al., 2013). In contrast, the WT showed a rapid rise in PIFE (Fig. 1 B). Not surprisingly, the rise for the WT was slower than previously observed with excess GTP (Fig. S2; Winsor et al., 2017), presumably due to slowed nucleotide binding under the condition of limiting GTP. More importantly, the rise in the WT PIFE signal was quickly followed by its decline and return to baseline (Fig. 1 B), consistent with a single round of dimer assembly and disassembly and return of the atlastin to the monomer state. This was in contrast with previous multiple-turnover assays with saturating GTP in which the PIFE signal persisted for much longer periods (Fig. S2; Winsor et al., 2017). To confirm that the observed decline in PIFE corresponded with dimer disassembly, we independently monitored G domain, or H/H, dimerization using cyt-DATL labeled with Förster resonance energy transfer (FRET) donor and acceptor fluorophores on an engineered G domain residue (Fig. 1 C) as done previously for cyt-hATL1 (Byrnes et al., 2013; O’Donnell et al., 2017) and for cyt-DATL (Winsor et al., 2017). The rise in PIFE was similar to the rise in FRET (Fig. 1 D), with H/H dimerization just preceding crossover. This was just as seen previously under multiple turnover conditions (O’Donnell et al., 2017; Winsor et al., 2017). Importantly, the loss of PIFE under this single-turnover condition was also largely coincident with a loss of FRET (Fig. 1 D), reinforcing the idea that the decline in the observed PIFE signal reflects dimer disassembly. Interestingly, the end state of disassembly as reported by PIFE had a lower fluorescence than the initial state (Fig. 1 B), possibly due to a difference in the positioning of the 3HB in the GDP-bound monomer as compared with the nucleotide-free form (Bian et al., 2011; Byrnes and Sondermann, 2011; Byrnes et al., 2013).
Current models in the literature predicted fast GTP hydrolysis that either precedes or is concurrent with dimerization. Indeed, a recent study reported a hydrolysis rate of 27 s−1 for cyt-hATL1 (O’Donnell et al., 2017). Therefore, we anticipated that the hydrolysis of GTP by WT cyt-DATL under the single-turnover condition used in this study might be complete within the first 100 ms. In contrast with this expectation, we observed incomplete hydrolysis of GTP even after 10 s (Fig. 1 E), a time when dimer disassembly was well underway by both PIFE and FRET indicators (Fig. 1 D). Overlaying the averaged normalized hydrolysis data (from Fig. 1 E and additional replicates) onto the normalized PIFE trace of crossover revealed crossover dimerization occurring well before nucleotide hydrolysis (Fig. 1 F). We also measured Pi release using Escherichia coli Pi-binding protein (PBP; Kubena et al., 1986) labeled with the fluorescent coumarin derivative MDCC to produce the Pi sensor MDCC-PBP (Brune et al., 1994). Pi binding by this sensor is with high affinity (KD ∼100 nM) and has been established to cause a rapid (317 s−1) conformational change in MDCC-PBP that increases its fluorescence intensity (Brune et al., 1994). Furthermore, Pi binding is considered essentially diffusion limited (>3 × 108 M−1s−1; Brune et al., 1994), meaning that any Pi released is detected almost instantaneously. In our assay, Pi release closely coincided with GTP hydrolysis (Fig. 1 F). The absence of a discernable lag between GTP hydrolysis and Pi release implied that Pi release is fast and limited by nucleotide hydrolysis. Moreover, fits of the rising phase of Pi release and the falling phase of PIFE from 7–60 s followed first-order kinetics with rates of 0.20 s−1 and 0.14 s−1, respectively, consistent with a close coupling between dimer disassembly and Pi release. Overall, these kinetic data demonstrated that dimerization and crossover precede GTP hydrolysis and that hydrolysis, occurring substantially later, is followed by rapid Pi release and associated with dimer disassembly.
These findings indicated that GTP hydrolysis could not be required for crossover dimerization, contradicting the interpretation of earlier results obtained using either nonhydrolysable GTP analogues or the R48A/R77A mutation to inhibit GTP hydrolysis. We wondered whether the previously reported inability of nonhydrolysable GTP analogues to support rapid crossover dimerization (Byrnes et al., 2013; Liu et al., 2015; Winsor et al., 2017) and fusion (Orso et al., 2009; Saini et al., 2014) could be due to issues other than absence of hydrolysis. An inability of the nucleotide analogues to bind atlastin was unlikely because in previous studies, the analogues were consistently present in large excess over their reported dissociation constants of 40 µM and 2 µM for GMPPNP and GTPγS, respectively (Byrnes et al., 2013; O’Donnell et al., 2017). Alternatively, we reasoned that the analogues, though capable of binding, might be incapable of promoting one or more GTP-induced G domain conformational changes required for rapid dimerization and fusion. To test this, we turned to intrinsic tryptophan fluorescence, which utilizes the sensitivity of tryptophan to its environment to monitor protein conformational change in a label-free system (Ghisaidoobe and Chung, 2014). The soluble domain of DATL contains four tryptophan residues, all restricted to the G domain, making it likely that upon nucleotide binding, one or more will experience changes in its environment resulting in an observable fluorescence change.
As anticipated, tryptophan fluorescence emission by WT cyt-DATL was strongly quenched within the first 100 ms of mixing with GTP but not with either buffer or GDP, consistent with a GTP-induced G domain conformational change (Fig. 2 A). As expected for a conformational change triggered by GTP binding, the rate of fluorescence quenching depended on nucleotide concentration (Fig. 2 B). To ensure that the fluorescence change observed upon GTP addition was not caused by dimerization or crossover, cyt-DATL in this particular assay was kept at 125 nM, below its previously reported dimerization constant of 0.5 µM (Winsor et al., 2017). Under this condition, crossover dimerization as monitored by PIFE showed a markedly slower kinetic (Fig. 2 C), indicating that the observed tryptophan fluorescence quenching reflects a G domain conformational change that occurs before crossover dimerization (see also Fig. 6). Interestingly, under the same condition, GTPγS caused a definite quenching of fluorescence (Fig. 2 D), indicating that GTPγS is also capable of promoting a G domain conformational change. However, both the rate and amplitude of quenching by the analogue appeared distinct from that induced by GTP, leaving uncertain whether the conformational change is the same as that induced by GTP. Meanwhile, GMPPNP caused no little or no observable change in tryptophan fluorescence even at the highest concentration of nucleotide used (Fig. 2 D). Thus, cyt-DATL seems able to undergo a G domain conformational change on binding GTPγS but not on GMPPNP binding, at least not on the same time scale. We also tested the hydrolysis-defective R48A cyto-DATL variant, which remarkably showed no quenching at most GTP concentrations used. Even at the highest GTP concentration, the quenching was distinct from the WT, suggesting an incomplete or different conformational shift upon nucleotide binding (Fig. 2 E). This difference was not likely due to an inability of the R48A variant to bind GTP because the equivalent R77A cyt-hATL1 variant has been reported to bind GTP as well as the WT (Byrnes et al., 2013). Furthermore, the difference was unlikely a reflection of lack of nucleotide hydrolysis by the R48A variant as the G domain conformational change, at least for the WT, occurred well before dimerization (Fig. 2 C), and hence by inference (Fig. 1 F), well before hydrolysis (see also Figs. 3 D and 6). Together, these data raised the possibility that the inability of GMPPNP as well as the R48A variant to promote rapid crossover dimerization and fusion might be due to their inability to induce or to undergo a required GTP-dependent G domain conformational change rather than to a lack of nucleotide hydrolysis.
Figure 2.

Neither GMPPNP nor the R48A mutation replicates a GTP-induced atlastin G domain conformational change. (A–E) Intrinsic tryptophan fluorescence of 125 nM WT, D127N, or R48A cyt-DATL after addition of the indicated concentrations of the indicated nucleotides under stopped flow at 25°C. (A) WT after mixing with 250 µM GTP, GDP, or buffer. (B) WT tryptophan fluorescence quenching with the indicated concentrations of GTP. (C) Crossover dimerization is slower than tryptophan fluorescence quenching under the same conditions. WT tryptophan fluorescence trace with 250 µM GTP (from A) replotted relative to WT crossover PIFE under the same conditions. (D) WT with 250 µM GTP, GMPPNP, or GTPγS. (E) R48A with the indicated concentrations of GTP. All concentrations are final after mixing. All traces are the average of three to five individual traces.
Figure 6.

DATL and hATL1 undergo a similar sequence of events during their GTPase cycle. (A and B) Timing of GTP-induced events for cyt-DATL (A) and cyt-hATL1 (B). dGTP binding measured using mant-dGTP and intrinsic tryptophan fluorescence quenching (Trp fluor) after GTP addition. All reactions were performed with stopped flow under single-turnover conditions with 15 µM cyt-DATL or cyt-hATL1 and 7.5 µM GTP or mant-dGTP (final concentrations). H/H FRET, C/O PIFE, GTP hydrolysis, and Pi release data for cyt-DATL (A) are replotted from Fig. 1 (D and F). C/O PIFE, GTP hydrolysis, and Pi release data for cyt-hATL1 (B) are replotted from Fig. 5 C. All traces were normalized (initial value = 0; maximum value = 1). Tryptophan fluorescence data are plotted as (1-normalized fluorescence) for ease of comparison. All traces are the average of three to five individual traces.
Figure 3.

A novel D127N active site mutation in cyt-DATL inhibits GTP hydrolysis but not the GTP-induced G domain conformational change or crossover dimer formation. (A) Sequence alignment of hGBP1, hATL1, DATL, and human dynamin1 (hDYN1) showing the positions of D127 and R93 in DATL relative to other signature GTPase residues. Residues conserved across GTPases are in green, catalytic arginine is in red, and D127 and R93 are in blue. (B) Side chains of active site residues in hGBP1 (PDB ID 2B92) and hATL1 (PDB ID 4IDQ) bound to GDP ⋅ AlF4 rendered in PyMOL. Magnesium ion and waters are shown as green and red spheres, respectively. (C) Steady-state GTPase assay of Pi release by WT, D127A, D127N, R93A, and R93Q cyt-DATL upon addition of GTP (n = 3; ± SEM). (D) Stopped-flow intrinsic tryptophan fluorescence quenching by D127N cyt-DATL after mixing with the indicated concentrations of GTP (as described in Fig. 2). (E and F) Stopped-flow PIFE of 2 µM WT or D127N cyt-DATL after addition of 1 mM GTP (E) or 1 mM GMPPNP (F). Traces in E and F were normalized (minimum value = 0; maximum value = 1), are the average of three runs, and are representative of two independent protein preparations.
While the above results for R48A cyt-DATL pointed to a possible unforeseen role for the R48 residue in promoting an early GTP-induced G domain conformational change, the R48 residue has also been suggested to play a direct role in G domain dimerization (Bian et al., 2011). In the loose (form 1) hATL1 crossover dimer structure (Bian et al., 2011; Byrnes and Sondermann, 2011), the side chain of the R48-equivalent residue R77 forms a salt bridge with E224 of the partner G domain. Then, in the tight (form 3) dimer structure, R77 faces away from the dimer interface and in toward the bound nucleotide, neutralizing the negative charge on the transition state of GTP hydrolysis (Byrnes et al., 2013). Thus, R48/R77 may mediate a transient G domain contact during dimerization before catalyzing nucleotide hydrolysis. Indeed, in human guanylate binding protein1 (hGBP1), a GTPase whose dynamin-related G domain is more closely related to atlastin than to any other dynamin-related G domain (Zhao et al., 2001), the equivalent of the R48 residue moves from the dimer interface toward the active site to facilitate nucleotide hydrolysis (Ghosh et al., 2006). hGBP1 is a peripheral membrane protein with antiviral activity and a cellular role distinct from that of atlastin (Praefcke and McMahon, 2004) and also differs mechanistically from atlastin in its ability to hydrolyze GTP successively to GDP and GMP (Schwemmle and Staeheli, 1994; Ghosh et al., 2006). Nonetheless, we suspected that there might be parallels between hGBP1 and atlastin and that the R48A DATL mutation might impair multiple steps in the reaction cycle.
The potential issues with the R48A mutation raised a serious concern that the inability of the R48A DATL variant to undergo rapid crossover dimerization might be due to one or more defects separate from its inability to hydrolyze GTP. This, combined with the inability of at least one of the two previously used nonhydrolysable analogues to induce a GTP-induced G domain conformational change, underscored a need to reevaluate the role of GTP hydrolysis in the atlastin mechanism. To address this need, we set out to identify an alternate mutation in atlastin that would inhibit hydrolysis more selectively. We focused at the outset on DATL because an important objective would be to test any new hydrolytic mutation not only for its effect on dimerization but also on fusion activity. And whereas DATL has robust activity in an in vitro fusion assay (Orso et al., 2009), hATL1 does not (Wu et al., 2015). For screening purposes, we took advantage of existing structural and biochemical data on hGBP1 (Ghosh et al., 2006). In hGBP1, a switch 1 serine residue, S73 (Fig. 3 A), interacts with the water nucleophile in the GDP ⋅ AlF4 transition state crystal structure (Fig. 3 B), and its mutation to alanine inhibits nucleotide hydrolysis (Ghosh et al., 2006). This serine residue is not conserved in the switch 1 region of either hATL1 or DATL (Fig. 3 A; S73 and equivalent residues are highlighted in blue). However, S73 in hGBP1 is also within hydrogen bonding distance to an aspartate residue D103 in switch 2 (Fig. 3 B). The latter aspartate residue is not only conserved in both hATL1 and DATL (Fig. 3 A; also highlighted in blue), but the corresponding residue in hATL1 (D152 in hATL1 and D127 in DATL) additionally contacts a water molecule that may assist in catalysis (Fig. 3 B). Targeting this conserved aspartate residue D127 in DATL as well as the nonconserved S73 equivalent residue R93 for mutagenesis, we found that either the D127N or D127A substitution in cyt-DATL abolished steady-state GTP hydrolysis (Fig. 3 C). In comparison, the R93A substitution had no effect, and R93Q had a modest effect (Fig. 3 C). Notably, neither D127 nor R93 (D152 and R118 in hATL1) is at the dimer interface in any of the hATL1 crystal structures (Bian et al., 2011; Byrnes and Sondermann, 2011; Byrnes et al., 2013), making them less likely to be involved directly in dimerization. Of the two D127 substitutions that blocked steady-state GTP hydrolysis (Fig. 3 C), we chose D127N for further analysis as it reflected a more conservative substitution; however, similar results have been obtained with D127A.
Even though it lacked any detectable GTPase activity at steady state (Fig. 3 C), D127N cyt-DATL underwent GTP-induced tryptophan fluorescence quenching much like the WT, with a similar GTP concentration dependence (compare Fig. 3 D with Fig. 2 B). This not only suggested that the D127N mutation might be more selective than the R48A mutation, but it also reinforced the idea that the quenching of tryptophan fluorescence reflects a G domain conformational change separate from nucleotide hydrolysis. To assess the dimerization and crossover properties of D127N cyt-DATL, we first looked at its crossover PIFE kinetics under conditions of saturating GTP. Here, the mutant variant showed a GTP-induced crossover kinetic that was somewhat slower than the WT but remarkably robust nevertheless (Fig. 3 E). The rapid dimerization and crossover by D127N, despite its undetectable steady-state GTPase activity (Fig. 3 C), further reinforced our conclusion above (based on Fig. 1) that crossover formation is not coupled directly to GTP hydrolysis. It also confirmed that the slow rate of dimerization and crossover seen previously with R48A (Byrnes et al., 2013) was most likely the result of a defect separate from hydrolysis, either in a G domain conformational change preceding dimerization and/or dimerization itself. Additionally, crossover dimerization by D127N cyt-DATL was profoundly slowed when GMPPNP was substituted for GTP, similarly to that observed previously for the WT (Fig. 3 F; Byrnes et al., 2013; Winsor et al., 2017). The substantially slower D127N crossover kinetic with GMPPNP relative to GTP (Fig. 3 F compared with Fig. 3 E) even in the apparent absence of hydrolysis also reinforced the above tryptophan fluorescence quenching data, indicating that GMPPNP is an imperfect mimic of GTP for atlastin (Fig. 2 D).
Although our steady-state assay of GTP hydrolysis suggested a complete block to hydrolysis for D127N cyt-DATL (Fig. 3 C), our single-turnover assay revealed a low level of hydrolysis activity (Fig. 4 A). However, the hydrolysis activity of D127N was extremely slow in comparison with WT. Whereas WT cyt-DATL hydrolyzed nucleotides to near completion by 10–20 s after GTP addition (Fig. 4 A; see also Fig. 1, E and F), D127N cyt-DATL required 20–40 min to complete a single round (Fig. 4 A). This kinetic block to hydrolysis provided an ideal platform to test whether the hydrolysis of GTP might be associated with dimer disassembly. As anticipated, dimerization and crossover by D127N cyt-DATL was rapid even under the single-turnover condition used above, with crossover formation largely completed by 10 s (Fig. 4 B), a time when the hydrolysis of GTP by this variant was negligible (Fig. 4 A). The disassembly kinetic of D127N, by contrast, was profoundly slowed as compared with WT (Fig. 4 B) and was incomplete even after 30 min (Fig. 4 C). Notably, the time to complete disassembly was similar to the time to complete hydrolysis (Fig. 4 C), in agreement with a role for GTP hydrolysis in promoting dimer disassembly as suggested above by the WT data (Fig. 1 F). Also, as observed for WT, Pi release by D127N cyt-DATL coincided well with nucleotide hydrolysis (Fig. 4 C), implying that GTP hydrolysis is rate limiting for Pi release not only in the WT but also in the variant.
Figure 4.

GTP hydrolysis is not required for crossover dimerization or initial fusion. (A–C) WT and D127N DATL single-turnover kinetics. All reactions contained 15 µM cyt-DATL and 7.5 µM GTP (final concentrations) and were performed at 25°C except where indicated. (A) GTP hydrolysis by WT or D127N cyt-DATL at room temperature. Reactions were quenched at the indicated times, and total Pi produced was measured after 10-fold dilution into 1.5 µM MDCC-PBP (n = 3; ±SD). (B) Stopped-flow PIFE of WT or D127N cyt-DATL after addition of GTP. The WT trace is the same as in Fig. 1 B. (C) Comparison of D127N crossover PIFE (C/O), GTP hydrolysis, and Pi release. D127N C/O PIFE was replotted from B after normalization. GTP hydrolysis was replotted from A. Single-turnover D127N Pi release was measured by including 15 µM MDCC-PBP (final) in the stopped flow. Traces were normalized (initial value = 0; maximum value = 1). (D and E) Fusion assay. (D) Full-length WT, R48A, or D127N DATL was reconstituted into donor and acceptor vesicles at a 1:1,000 protein/lipid ratio, and fusion was monitored as the dequenching of NBD-labeled lipid present in the donor vesicles over time at 28°C after addition of 1 mM GTP (average of three runs plotted). (E) Magnified view of the early time points of traces boxed in D. All stopped flow, GTP hydrolysis, and fusion kinetics are representative of two independent protein preparations.
The robust crossover kinetics of D127N, along with our observation that GTP hydrolysis is associated with dimer disassembly, a step believed to occur after membrane fusion, led us to predict that D127N DATL might be capable of at least one round of fusion catalysis. However, due to its >100-fold slower nucleotide hydrolysis rate (Fig. 4 A), it should be greatly hindered in dimer disassembly and subunit recycling. Consequently, we also predicted that this variant, unlike the WT, would be capable of supporting one but not multiple rounds of fusion. To test our predictions, we looked at the membrane fusion capability of D127N versus R48A and WT in the presence or absence of saturating GTP. We incorporated full-length versions of each DATL variant into synthetic vesicles and used a standard lipid-mixing assay to determine the relative rates of fusion. As expected from previous research (Pendin et al., 2011), R48A showed no fusion activity at all, while WT showed robust GTP-dependent activity (Fig. 4 D). Remarkably, and consistent with the lack of a major dimerization defect, D127N DATL catalyzed GTP-dependent fusion at a rate that was similar to the WT, at least at early times (Fig. 4 E). Thus, neither crossover formation nor fusion appears to require prior hydrolysis of GTP. Importantly, at later times, the lipid-mixing activity by D127N ceased (Fig. 4 D), consistent with our prediction that the strong impairment in nucleotide hydrolysis and dimer disassembly would impede the recycling of D127N atlastin subunits required for multiple rounds of fusion catalysis.
As mentioned above, hATL1, for reasons not yet understood, is incapable of catalyzing in vitro fusion (Wu et al., 2015). Despite this, the hATL1 soluble domain shares 55% amino acid identity and 75% amino acid similarity with that of DATL. Furthermore, cyt-hATL1 has been shown to behave similarly to cyt-DATL in several distinct assays (Byrnes et al., 2013; O’Donnell et al., 2017; Winsor et al., 2017). Therefore, we reasoned that GTP hydrolysis might also promote the disassembly, rather than the assembly, of the cyt-hATL1 crossover dimer. To evaluate this, we adapted our PIFE assay to cyt-hATL1 by labeling it with Cy3 on an engineered 3HB cysteine residue (N368C) equivalent to that used previously for cyt-DATL (Winsor et al., 2017). Under the same single-turnover condition as used above for cyt-DATL, GTP induced a rapid rise and fall in the cyt-hATL1 PIFE signal (Fig. 5 A). Though the relative amplitudes of the peak and the end state for the monomer differed somewhat from that observed for cyt-DATL, the rise and fall of the PIFE signal was on a similar time scale, consistent with a single round of crossover dimer formation and disassembly for cyt-hATL1 (Fig. 5 A). We next examined the timing of GTP hydrolysis and Pi release for cyt-hATL1. Importantly, at a time when dimer disassembly was already well underway (∼5 s), hydrolysis was only ∼60% complete for cyt-hATL1 (Fig. 5, B and C). Overlaying the averaged hydrolysis data (from Fig. 5 B and additional replicates) onto the normalized PIFE trace of crossover revealed crossover dimerization occurring well before nucleotide hydrolysis (Fig. 5 C) in a manner similar to that observed above for cyt-DATL. We also examined the timing of Pi release using MDCC-PBP as above. Similar to cyt-DATL, Pi release for cyt-hATL1 closely coincided with GTP hydrolysis (Fig. 5 C), again indicating that the release of Pi is fast and limited by nucleotide hydrolysis. Interestingly, we noticed that GTP hydrolysis, Pi release, and dimer disassembly were all somewhat faster for cyt-hATL1 than for cyt-DATL, the significance of which remains to be determined. Nonetheless, and more importantly, GTP hydrolysis and Pi release coincided more closely with dimer disassembly than with its formation, just as observed for cyt-DATL. Fits of the rising phase of Pi release and the falling phase of PIFE followed first-order kinetics with rates of 0.33 s−1 and 0.24 s−1, respectively, again consistent with a close coupling between dimer disassembly and Pi release. Notably, the approximate turnover time of ∼10 s for hydrolysis by hATL1 observed in this study contrasted sharply with the recently reported burst rate of 27 s−1 (O’Donnell et al., 2017). The source of the >100-fold discrepancy in hydrolysis rates is unclear, but it may be due to the saturating level of GTP used previously (O’Donnell et al., 2017), which may have made it difficult to separate a burst phase from the steady state, and which may also have contributed a high background of GDP and Pi.
Figure 5.

Crossover dimerization of hATL1 also precedes GTP hydrolysis and Pi release. (A–C) Single-turnover kinetics of cyt-hATL1. (A) Stopped-flow PIFE of WT cyt-hATL1 after addition of GTP. (B) Hydrolysis of GTP (containing a-32P-GTP) by WT cyt-hATL1 at room temperature. Reactions were acid quenched at the indicated times (one of three replicates shown). (C) Comparison of WT crossover PIFE (C/O), GTP hydrolysis, and Pi release. Traces were normalized (initial value = 0; maximum value = 1). Normalized PIFE (C/O) trace is from A. GTP hydrolysis was the average of three replicates from B (±SEM). Pi release was measured by including 15 µM MDCC-PBP (final) in the stopped flow. All reactions contained 15 µM cyt-hATL1 and 7.5 µM GTP (final concentrations) and were performed at 25°C except where indicated. All traces are the average of three to five individual traces and are representative of two independent protein preparations.
These results indicate that the apparent close coupling between GTP hydrolysis and dimer disassembly observed in this study for cyt-DATL is most likely a feature that is conserved among atlastins. We wondered whether the relative timing of other events such as GTP binding and the G domain conformational change identified in this study would also be conserved. To assess this, we additionally monitored both nucleotide binding and tryptophan fluorescence quenching for both WT cyt-DATL and WT cyt-hATL1 under the same single-turnover condition used above (Figs. 1 and 5). GTP binding was monitored using N-methylanthraniloyl (mant)-labeled nucleotides, which undergo fluorescence enhancement upon protein binding (Moore et al., 1993). We used the deoxyribonucleotide mant-dGTP instead of the ribonucleotide mant-GTP because the former is uniformly labeled at the 3′ position on the ribose ring, whereas the latter is a mixture of 2′ and 3′OH-labeled nucleotides. An independent assay showed that the two versions bound cyt-DATL with similar kinetics (Fig. S3). Tryptophan fluorescence was monitored as above except that here, the atlastin and GTP concentrations were those of the single-turnover condition. Overlaying the nucleotide binding and tryptophan fluorescence quenching data for cyt-DATL onto the previously observed traces for crossover dimerization, GTP hydrolysis and Pi release (Fig. 1 F) revealed the complete sequence of events for cyt-DATL (Fig. 6 A). GTP (dGTP) binding was very fast, and this was followed by a significantly slower GTP binding–induced G domain conformational change. Notably, the G domain conformational change, as reported by tryptophan fluorescence quenching, preceded dimerization by a significant margin, implying the existence of a further, yet-to-be-identified G domain conformational change preceding dimerization. Remarkably, the same analysis for cyt-hATL1 revealed a similar sequence of events for the human atlastin, with very fast GTP binding followed by a slower G domain conformational change, which also preceded crossover dimerization by a significant margin (Fig. 6 B). Again, as mentioned above, it was noticeable that GTP hydrolysis, Pi release, and dimer disassembly were all somewhat faster for cyt-hATL1 than for cyt-DATL.
Altogether, our findings can be interpreted through a model (Fig. 7) in which GTP binding on its own is sufficient to trigger a series of conformational changes in the atlastin G domain that renders rapid crossover dimer formation and initiation of lipid mixing an energetically downhill reaction. The nucleotide analogue GMPPNP and the R48A mutation appear to inhibit this series of GTP-induced conformational changes early on, while whether GTPγS promotes the same or different set of changes will need to be further investigated. In either case, once the series of G domain conformational changes have been completed, crossover dimerization ensues unhindered, driving lipid mixing and fusion. Finally, GTP hydrolysis and the release of Pi return atlastin to a conformation that strongly favors the monomer state, thus enabling the recycling of atlastin subunits for reuse.
Figure 7.
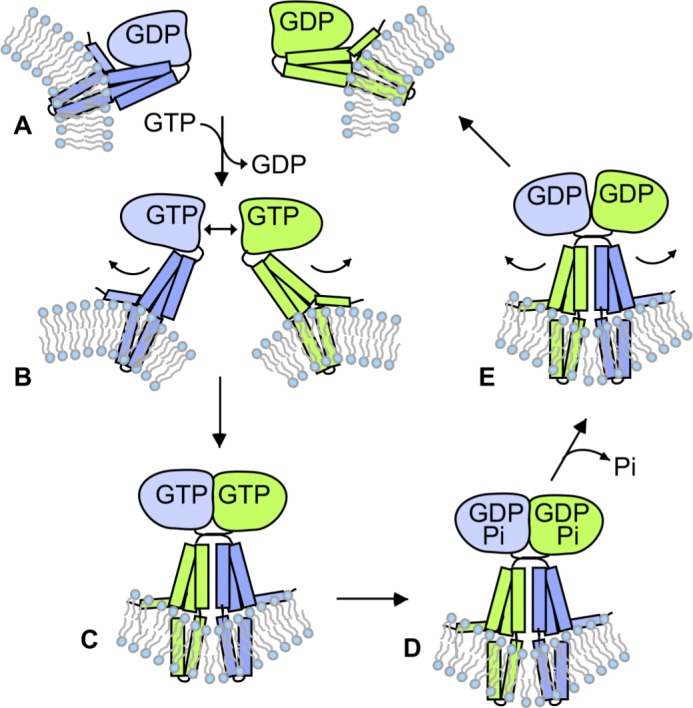
Model depicting the role of GTP hydrolysis in atlastin-catalyzed fusion. (A–E) GDP-bound monomers (A) undergo a conformational change upon GTP binding (B) that triggers dimerization and crossover for fusion (C). After fusion, hydrolysis of GTP (D) and release of Pi trigger dimer disassembly (E) to restart the cycle (A). Note that the order of dimer dissociation and GDP release is not necessarily being specified in this figure, and GDP could be released before dimer disassembly.
Discussion
In this study, we have applied a comprehensive kinetic analysis to monitor the discrete conformational states of the atlastin GTPase-soluble domain as it undergoes dimerization and disassembly during a single round of GTP binding and hydrolysis. For the first time, our results demonstrate that GTP hydrolysis promotes the breaking apart of the atlastin crossover dimer rather than its formation. Our conclusion is seemingly in opposition to those of previous studies including our own (Byrnes et al., 2013; McNew et al., 2013; Saini et al., 2014; Liu et al., 2015; Hu and Rapoport, 2016; O’Donnell et al., 2017; Winsor et al., 2017). However, we suggest that the contradictions reside not in the data but only in their interpretation. For instance, the inability of the R48A DATL variant either to undergo rapid crossover dimerization (Byrnes et al., 2013) or to promote fusion (Pendin et al., 2011) was previously interpreted as evidence that GTP hydrolysis is required for both. However, we now show that a newly identified hydrolysis-deficient mutant variant D127N DATL is quite capable of both dimerization and fusion despite extremely slowed nucleotide hydrolysis, suggesting that the previously observed failures of the R48A variant (Pendin et al., 2011; Byrnes et al., 2013) most likely stemmed from defects other than a lack of nucleotide hydrolysis. One such defect may be an inability to undergo an early GTP-induced G domain conformational change as reflected by tryptophan fluorescence quenching (Fig. 2 E), and another may be an impairment of the dimerization interface (Bian et al., 2011). Similarly, the inefficacy of the nonhydrolysable GTP analogues GMPPNP and GTPγS to promote either rapid dimerization or fusion by WT atlastin (Orso et al., 2009; Byrnes et al., 2013; Saini et al., 2014; Winsor et al., 2017) was previously interpreted as evidence that GTP hydrolysis is required for both rapid dimerization and fusion. However, we now show that GMPPNP is unable to replicate a GTP-induced G domain conformational change that precedes dimerization (Fig. 2 D), potentially explaining why it is incapable of inducing either rapid crossover dimerization (Byrnes et al., 2013; Winsor et al., 2017) or fusion (Orso et al., 2009; Saini et al., 2014). GTPγS may be capable of promoting a conformational change, but whether the change is the same as that induced by GTP is debatable. As a whole, our findings tell a cautionary tale regarding the over interpretation of the effects of inhibitors that may not be entirely specific.
The kinetic data obtained in this study exclude most previously proposed models. For instance, one model had atlastin monomers coming together in the transition state of GTP hydrolysis (Liu et al., 2015). Since the transition state is expected to be fleeting, such a model would predict the hydrolysis of GTP and crossover to be near concurrent, whereas we observed a pronounced lag between crossover dimerization and nucleotide hydrolysis for both cyt-DATL and cyt-hATL1 (Figs. 1 and 5). Another model (our own) had atlastin monomers coming together weakly in the GTP-bound state but had stable dimerization requiring GTP hydrolysis (Saini et al., 2014; Winsor et al., 2017). Such a model would also predict the hydrolysis of GTP to be concurrent with crossover dimerization, whereas again, we observed hydrolysis to occur only after dimerization. Finally, a third model had GTP hydrolysis occurring within the monomer of atlastin to produce a GDP ⋅ Pi–bound dimerization-competent conformation (Byrnes et al., 2013; O’Donnell et al., 2017). That model would predict GTP hydrolysis to be faster than dimerization, the opposite of what we observed in this study.
In our new model (Fig. 7), GTP binding is sufficient to trigger a series of G domain conformational changes to promote atlastin dimerization. The changes likely start with an immediate release of the 3HB from the G domain following GTP binding not studied explicitly here but described previously (O’Donnell et al., 2017; Winsor et al., 2017). This may then be followed by further G domain rearrangements that serve to produce a conformation for whom dimer formation is now strongly favored. Based on its timing, we suggest that the intrinsic tryptophan fluorescence quenching observed in this study, slower than either GTP binding or the previously observed 3HB release, reflects one aspect of the proposed conformational rearrangement. Also, based on the observation that tryptophan fluorescence quenching significantly precedes dimerization, we suggest that at least one additional conformational rearrangement follows and is required to fully form the dimerization-competent state. Finally, our new model proposes that once GTP-bound dimerization-competent atlastin monomers encounter one another, trans-dimerization and crossover are highly favored, leading to lipid mixing and fusion catalysis. Membrane insertion of the tail amphipathic helix, not studied here but previously shown to be required for fusion (Moss et al., 2011; Liu et al., 2012; Faust et al., 2015), is also assumed.
A salient feature of our new model is that it provides a plausible mechanism by which the stable post-fusion crossover dimer of atlastin can be disassembled following each round of fusion catalysis. The theoretical energy barrier for fusion has been estimated to be >40 kBT (Cohen and Melikyan, 2004), and the binding energy of the SNARE coiled-coil bundle has been experimentally determined to be within range at 30–40 kBT (Yersin et al., 2003; Liu et al., 2006; Gao et al., 2012). The binding energy for atlastin has yet to be quantitatively measured, but we previously showed that the WT DATL soluble domain crossover dimer is sufficiently stable that it does not undergo detectable disassembly over a period of several hours as long as GTP hydrolysis is prevented (Winsor et al., 2017). Because atlastin catalyzes a fusion reaction fundamentally similar to that catalyzed by SNAREs, we would anticipate a substantial binding energy for the atlastin crossover dimer in the range of that of the SNARE postfusion complex, though it could be less if more atlastin molecules than SNAREs were to participate in a single fusion event. Based on these considerations, it is not surprising that the majority of the free energy release associated with GTP hydrolysis would be devoted to breaking apart the postfusion dimer.
If, as we suggest, the role of GTP hydrolysis were to trigger dimer disassembly, it would be critical for the hydrolysis of GTP not to precede crossover dimerization. Accordingly, we suggest that it is dimerization itself that promotes the rearrangement of catalytic residues that stimulate nucleotide hydrolysis. As previously suggested for hGBP1 (Ghosh et al., 2006), this rearrangement may include the movement of the catalytic R48 side chain (R77 in hATL1) away from the dimer interface toward the bound GTP to stabilize the negatively charged transition state. Interestingly, we previously showed that a mutant variant of atlastin, which fails to undergo crossover but retains G domain dimerization capability, still retains robust GTP hydrolysis activity (Morin-Leisk et al., 2011; Saini et al., 2014). This suggests that dimerization through the G domain is more important than dimerization through the 3HBs for triggering GTP hydrolysis. At the same time, while crossover may not be strictly required for triggering GTP hydrolysis, we presume that it would accompany G domain dimerization so as to also precede nucleotide hydrolysis. This would ensure that fusion occurs before dimer disassembly. Consistent with this idea, G domain dimerization and 3HB crossover occur in rapid succession, at least in the soluble phase (Fig. 1 D; O’Donnell et al., 2017; Winsor et al., 2017). Altogether, the proposed dependence of nucleotide hydrolysis on G domain dimerization in our model would allow for the recycling of atlastin monomers to be built in as the terminal step in each fusion reaction cycle.
How the load of a membrane anchor might influence the rates of the individual steps of the atlastin reaction cycle remains untested. Crossover formation between atlastins in opposing membranes would be predicted to occur considerably more slowly than in the soluble phase because fusion, with its substantial energy barrier, would presumably have to occur in order for crossover dimerization to progress to completion. Indeed, it is noteworthy that the kinetic of lipid mixing and fusion is substantially slower than that of crossover dimerization in the soluble phase. A difference in reaction rates under load may also explain why the D127N DATL variant appeared incapable of undergoing any recycling, thus ceasing fusion after an initial period of ∼60 s, even though it was capable of at least slow hydrolysis of GTP in the soluble phase. We suspect that the conformational constraints imposed by the membrane anchor may diminish nucleotide hydrolysis by the variant even more than in the soluble phase. Further investigation of the reaction kinetics of both WT and mutant variants of atlastin under the load of membranes, albeit challenging, is likely to reveal additional insights into the fusion mechanism.
The initial discovery of the atlastin GTPase as a membrane fusion catalyst (Orso et al., 2009; Bian et al., 2011; Moss et al., 2011; Pendin et al., 2011) naturally raised the question of whether its mechanism of fusion would bear any resemblance to that of previously studied fusion catalysts. We recently showed that formation of the atlastin crossover dimer energizes fusion (Winsor et al., 2017) in a manner analogous to coiled-coil bundle formation energizing SNARE dependent fusion (Weber et al., 1998; Jahn and Scheller, 2006). Our findings extend that analogy and demonstrate just how conceptually similar the atlastin fusion mechanism is to the SNARE mechanism, the main difference being that whereas SNAREs rely on the ATPase NSF for subunit recycling after fusion (Otto et al., 1997; Jahn and Scheller, 2006; Zhao and Brunger, 2016), the recycling mechanism for atlastin is built into the fusion apparatus. Notably, the picture of the atlastin reaction cycle that emerges from this study also bears resemblance to other G proteins activated by dimerization (glutamate decarboxylases [GADs]; Gasper et al., 2009), for whom GTP binding–induced dimerization triggers both the biological function of the GTPase and nucleotide hydrolysis-induced dimer disassembly to initiate a new reaction cycle (Gasper et al., 2009). An analysis of the evolutionary history of atlastin and other GADs such as the signal recognition particle (SRP) and SRP receptor (Shan et al., 2007) has revealed that they are the products of an early split in the diversification of P-loop NTPases (Leipe et al., 2002; Shan, 2016). Therefore, the general mechanism of activation of G proteins by GTP-dependent dimerization and inactivation upon nucleotide hydrolysis appears to have evolved multiple times during evolution to regulate a wide variety of biological processes.
Materials and methods
Reagents and constructs
The soluble domain cyt-DATL construct was 6×His tagged at the N terminus by cloning aa 1–415 of DATL into NheI and EcoRI sites of the pRSETB vector as described previously (Saini et al., 2014; Winsor et al., 2017) except with an engineered cysteine H410C for the PIFE assays or an engineered cysteine S270C for the H/H FRET assay. Catalytic mutations in DATL (R48A or D127N) were made in the H410C PIFE construct background. The soluble domain of hATL1 (aa 1–446) was 6×His tagged at the N terminus in the same way as DATL and had an engineered cysteine N368C for PIFE assays. All assays not relying on protein labeling used unlabeled versions of the relevant PIFE constructs. The full-length DATL WT parent construct, also 6×His tagged at the N terminus using the same cloning strategy, had the following cysteine substitutions: G343C, C429L, C452L, C501A, and C350A as described previously (Saini et al., 2014; Winsor et al., 2017). The cysteine-substituted full-length protein was previously shown to have fusion activity similar to the WT (Saini et al., 2014). All amino acid–substituted constructs were generated using PCR mutagenesis and fully sequence confirmed (GENEWIZ). Unlabeled nucleotides were purchased from Sigma-Aldrich, reconstituted to 100-mM stocks in 10 mM Tris, pH 8.0, and 1 mM EDTA, and stored at −80°C. Mant-labeled nucleotides were purchased from Jena Bioscience. Lipids were purchased from Avanti Polar Lipids. Cy3 maleimide was from GE Healthcare, and Alexa Fluor 488/647 was from Thermo Fisher Scientific.
Protein expression and purification
Expression of cyt-DATL or cyt-hATL1 was in (DE3) pLysS E. coli grown at 25°C to OD ∼0.6 and induced with 0.5 mM IPTG overnight at 20°C. Cells were harvested, washed once with cold PBS, and flash frozen. All purification steps were conducted on ice or at 4°C with chilled buffers. Cell pellets (from 2 liters culture) were resuspended in 20 ml lysis buffer (50 mM Tris, pH 8.0, 5 mM MgCl2, 10 mM imidazole, 500 mM NaCl, 10% glycerol, 0.5 mM PMSF, 1 µg/ml pepstatin, 1 µM leupeptin, and 2 mM 2-mercaptoethanol [2-ME]) per liter culture and sonicated 4 × 5 min on a 50% duty cycle with output control setting 3 with a microtip on a Branson sonifier 250 (Branson Ultrasonics) with 5-min rests between rounds. Samples were then spun at 10,500 rpm in a SA600 rotor (Sorvall; Thermo Fisher Scientific) for 30 min, and the supernatant was spun at 50,000 rpm in a Ti70 rotor (Beckman Coulter) for 1 h. The final supernatant was mixed with 0.25 ml Ni-NTA agarose (QIAGEN) for 2 h, poured into a column support, and washed with 30 ml wash buffer (50 mM Tris, pH 8.0, 5 mM MgCl2, 20 mM imidazole, 100 mM NaCl, 10% glycerol, and 2 mM 2-ME). Protein was eluted in 0.25-ml fractions with elution buffer (50 mM Tris, pH 8.0, 5 mM MgCl2, 250 mM imidazole, 100 mM NaCl, 10% glycerol, and 2 mM 2-ME). Fractions were flash frozen and stored at −80°C. Samples of each fraction were resolved by SDS-PAGE and determined to be >95% pure. Expression of full-length DATL was also grown in (DE3) pLysS E. coli at 25°C but induced at OD ∼0.4 with 0.2 mM IPTG at 18°C for 2.5 h. Harvesting was as above, and all purification steps were on ice or at 4°C. Cell pellets (from 4 liters culture) were resuspended in 10 ml lysis buffer containing 4% Triton X-100 (Sigma-Aldrich) per liter culture, sonicated, and cleared of insoluble material as above. The final supernatant was diluted to 1% Triton X-100 using lysis buffer and then loaded (from the bottom) overnight onto a prepoured 0.5-ml Ni-NTA agarose column. The following day, the column was washed with 30 ml lysis buffer with 1% Triton X-100 and then with 30 ml wash buffer with 0.1% Anapoe X-100 (Affymetrix). The protein was eluted in 0.5-ml fractions with elution buffer containing 0.1% Anapoe X-100. Fractions were flash frozen and stored at −80°C. Samples of each fraction were resolved by SDS-PAGE and determined to be ≥50% pure.
Steady-state GTPase assay
GTPase activity of cyt-DATL or cyt-hATL1 was measured under steady-state conditions (Fig. 3 C) using the EnzChek Phosphate Assay Kit (Molecular Probes; Thermo Fisher Scientific) on a TECAN Safire2 plate reader (Tecan). Purified cyt-DATL protein was desalted into SEC buffer (25 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2 mM EGTA, 5 mM MgCl2, and 2 mM 2-ME) without magnesium. A standard reaction involved mixing 1 U/ml purine nucleoside phosphorylase (PNP), 0.2 mM 2-amino-6-mercapto-7-methylpurine riboside, 2 µM cyt-DATL, and 0.5 mM GTP in 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 2 mM 2-ME in a total volume of 0.2 ml at 28°C. The reaction was started with the addition of 5 mM MgCl2. Therefore, buffers provided within the EnzChek kit were altered so that they did not contain any magnesium.
MDCC-PBP purification
MDCC-PBP was prepared following a previously published protocol (Solscheid et al., 2015). Expression from the plasmid PstS1 encoding PBP A197C (Addgene) was grown in (DE3) pLysS E. coli at 37°C to OD ∼0.8 and induced with 0.5 mM IPTG for 4 h at 37°C. Cells were harvested, washed twice in cold 10 mM Tris, pH 8.0, and 30 mM NaCl, resuspended in 20 mM Tris, pH 8.0 (20 ml per 500-ml culture), and flash frozen. All purification steps were on ice or at 4°C. Cells (from 0.5 liters culture) were thawed with addition of 1 mM DTT and 0.5 mM PMSF, homogenized, sonicated 3 × 0.5 min on a 50% duty cycle with the #3 setting, and cleared by low- and high-speed centrifugation as above. The resulting supernatant was adjusted to have the same conductivity as buffer A (10 mM Tris, pH 8.0) and loaded onto a 5-ml Q Sepharose (GE Healthcare) column preequilibrated in buffer A. After washing with 30 ml buffer A, protein was eluted in 2.5-ml fractions with a 50-ml gradient (0–200 mM NaCl) in buffer A. Peak fractions were pooled, concentrated with a C-10 Amicon centriprep (Sigma-Aldrich), flash frozen, and stored at −80°C. For MDCC labeling, two to three PBP preparations were pooled and dialyzed against 20 mM Tris, pH 8.0, and 100 µM EDTA and then placed in a 15-ml Falcon tube to achieve 100 µM PBP in a volume of 8 ml, and a Pi mop was added (200 µM MEG and 0.2 U/ml microbial PNPase [Sigma-Aldrich] final) at room temperature. After 10 min, MDCC (Thermo Fisher Scientific) was added to 150 µM from a DMSO stock, and then the tube was wrapped in foil and rotated for 30 min at room temperature. Precipitate was removed by centrifugation in a TLA 100.3 (Beckman Coulter) rotor at 100,000 rpm for 10 min at 4°C. The supernatant was adjusted to have the conductivity of buffer A, and the sample was loaded onto a 20-ml Q Sepharose column equilibrated in buffer A. MDCC-PBP was eluted with a 400-ml (0–50 mM NaCl) gradient in buffer A in 4-ml fractions. Peak fractions were concentrated to 1 ml with a C-10 centriprep, flash frozen, and stored at −80°C.
Single-turnover GTPase assays
GTP hydrolysis activity under single-turnover conditions (Figs. 1 E, 4 A, and 5 B) was measured using 15 µM Cyt-DATL (or cyt-hATL1) and 7.5 µM GTP in SEC buffer at room temperature. For WT cyt-DATL or cyt-hATL1, which has fast turnover times, GTP hydrolysis assays used radiolabeled GTP (Figs. 1 E and 5 B) with 15 µM cyt-DATL (or cyt-hATL1) and 7.5 µM GTP (containing 1 µCi/µl α-32P-GTP; PerkinElmer) in SEC buffer at room temperature. At the indicated times, samples were quenched by addition of 1 vol of 1 M perchloric acid neutralized with addition of 0.75 vol of 1 M KOAc and cleared by centrifugation at 4,000 rpm in a microcentrifuge for 2 min to remove denatured protein. 3 µl of each supernatant was loaded onto a PEI cellulose thin-layer chromatography plate (Macherey-Nagel), and nucleotides were resolved with a solution of 1 M LiCl2 and 1 M formic acid for 2 h. After drying, samples were visualized using a phosphorimager (Typhoon; Amersham). Alternately, for the D127N cyt-DATL variant, which has a very slow turnover time, the hydrolysis assay used MDCC-PBP (Fig. 4 A). Reactions were stopped, and any bound Pi was released from protein at the indicated times by boiling for 2 min and then cleared with a 2-min, 16,000 g microcentrifuge spin. The supernatant of each reaction (20 µl) was diluted 10-fold into SEC buffer containing MDCC-PBP to achieve a final concentration of 1.5 µM MDCC-PBP. MDCC fluorescence was measured in a Tecan Safire2 plate reader at 465 nm after excitation at 430 nm. A Pi standard curve showed the assay to be linear with Pi concentration over the range of Pi produced under these conditions.
Cy3 labeling for PIFE and FRET
Cyt-DATL containing an engineered cysteine H410C or cyt-hATL1 containing an engineered cysteine N368C was desalted over a 4-ml Sephadex G-25 (Sigma-Aldrich) column preequilibrated with labeling buffer (25 mM Tris, pH 7.0, 100 mM NaCl, 5 mM MgCl2, 2 mM EGTA, 1 mM imidazole, and 500 µM TCEP). Cy3 maleimide was added from a DMSO stock at a 1:1 protein/dye molar ratio and incubated for 2 h at room temperature before being centrifuged at 100,000 rpm in a TLA100 rotor for 10 min at 4°C to remove any precipitate. Labeled cyt-DATL or cyt-hATL1 was then desalted twice as above to remove free Cy3. Typical labeling efficiencies were 20–30%. Differences in labeling efficiency did not alter the rate of crossover formation (Winsor et al., 2017). Labeling with Alexa Fluor 488/647 maleimide for the H/H FRET assay followed essentially the same procedure except that it used cyt-DATL containing an engineered cysteine S270C and the labeling time was only 30 min.
PIFE assays of crossover
For multiple-turnover assays with either GTP or GMPPNP (Fig. 3, E and F), 2 µM cyt-DATL labeled with Cy3 as described above was mixed with 1 mM nucleotide (final concentrations after mixing) in SEC buffer at 25°C using a stopped-flow accessory mounted on a PTI QuantaMaster-400 fluorimeter (Horiba Instruments Inc.), and 570 nm fluorescence was monitored at 100-ms intervals after 540-nm excitation. Data were acquired using the FelixGX software (Horiba Instruments Inc.) and normalized using the equation (Fluorescence – Minimum fluorescence observed)/(Maximum fluorescence observed – Minimum fluorescence observed), where fluorescence is F/Fo. Single-turnover PIFE assays (Figs. 1 B, 4 B, and 5 A) were essentially the same as above except that the final concentrations after mixing were 15 µM cyt-DATL (or cyt-hATL1) and 7.5 µM GTP in SEC buffer, and data were acquired at 50-ms intervals. Data were plotted either as F/Fo or as normalized F/Fo using the equation (Fluorescence – Initial fluorescence)/(Maximum fluorescence observed – Initial fluorescence). All data shown are the average of three to five runs per condition. All data analysis was in Microsoft Excel. Each of the three to five PIFE traces was nearly identical to the others, and each result was reproduced with at least two independent protein preparations.
FRET assay for H/H dimerization
H/H dimerization kinetics under single-turnover conditions used cyt-DATL (S270C) labeled with Alexa Fluor 488 (donor) and Alexa Fluor 647 (acceptor) mixed at a 1:2 donor/acceptor ratio (15 µM total cyt-DATL final) with 7.5 µM GTP (final) in SEC buffer at 25°C. Measurements were acquired as for PIFE. Both donor and acceptor fluorescence emission with 490 nm donor excitation was monitored at 50-ms intervals at 520 and 670 nm respectively, though only the acceptor emission trace is shown (Fig. 1 D). The data shown are the average F/Fo traces of three runs without normalization, and each run was nearly identical to the others.
Pi release kinetics
Assay conditions followed the above for single-turnover PIFE except that unlabeled H410C cyt-DATL (or N368C cyt-hATL1) was mixed with MDCC-PBP before mixing with GTP so that the final concentrations after mixing were 15 µM cyt-DATL, 15 µM MDCC-PBP, and 7.5 µM GTP in SEC buffer. MDCC fluorescence at 465 nm was monitored at 50-ms intervals at 25°C with 430 nm excitation (Figs. 1 F, 4 C, and 5 C). All data shown are the average of three runs per condition, where each run was nearly identical to the others. Each result was reproduced with at least two independent protein preparations.
Tryptophan fluorescence quenching assay
In Fig. 2, 125 nM unlabeled H410C cyt-DATL was mixed with various concentrations of nucleotide (all indicated concentrations were final after mixing), and tryptophan fluorescence was monitored at 318 nm every 20 ms after excitation at 295 nm. Assays were performed in SEC buffer at 25°C using stopped-flow conditions as described above for PIFE. All data were the average of three to five traces. For single-turnover conditions (Fig. 6), 15 µM unlabeled H410C cyt-DATL (or unlabeled N368C cyt-hATL1) and 7.5 µM GTP were used, and data were plotted as 1 − (normalized F/Fo) for clarity. In all cases, the fluorescence baseline for buffer at each nucleotide concentration without cyt-DATL (or cyt-hATL1) was subtracted from the fluorescence with cyt-DATL (or cyt-hATL1) before F/Fo calculations.
Mant-nucleotide binding assay
GTP-binding kinetics was assessed using mant-labeled nucleotide. Mant-dGTP was used as a proxy for mant-GTP because the latter is a mixture of 2′- and 3′-ribose–labeled nucleotides and exhibits biphasic binding behavior due a slower binding kinetic by the 2′-labeled nucleotide. In contrast, mant-dGTP is uniformly labeled at the 3′ position and exhibits binding that fits well with a single exponential decay equation. Nonetheless, the two nucleotides bound cyt-DATL with similar overall kinetics (Fig. S3). The assay (Fig. 6) used single-turnover conditions with 15 µM unlabeled H410C cyt-DATL (or unlabeled N368C cyt-hATL1) and 7.5 µM mant-dGTP in SEC at 25°C under stopped flow as described above for PIFE. Fluorescence at 425 nm was monitored at 10-ms intervals with excitation at 295 nm.
Preparation of liposomes and lipid-mixing fusion assay
Lipids in chloroform dried down by rotary evaporation were hydrated by resuspension in A100 buffer (25 mM Hepes, pH 7.4, 100 mM KCl, 10% glycerol, 1 mM EDTA, and 2 mM 2-ME) at a final 10-mM lipid concentration and subjected to 11 freeze–thaw cycles in liquid N2 and 42°C water bath. Liposomes (100–300-nm diameter) were formed by extrusion through 100-nm polycarbonate filters using the LipoFast LF-50 extruder (Avestin) and checked for size by dynamic light scattering (Zen3600). Purified full-length D-ATL was incorporated at a 1:1,000 protein/lipid ratio into labeled and unlabeled liposome populations at an effective detergent/lipid ratio of ∼0.7 by incubating protein and lipid at 4°C for 1 h followed by four detergent-removal incubations by SM-2 Bio-Beads (Bio-Rad) at 1 g beads per 70 mg Anapoe X-100. Insoluble protein aggregates were pelleted by centrifugation in a microcentrifuge for 10 min at 16,000 g. Thereafter, reconstituted proteoliposomes were adjusted to 50% Nycodenz (Axis-Shield) and separated from unincorporated protein by flotation through a 5-ml Nycodenz step gradient (50%/45%/0%) in A100 buffer without glycerol in a SW50.1 (Beckman Coulter) rotor overnight. Finally, the floated fraction was desalted over a 2.4-ml Sephadex G-25 column into A100 buffer, stored at 4°C, and used within 72 h or flash frozen and stored at −80°C. Unlabeled liposomes consisted of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (PC) and 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (PS) at an 85:15 ratio. Labeled liposomes consisted PC:PS:1,3-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD):1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) at an 82:15:1.5:1.5 molar ratio. For the fusion assay, proteoliposomes (0.6 mM total lipid) were incubated in A100 buffer containing 5 mM MgCl2 at a 1:3 labeled/unlabeled ratio. Following a 10-min incubation at 28°C in a Tecan M1000 plate reader, 2 mM GTP was injected using the automated injector attachment and fluorescence dequenching of NBD monitored at 5-s intervals at 538 nm after excitation at 460 nm. After 10 min, 0.5% Anapoe X-100 was added for determination of the maximum possible dequenching signal. Data were plotted using the equation (Fluorescence observed – Minimum fluorescence observed)/(Maximum fluorescence – Minimum fluorescence), and the average of three runs is shown (Fig. 4, D and E). The results were reproduced with at least two independent protein preparations and liposome incorporations.
Online supplemental material
Fig. S1 shows that cyt-DATL labeled on H410C has the same sensitivity to crossover as cyt-DATL labeled on G343C. Fig. S2 shows a comparison of cyt-DATL PIFE kinetics under single- and multiple-turnover conditions. Fig. S3 shows that mant-dGTP and mant-GTP bind cyt-DATL with similar kinetics.
Supplementary Material
Acknowledgments
We thank A. Linstedt for discussions throughout, G. Rule for suggestions on active site mutations, M. Webb for advice on producing MDCC-PBP, and M. Bruchez and J. Minden for sharing equipment.
This work was supported by a grant from the National Institutes of Health/National Institute for General Medical Sciences R01GM107285 (to T.H. Lee) and supplement R01GM107285-02S1.
The authors declare no competing financial interests.
Author contributions: All authors contributed to experimental design and data analysis. T.H. Lee and J. Winsor wrote the manuscript with input from all authors.
References
- Anwar K., Klemm R.W., Condon A., Severin K.N., Zhang M., Ghirlando R., Hu J., Rapoport T.A., and Prinz W.A.. 2012. The dynamin-like GTPase Sey1p mediates homotypic ER fusion in S. cerevisiae. J. Cell Biol. 197:209–217. 10.1083/jcb.201111115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian X., Klemm R.W., Liu T.Y., Zhang M., Sun S., Sui X., Liu X., Rapoport T.A., and Hu J.. 2011. Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. USA. 108:3976–3981. 10.1073/pnas.1101643108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune M., Hunter J.L., Corrie J.E., and Webb M.R.. 1994. Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry. 33:8262–8271. 10.1021/bi00193a013 [DOI] [PubMed] [Google Scholar]
- Byrnes L.J., and Sondermann H.. 2011. Structural basis for the nucleotide-dependent dimerization of the large G protein atlastin-1/SPG3A. Proc. Natl. Acad. Sci. USA. 108:2216–2221. 10.1073/pnas.1012792108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes L.J., Singh A., Szeto K., Benvin N.M., O’Donnell J.P., Zipfel W.R., and Sondermann H.. 2013. Structural basis for conformational switching and GTP loading of the large G protein atlastin. EMBO J. 32:369–384. 10.1038/emboj.2012.353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.A., and Scheller R.H.. 2001. SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2:98–106. 10.1038/35052017 [DOI] [PubMed] [Google Scholar]
- Chen J., Stefano G., Brandizzi F., and Zheng H.. 2011. Arabidopsis RHD3 mediates the generation of the tubular ER network and is required for Golgi distribution and motility in plant cells. J. Cell Sci. 124:2241–2252. 10.1242/jcs.084624 [DOI] [PubMed] [Google Scholar]
- Cohen F.S., and Melikyan G.B.. 2004. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol. 199:1–14. 10.1007/s00232-004-0669-8 [DOI] [PubMed] [Google Scholar]
- Daumke O., and Praefcke G.J.. 2011. Structural insights into membrane fusion at the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 108:2175–2176. 10.1073/pnas.1019194108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert D.M., and Kim P.S.. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810. 10.1146/annurev.biochem.70.1.777 [DOI] [PubMed] [Google Scholar]
- Faust J.E., Desai T., Verma A., Ulengin I., Sun T.L., Moss T.J., Betancourt-Solis M.A., Huang H.W., Lee T., and McNew J.A.. 2015. The Atlastin C-terminal tail is an amphipathic helix that perturbs the bilayer structure during endoplasmic reticulum homotypic fusion. J. Biol. Chem. 290:4772–4783. 10.1074/jbc.M114.601823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov V.A., and Zimmerberg J.. 2010. Cooperative elastic stresses, the hydrophobic effect, and lipid tilt in membrane remodeling. FEBS Lett. 584:1824–1829. 10.1016/j.febslet.2010.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Zorman S., Gundersen G., Xi Z., Ma L., Sirinakis G., Rothman J.E., and Zhang Y.. 2012. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 337:1340–1343. 10.1126/science.1224492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasper R., Meyer S., Gotthardt K., Sirajuddin M., and Wittinghofer A.. 2009. It takes two to tango: regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 10:423–429. 10.1038/nrm2689 [DOI] [PubMed] [Google Scholar]
- Ghisaidoobe A.B., and Chung S.J.. 2014. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: a focus on Förster resonance energy transfer techniques. Int. J. Mol. Sci. 15:22518–22538. 10.3390/ijms151222518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A., Praefcke G.J., Renault L., Wittinghofer A., and Herrmann C.. 2006. How guanylate-binding proteins achieve assembly-stimulated processive cleavage of GTP to GMP. Nature. 440:101–104. 10.1038/nature04510 [DOI] [PubMed] [Google Scholar]
- Hoppins S., and Nunnari J.. 2009. The molecular mechanism of mitochondrial fusion. Biochim. Biophys. Acta. 1793:20–26. 10.1016/j.bbamcr.2008.07.005 [DOI] [PubMed] [Google Scholar]
- Hu J., and Rapoport T.A.. 2016. Fusion of the endoplasmic reticulum by membrane-bound GTPases. Semin. Cell Dev. Biol. 60:105–111. 10.1016/j.semcdb.2016.06.001 [DOI] [PubMed] [Google Scholar]
- Hu J., Shibata Y., Zhu P.P., Voss C., Rismanchi N., Prinz W.A., Rapoport T.A., and Blackstone C.. 2009. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 138:549–561. 10.1016/j.cell.2009.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H., Kim H., and Myong S.. 2011. Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc. Natl. Acad. Sci. USA. 108:7414–7418. 10.1073/pnas.1017672108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R., and Scheller R.H.. 2006. SNAREs--engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7:631–643. 10.1038/nrm2002 [DOI] [PubMed] [Google Scholar]
- Kozlov M.M., McMahon H.T., and Chernomordik L.V.. 2010. Protein-driven membrane stresses in fusion and fission. Trends Biochem. Sci. 35:699–706. 10.1016/j.tibs.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubena B.D., Luecke H., Rosenberg H., and Quiocho F.A.. 1986. Crystallization and x-ray diffraction studies of a phosphate-binding protein involved in active transport in Escherichia coli. J. Biol. Chem. 261:7995–7996. [PubMed] [Google Scholar]
- Leipe D.D., Wolf Y.I., Koonin E.V., and Aravind L.. 2002. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 317:41–72. 10.1006/jmbi.2001.5378 [DOI] [PubMed] [Google Scholar]
- Liu T.Y., Bian X., Sun S., Hu X., Klemm R.W., Prinz W.A., Rapoport T.A., and Hu J.. 2012. Lipid interaction of the C terminus and association of the transmembrane segments facilitate atlastin-mediated homotypic endoplasmic reticulum fusion. Proc. Natl. Acad. Sci. USA. 109:E2146–E2154. 10.1073/pnas.1208385109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.Y., Bian X., Romano F.B., Shemesh T., Rapoport T.A., and Hu J.. 2015. Cis and trans interactions between atlastin molecules during membrane fusion. Proc. Natl. Acad. Sci. USA. 112:E1851–E1860. 10.1073/pnas.1504368112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Montana V., Bai J., Chapman E.R., Mohideen U., and Parpura V.. 2006. Single molecule mechanical probing of the SNARE protein interactions. Biophys. J. 91:744–758. 10.1529/biophysj.105.073312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew J.A., Sondermann H., Lee T., Stern M., and Brandizzi F.. 2013. GTP-dependent membrane fusion. Annu. Rev. Cell Dev. Biol. 29:529–550. 10.1146/annurev-cellbio-101512-122328 [DOI] [PubMed] [Google Scholar]
- Moore K.J., Webb M.R., and Eccleston J.F.. 1993. Mechanism of GTP hydrolysis by p21N-ras catalyzed by GAP: studies with a fluorescent GTP analogue. Biochemistry. 32:7451–7459. 10.1021/bi00080a016 [DOI] [PubMed] [Google Scholar]
- Morin-Leisk J., Saini S.G., Meng X., Makhov A.M., Zhang P., and Lee T.H.. 2011. An intramolecular salt bridge drives the soluble domain of GTP-bound atlastin into the postfusion conformation. J. Cell Biol. 195:605–615. 10.1083/jcb.201105006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss T.J., Andreazza C., Verma A., Daga A., and McNew J.A.. 2011. Membrane fusion by the GTPase atlastin requires a conserved C-terminal cytoplasmic tail and dimerization through the middle domain. Proc. Natl. Acad. Sci. USA. 108:11133–11138. 10.1073/pnas.1105056108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell J.P., Cooley R.B., Kelly C.M., Miller K., Andersen O.S., Rusinova R., and Sondermann H.. 2017. Timing and Reset Mechanism of GTP Hydrolysis-Driven Conformational Changes of Atlastin. Structure. 25:997–1010. 10.1016/j.str.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orso G., Pendin D., Liu S., Tosetto J., Moss T.J., Faust J.E., Micaroni M., Egorova A., Martinuzzi A., McNew J.A., and Daga A.. 2009. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 460:978–983. 10.1038/nature08280 [DOI] [PubMed] [Google Scholar]
- Otto H., Hanson P.I., and Jahn R.. 1997. Assembly and disassembly of a ternary complex of synaptobrevin, syntaxin, and SNAP-25 in the membrane of synaptic vesicles. Proc. Natl. Acad. Sci. USA. 94:6197–6201. 10.1073/pnas.94.12.6197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendin D., Tosetto J., Moss T.J., Andreazza C., Moro S., McNew J.A., and Daga A.. 2011. GTP-dependent packing of a three-helix bundle is required for atlastin-mediated fusion. Proc. Natl. Acad. Sci. USA. 108:16283–16288. 10.1073/pnas.1106421108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praefcke G.J., and McMahon H.T.. 2004. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 5:133–147. 10.1038/nrm1313 [DOI] [PubMed] [Google Scholar]
- Saini S.G., Liu C., Zhang P., and Lee T.H.. 2014. Membrane tethering by the atlastin GTPase depends on GTP hydrolysis but not on forming the cross-over configuration. Mol. Biol. Cell. 25:3942–3953. 10.1091/mbc.e14-08-1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwemmle M., and Staeheli P.. 1994. The interferon-induced 67-kDa guanylate-binding protein (hGBP1) is a GTPase that converts GTP to GMP. J. Biol. Chem. 269:11299–11305. [PubMed] [Google Scholar]
- Shan S.O. 2016. ATPase and GTPase Tangos Drive Intracellular Protein Transport. Trends Biochem. Sci. 41:1050–1060. 10.1016/j.tibs.2016.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan S.O., Chandrasekar S., and Walter P.. 2007. Conformational changes in the GTPase modules of the signal reception particle and its receptor drive initiation of protein translocation. J. Cell Biol. 178:611–620. 10.1083/jcb.200702018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel J.J., and Wiley D.C.. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. 10.1146/annurev.biochem.69.1.531 [DOI] [PubMed] [Google Scholar]
- Söllner T.H. 2004. Intracellular and viral membrane fusion: a uniting mechanism. Curr. Opin. Cell Biol. 16:429–435. 10.1016/j.ceb.2004.06.015 [DOI] [PubMed] [Google Scholar]
- Solscheid C., Kunzelmann S., Davis C.T., Hunter J.L., Nofer A., and Webb M.R.. 2015. Development of a Reagentless Biosensor for Inorganic Phosphate, Applicable over a Wide Concentration Range. Biochemistry. 54:5054–5062. 10.1021/acs.biochem.5b00449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof T.C., and Rothman J.E.. 2009. Membrane fusion: grappling with SNARE and SM proteins. Science. 323:474–477. 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton R.B., Fasshauer D., Jahn R., and Brunger A.T.. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 395:347–353. 10.1038/26412 [DOI] [PubMed] [Google Scholar]
- Tamm L.K., Crane J., and Kiessling V.. 2003. Membrane fusion: a structural perspective on the interplay of lipids and proteins. Curr. Opin. Struct. Biol. 13:453–466. 10.1016/S0959-440X(03)00107-6 [DOI] [PubMed] [Google Scholar]
- Weber T., Zemelman B.V., McNew J.A., Westermann B., Gmachl M., Parlati F., Söllner T.H., and Rothman J.E.. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92:759–772. 10.1016/S0092-8674(00)81404-X [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Dessen A., Calder L.J., Harrison S.C., Skehel J.J., and Wiley D.C.. 1999. Structural basis for membrane fusion by enveloped viruses. Mol. Membr. Biol. 16:3–9. 10.1080/096876899294706 [DOI] [PubMed] [Google Scholar]
- Winsor J., Hackney D.D., and Lee T.H.. 2017. The crossover conformational shift of the GTPase atlastin provides the energy driving ER fusion. J. Cell Biol. 216:1321–1335. 10.1083/jcb.201609071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Hu X., Bian X., Liu X., and Hu J.. 2015. Comparison of human and Drosophila atlastin GTPases. Protein Cell. 6:139–146. 10.1007/s13238-014-0118-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L., Sun S., Wang W., Shi J., Hu X., Wang S., Su D., Rao Z., Hu J., and Lou Z.. 2015. Structures of the yeast dynamin-like GTPase Sey1p provide insight into homotypic ER fusion. J. Cell Biol. 210:961–972. 10.1083/jcb.201502078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yersin A., Hirling H., Steiner P., Magnin S., Regazzi R., Hüni B., Huguenot P., De los Rios P., Dietler G., Catsicas S., and Kasas S.. 2003. Interactions between synaptic vesicle fusion proteins explored by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 100:8736–8741. 10.1073/pnas.1533137100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Wu F., Shi J., Zhu Y., Zhu Z., Gong Q., and Hu J.. 2013. ROOT HAIR DEFECTIVE3 family of dynamin-like GTPases mediates homotypic endoplasmic reticulum fusion and is essential for Arabidopsis development. Plant Physiol. 163:713–720. 10.1104/pp.113.224501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., and Brunger A.T.. 2016. Recent Advances in Deciphering the Structure and Molecular Mechanism of the AAA+ ATPase N-Ethylmaleimide-Sensitive Factor (NSF). J. Mol. Biol. 428(9, 9 Pt B):1912–1926. 10.1016/j.jmb.2015.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Alvarado D., Rainier S., Lemons R., Hedera P., Weber C.H., Tukel T., Apak M., Heiman-Patterson T., Ming L., et al. 2001. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 29:326–331. 10.1038/ng758 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.