

Abstract
C-C motif chemokine receptor 2 (CCR2) is a major chemokine axis that recruits myeloid cells including monocytes and macrophages. Thus far, CCR2−/− mice have not been found to be susceptible to infection with Mycobacterium tuberculosis (Mtb). Here, using a prototype W-Beijing family lineage 2 Mtb strain, HN878, we show that CCR2−/− mice exhibit increased susceptibility to tuberculosis (TB). Following exposure to Mtb HN878, alveolar macrophages (AMs) are amongst the earliest cells infected. We show that AMs accumulate early in the airways following infection and express CCR2. During disease progression, CCR2-expressing AMs exit the airways and localize within the TB granulomas. RNA-sequencing of sorted airway and non-airway AMs from infected mice show distinct gene expression profiles, suggesting that upon exit from airways and localization within granulomas, AMs become classically activated Absence of CCR2+ cells specifically at the time of AM egress from the airways resulted in enhanced susceptibility to Mtb infection. Furthermore, infection with an Mtb HN878 mutant lacking phenolic glycolipid (PGL) expression still resulted in increased susceptibility in CCR2−/− mice. Together, these data show a novel role for CCR2 in protective immunity against clinically relevant Mtb infections.
Keywords: Alveolar macrophages, C-C motif chemokine receptor 2, tuberculosis, chemokines, lung, innate immunity
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of pulmonary tuberculosis (TB),infects approximately one-third of the world’s population and results in 1.4 million deaths annually (W.H.O., 2016). M. bovis bacillus Calmette-Guérin is the only licensed vaccine against TB, however it is not very effective at protecting against adult pulmonary TB. A major hurdle in design of new and effective vaccines against TB is our poor understanding of the early immune mechanisms mediating protective immunity against Mtb infection.
A major chemokine axis that recruits innate immune cells to the lungs is the C-C motif chemokine receptor 2 (CCR2). CCR2−/− mice aerosol-infected with Euro-American lineage 4 Mtb strains showed defective accumulation of myeloid dendritic cell (mDC) and macrophage/monocyte populations in the Mtb-infected lung, with coincident delayed T cell responses1,2. Despite decreased innate cellular recruitment, CCR2−/− mice were surprisingly not more susceptible to Mtb infection1, propagating the idea that the CCR2 axis is dispensable for protective immunity to infection with Mtb3.
A major ligand for CCR2 is C-C motif chemokine ligand 2 (CCL2), along with the ligands CCL7 and CCL12. In human populations, meta-analysis of the identified −2518 A/G single nucleotide polymorphism (SNP) in the promoter region of the CCL2 gene show significantly elevated risk for pulmonary TB4. Recent studies have highlighted differences in cytokine induction and immune requirements for Mtb control to be dependent on the infecting Mtb strain5,6. Studies using the zebrafish granuloma model have described that M. marinum expressing virulence factors such as PGLs drive increased expression of CCL2, and mediate recruitment of CCR2+ permissive monocytes to promote pathogenesis7,8. Thus, murine studies suggest a dispensable role for CCR2, zebrafish and M. marinum infection models suggest a pathological role for CCR2, while human studies propose a critical but as yet undefined role for the ligand, CCL2.
Alveolar macrophages (AMs) are tissue resident phagocytes localized to the airway and are believed to be the first contact and primary reservoir for replication of Mtb following inhalation of the bacteria9,10. During acute Mtb infection, AMs can exacerbate spreading of bacteria and formation of necrotic granulomas10. However, despite the consensus that AMs are the first innate cells that interact with Mtb, not much is known about how AMs participate to mediate control of Mtb infection. In the current study, we show that AMs are amongst the earliest infected cells upon exposure to W-Beijing Mtb HN878, they accumulate in the airways and express CCR2. During disease progression, we demonstrate that AMs exit the airways and localize within the TB granulomas. Importantly, following Mtb HN878 infection, sorted non-airway AMs highly express genes belonging to classical macrophage activation, when compared to airway AMs that express a unique transcriptional signature. Depletion of CCR2-expressing cells, specifically at the timing of CCL2 induction and AM egress from the airways,resulted in increased susceptibility to Mtb infection, with the accumulation of neutrophils and loss of Mtb control. Additionally, we provide new evidence that mutant HN878 lacking PGL expression still resulted in increased susceptibility in CCR2−/− mice. Together, our data provide novel evidence for a protective role for CCR2 in mediating alveolar macrophage localization and immunity against emerging Mtb infections.
Results
CCR2 is required for AM accumulation and protective granuloma formation following infection with emerging W-Beijing Mtb
Published studies thus far have shown a redundant role for CCR2 in Mtb infection, specifically using Euro-American lineage strains such as H37Rv and Erdman1,2. These studies have documented either negligible1 or small11 increases in Mtb burden in CCR2−/− mice, unless CCR2−/− mice were infected with high doses of Mtb administered intravenously12. Our data confirms these findings as CCR2−/− mice showed similar lung Mtb burden when compared with C57BL/6J (B6) mice following infection with Mtb H37Rv (H37Rv) (Fig. 1a). Additionally, CCR2−/−4 mice only showed a small increase in lung burden when infected with another Euro-American clinical Mtb strain, CDC1551 (Fig. 1b). Infection with neither H37Rv nor CDC1551 resulted in increased dissemination to the spleen in CCR2−/− infected mice (Fig. 1a,b). In addition, infection with an Indo-Oceanic lineage clinical Mtb strain, T17x13, also resulted in a small increase in lung and spleen Mtb burden in CCR2−/− mice, when compared to B6 infected mice (Fig. 1c). In sharp contrast, when infected with lineage 2 Mtb HN878 (HN878), CCR2−/− mice showed significantly increased Mtb burden at early days post infection (dpi) which was also maintained during chronic infection (Fig. 1d). Indeed, CCR2−/− HN878-infected mice also exhibited increased dissemination to the spleen, when compared to HN878-infected B6 mice (Fig. 1d). Severe susceptibility with increased lung and spleen Mtb burden was also observed when CCR2−/− mice were infected with a pyrazinamide-resistant lineage 2 Mtb clinical strain, HN563 (Fig. 1e). These results together show a previously undocumented susceptibility of CCR2−/− mice to Mtb infection, projecting a protective role for CCR2 in virulent, emerging W-Beijing Mtb infections.
Figure 1: CCR2−/− mice show increased susceptibility to low dose aerosol HN878 infection.
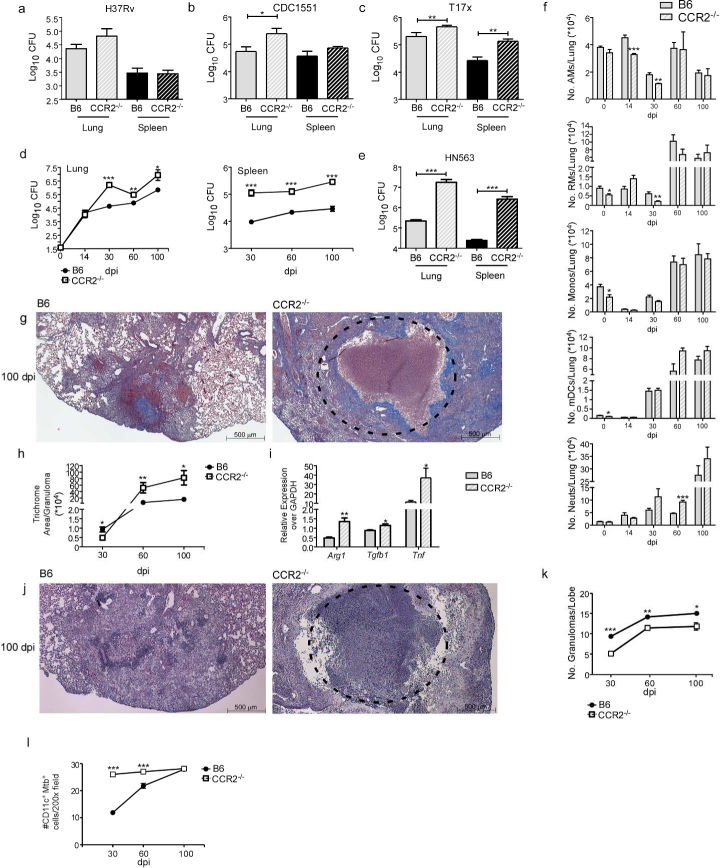
B6 and CCR2−/− mice were aerosol-infected with ~100 CFU of (a) H37Rv, (b) CDC1551, (c) T17x, (d) HN878, or (e) HN563. Bacterial burden in the lung and spleen was determined by plating (a-c and e) at 30 dpi or (d) at different dpi. (f) Lung myeloid cell populations were enumerated in B6 and CCR2 −/− HN878-infected mice using flow cytometry at indicated dpi. (g-k) Pulmonary histology was assessed on FFPE lung sections from 30, 60 and 100 dpi samples stained with (g) Trichrome staining or (j) H&E staining. (h) Inflammatory area expressing collagen was quantified using Visiomorph image processing software to determine lung fibrosis.(i) RNA was extracted from B6 and CCR2−/− HN878-infected lungs and relative mRNA expression of specific genes was determined by qRT-PCR. Gapdh was used as internal control.(k) Inflammation was quantified using the morphometric tool of the Zeiss Axioplan microscope to determine the total number of granulomas per lobe. (l) The total number of CD11c+ Mtb containing cells per 200X was determined by counting in FFPE lung sections of B6 and CCR2−/− mice. AMs=Alveolar Macrophages, RMs=Recruited Macrophages, Monos=Monocytes, mDCs=Myeloid Dendritic Cells, Neuts=Neutrophils. n=5, (a-e) Student’s t-test between B6 and CCR2−/−, (f) 2-way ANOVA with Bonferroni’s post test. (h-l) Student’s t-test was used to determine differences per time point.
To delineate the cellular mechanisms by which CCR2 mediates protective immunity against HN878 infection, we determined myeloid cell recruitment to the lungs of infected B6 and CCR2−/− Mtb-infected mice (Supplementary Fig. 1 - gating strategy by flow cytometry). Following H37Rv infection, we observed a trend towards decreased accumulation of monocytes, AMs, recruited macrophages (RMs), mDCs, and neutrophils in the lungs of CCR2−/− H37Rv-infected mice when compared to B6 H37Rv-infected mice (Supplementary Fig. 2a-e) confirming previous findings1,12,14. CCR2−/− mice also exhibited lower numbers of RMs, mDCs, and monocytes in the uninfected lungs (Fig. 1f, day 0). Upon infection with HN878, CCR2−/− mice showed significantly decreased early AM and RM accumulation and maintained a small reduction in monocyte accumulation, when compared with B6 HN878-infected lungs (Fig. 1f). Although mDC numbers were lower in uninfected lungs of CCR2−/− mice, there were no significant changes in mDC numbers in CCR2−/− HN878-infected mice when compared to B6 HN878-infected mice (Fig. 1f). Also, the accumulation of activated lung IFN-γ-producing CD4+ T cells was comparable between 5 B6 and CCR2−/− mice infected with either H37Rv or HN878 (Supplementary Fig. 2f-h). These results demonstrate that early macrophage accumulation and protective immunity are dependent on CCR2 expression during HN878 infection, but that this requirement is dispensable for H37Rv infection.
The decreased early accumulation of macrophage populations in CCR2−/− HN878-infected mice coincided with significantly increased early accumulation of neutrophils, which was maintained during chronic infection (Fig. 1f). Incidentally, neutrophil accumulation is associated with failed immunity to Mtb infection and neutrophils are the predominant infected myeloid cell type15–18. In support of this, we observed increased fibrosis and formation of necrotic granulomas in lungs of HN878-infected CCR2−/− mice, when compared with lungs of B6 HN878-infected mice (Fig. 1g,h). Expression of mRNA for tissue remodeling markers such as Arginase-1 (Arg1), transforming growth factor (Tgfb1) and tumor necrosis factor alpha (Tnfa) were also increased in CCR2−/− HN878-infected lungs (Fig. 1i). Additionally, fewer granulomas were found in CCR2−/− Mtb HN878-infected lungs, when compared with well-formed granulomas present in B6 HN878-infected lungs (Fig. 1j,k). Finally, although there was decreased accumulation of AMs in CCR2−/− Mtb-infected lungs (Fig. 1f), more CD11c+ macrophages were infected with Mtb within granulomas of CCR2−/− mice, when compared to the AMs in granulomas from B6 mice (Fig. 1l). Together, these data imply that the CCR2 axis is required for formation of protective granulomas during TB. In the absence of CCR2, there are fewer macrophages and fewer protective granulomas formed, instead resulting in an influx of neutrophils and development of necrotic granulomas that do not effectively control Mtb thus leading to increased susceptibility.
AMs are preferentially infected with Mtb HN878 and require lung epithelial signaling for accumulation upon infection
To delineate the unique requirement for CCR2 expression during HN878 infection, we used H37Rv-GFP and HN878–GFP Mtb reporter strains and addressed if they similarly infect myeloid cell populations, and if Mtb infection modulated CCR2 expression on myeloid cells. Following in vivo infection with Mtb-GFP reporter strains, AMs more significantly uptake HN878 than H37Rv, suggesting a preferential localization of Mtb HN878 within AMs (Fig. 2a,b). Several lung myeloid subsets expressed CCR2 in H37Rv- and HN878-infected mice, including AMs and RMs, monocytes, neutrophils, and mDCs (Fig. 2c-e). Overall, significantly higher numbers of AMs, monocytes, and neutrophils expressing CCR2 were found in the lungs of HN878-infected mice, when compared to H37Rv-infected mice and uninfected mice (Fig. 2c-e). Following H37Rv infection, the RM population predominantly expressed CCR2 when compared to expression levels in uninfected mice (Fig. 2d). In contrast, during HN878 infection, the predominant myeloid cell type expressing CCR2 was the AM population (Fig. 2d). Further more,we observed significantly increased CCR2 expression on AMs on a per cell basis during HN878 infection, when compared with AMs during H37Rv infection (Fig. 2f). Thus, our data suggest that AMs preferentially uptake HN878 and specifically upregulate the expression of CCR2.
Figure 2: AMs are preferentially infected with HN878 and CCR2 expression on AMs is Mtb strain dependent.

(a) The flow cytometry gating strategy for myeloid populations. Briefly, AMs were defined as CD11b−CD11c+Siglec F+ cells. mDCs were defined as CD11b+CD11c+ cells. Neutrophils were defined as CD11b+CD11c−Gr-1hi cells, monocytes were defined as CD11b+CD11c−Gr-1lo cells, and recruited macrophages were defined as CD11b+CD11c−Gr-1− cells. Mtb-GFP+ cells were gated from individual subsets. (b) B6 mice were aerosol-infected with ~100 CFU of H37Rv-GFP or HN878-GFP and myeloid cell subsets infected with Mtb-GFP were determined by flow cytometry on 30 dpi (n=5). (c,d) B6 mice (n=5) were infected with aerosolized H37Rv or HN878 and the total number (c) and percentage (d) of CCR2+ myeloid lung cell populations were determined by flow cytometry and compared to uninfected controls. (e) Representative histograms of each subset displaying CCR2 expression by antibody staining compared to CCR2−/−. (f) Mean fluorescence intensity (MFI) of CCR2 expression on myeloid populations was normalized relative to MFI of unstained, uninfected controls. Un.=uninfected, AMs=Alveolar Macrophages, Monos=Monocytes, RMs=Recruited Macrophages, Neuts=Neutrophils, mDCs=Myeloid Dendritic Cells. (a-d) 2-Way ANOVA with Bonferroni posttest was used. (f) Student’s t-test.
To further elucidate the Mtb strain specific requirement for the CCR2 axis, we next determined the expression of CCR2 ligands in H37Rv- and HN878-infected lungs. Early expression of mRNA for Ccl2, Ccl7 and Ccl12 was significantly higher in HN878-infected lungs, when compared to levels in H37Rv-infected lungs (Fig. 3a). Additionally, CCL2−/− mice infected with Mtb HN878 exhibited increased Mtb bacterial burden (Supplementary Fig. 3a). However, since CCL2−/− mice did not fully reflect the heightened susceptibility of the CCR2−/− mice to HN878 infection, other ligands such as CCL7 and CCL12 may mediate CCR2 driven protection during Mtb HN878 infection. Given the relevance of CCL2 in human disease4,19, we chose to focus on CCL2 production in subsequent experiments. To determine the main cellular sources of CCL2 upon infection, we assessed CCL2 levels in supernatants of lung epithelial cells, DCs,and macrophages after in vitro infection with either H37Rv or HN878. We observed that CCL2 production was significantly higher upon infection with HN878 in epithelial cells and DCs (Fig. 3b). In contrast, in macrophages as previously shown5, HN878 infection resulted in decreased CCL2 production when compared to H37Rv infection (Fig. 3b).
Figure 3: Increased early CCR2 ligand expression is induced in the lung following HN878 infection.

(a) RT-PCR analysis was performed at 21 and 30 dpi to determine mRNA expression for Ccl2,Ccl7, and Ccl12 in lungs of H37Rv- or HN878-infected mice (n=5 per group, per time point). (b)C10 epithelia, BMDCs, and BMDMs were cultured and infected with indicated Mtb strains at an MOI of 1 for 48 hours (n=6). Supernatants were analyzed by multiplex or ELISA assay forCCL2. (c-e) IKK2 fl/fl Sftpc-Cre mice and littermate controls were infected with HN878 for 14 (n=7) and 30 (n=5) dpi and the accumulation of (c) AMs, (d) neutrophils, and (e) RMs were calculated by flow cytometry. (f) Bacterial burden in the lung was determined by plating at 14 (n=7 per group) and 30 dpi (n=5 per group) in IKK2 fl/fl Sftpc-Cre mice and littermate controls. (g) Confocal microscopy of lung sections stained for E-cadherin (red) and CCL2 (green) in IKK2 fl/fl Sftpc-Cre mice and littermate controls. AMs=Alveolar Macrophages, Neuts=Neutrophils, RMs=Recruited Macrophages. (a) 2-way ANOVA with Bonferroni posttest (b) 1-way ANOVA with Tukey’s posttest. (c-f) Student’s t-test was used to compare between groups per time point.
Epithelial cells induced CCL2 in response to infection with HN878, thus we hypothesized that epithelial cell signaling may be involved in chemokine induction and coordinate the localization or myeloid cells, including AMs to form granulomas. Thus, we utilized the IKK2 fl/fl Sftpc-Cre mice 20, which lack canonical IKK and NF-κB signaling in Sftpc-expressing cells, mainly lung epithelial cells21. Upon infection of IKK2 fl/fl Sftpc-Cre mice with Mtb HN878, we observed decreased AM (Fig. 3c) and neutrophil (Fig. 3d) accumulation in HN878-infected lungs, without any changes in RM accumulation (Fig. 3e) when compared to myeloid cell accumulation in infected littermate controls. Associated with decreased AM accumulation, IKK2 fl/fl Sftpc-Cre HN878-infected mice were more susceptible and exhibited increased lung Mtb CFU at 30 dpi (Fig. 3f). We found that total protein levels of C-C-chemokines such as CCL2 and CCL3, and CXC-chemokines such as CXCL2 were not different in lung homogenates of IKK2 fl/fl Sftpc-Cre mice, compared to littermate controls (Supplementary Fig. 3b). However, we specifically found that lungs of IKK2 fl/fl Sftpc-Cre mice exhibited fewer E-cadherin expressing epithelial cells producing CCL2 protein, when compared to littermate controls (Fig. 3g). The protein levels of CXCL1, a neutrophil attracting chemokine, were reduced in total lung homogenates of IKK2 fl/fl Sftpc-Cre mice (Supplementary Fig. 3b), likely resulting in the decreased neutrophil recruitment observed in infected mice (Fig. 3d). These findings suggest that early epithelial signaling has a role in coordinating the accumulation of myeloid cells ,specifically AMs and neutrophils, to the lungs during infection.
CCR2 expression mediates AMs to egress from airways and localize within TB granulomas
CCL2 is produced by HN878-infected epithelial cells and DCs, and thus we next examined if the CCR2 axis was involved in AM movement out of the airways. To address this,we specifically labeled myeloid cells within the airways by delivering fluorophore-conjugated CD45.2 antibody intratracheally (IT) to Mtb HN878-infected mice prior to harvest (Supplementary Fig. 4a-d). This technique allowed us to distinguish between Broncho-Alveolar Lavage (BAL) stain positive airway CCR2+ AMs (CD45.2+CCR2+CD11c+SiglecF+) and BAL stain negative non-airway CCR2+ AMs that are not exposed to the CD45.2 antibody (CD45.2−CCR2+CD11c+SiglecF+) (Fig. 4a). To validate the technique, we demonstrated that delivery of CD45.2 antibody into the airways does not leak into the interstitium in naïve mice (Supplementary Figure 4d). However, when lung injury is induced in mice by intratracheal instillation of HCl, we observed increased dispersion of CD45 antibody into the interstitium (Supplementary Figure 4d). Additionally, mice undergoing lung injury due to treatment with HCl showed increased frequency of total lung cells and myeloid cells stained with CD45.2 antibody when compared with CD45.2+ lung cells in naïve PBS treated mice (Supplementary Figure 4a) Furthermore, when BAL was collected immediately after delivery of CD45.2 antibody I.T., we found that the airway localized AMs were ~99% CD45.2+ in both uninfected and Mtb-infectedmice (Supplementary Figure 4e, left panel), though AMs were a smaller proportion of the total airway cells during infection (Supplementary Figure 4e, right panel).
Figure 4: CCR2 expression is required for AMs to egress from airways and localize within TB granulomas.
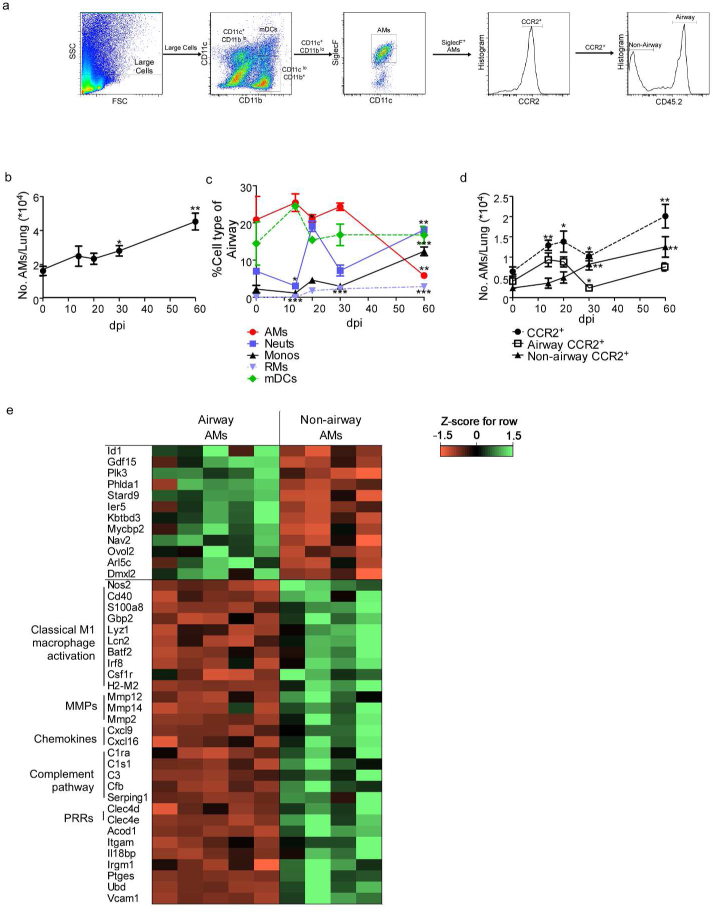
Single cell lung suspensions from uninfected and infected mice (n=5) were prepared and (a) the gating strategy for airway and non-airway AMs is shown. The percentage and number of specific cell subsets with airway label CD45.2 delivered IT is shown.
CD11c+CD11bloSiglecF+CD45.2+ cells were gated as airway AMs, while CD11c+CD11blo SiglecF+ CD45.2− cells were gated as non-airway AMs. (b) The total number of AMs over the time course of HN878 in B6 mice was determined by flow cytometry (n=5 per time point). (c) From total airway labelled cells (CD45.2+), the percentage of each myeloid cell type was determined in HN878-infected B6 mice by flow cytometry. (d) Total CCR2+ AMs, CCR2+ airway (CD45.2+) AMs, and CCR2+ non-airway (CD45.2−) AMs over the course of HN878 infection in B6 mice were determined by flow cytometry. (e) Z-score Pearson correlation-based clustering of differentially expressed genes of interest (all 12 significantly differentially expressed genes in airway AMs, and 29 genes of functional interest that were higher in non-airway AMs). AMs=Alveolar Macrophages, Neuts=Neutrophils, Monos=Monocytes, RMs=Recruited Macrophages, mDCs=Myeloid Dendritic Cells, MMPs=matrix metallopeptidases, PRRs=pattern recognition receptors. (b-d) Each time point was compared to baseline d0 counts using Student’s t-test. (e) The gene expression levels of a subset of significantly differentially expressed genes between airway and non-airway AMs (according to DESeq).
During Mtb infection, we observed an overall increase in AM accumulation in the lung (Fig. 4b). In addition, we found that early during infection, of the airway labeled myeloid cells,AMs were the most represented cell type, followed by DCs (Fig. 4c). However, as infection progressed, neutrophils along with monocytes were increasingly represented in the airway, while AMs were less represented (Fig. 4c). We observed increased CCR2+ AM accumulation early following infection, especially within the airways (Fig. 4d). As infection progressed, we observed decreased CD45.2+CCR2+ AMs localizing within the airways and increased non-airway CD45.2−CCR2+ AMs, suggesting either an egress of bonafide AMs from the airway, or the presence of recruited, monocyte-derived, alveolar macrophage-like cells within the tissue (Fig. 4d). The timing of these changes in localization coincided with the timing of increased CCR2 ligand expression in the lung at 21 dpi (Fig. 3a).
To determine the identity and functional relevance of the AM populations, we sorted for highly purified airway AMs (~99% purity, Supplementary. Fig. 5a) (CD45.2+CD11c+SiglecF+)from uninfected and infected mice, and non-airway AMs from infected mice (~95% purity, Supplementary Fig. 5a) (CD45.2−CD11c+SiglecF+) and carried out RNA sequencing. We confirmed that both populations were in fact bonafide AMs by examining the common AM gene signature22,23 between groups (i.e. Siglecf, Pparg, Tgfbr2, Csf2r, Mertk, Itgax, Lyz2, and Fcgr1). We also confirmed that the AM populations did not highly express genes associated with monocyte-derived interstitial or recruited macrophages24 (Ly6c1, Itgam, and CD163)(Supplementary Table 1, 2a-b). These data support our hypothesis that the both airway and non-airway AMs are not interstitial monocyte-derived macrophages24 but in fact bonafide AM populations.
According to the RNA-Seq analysis, non-airway AMs expressed significantly higher mRNA levels (according to DESeq225) for genes belonging to classical macrophage activation including inducible nitric oxide synthase (Nos2), CD40 antigen (Cd40), S100 calcium binding protein A8 (S100a8), Guanylate binding protein 2 (Gbp2), Lysozyme 1 (Lyz1) and Lipocalin (Lcn2), when compared with sorted airway AMs (Fig. 4e and Supplementary Table 3). Additionally, non-airway AMs also expressed significantly higher mRNA for genes such as Matrix metallopeptidases (Mmp2, Mmp14) and proinflammatory chemokines such as Cxcl9, Cxcl16. In addition, mRNA belonging to classical macrophage transcriptional signatures such as Basic leucine zipper transcription factor, ATF-like 2 (Batf2)26 and Interferon regulatory factor 8 (Irf8)27, were significantly higher in sorted non-airway AMs than airway AMs. Transcriptional profiles of non-airway AMs when compared with airway AMs showed significantly higher expression of pathways associated with infections, complement cascade, and the phagosome activation (Supplementary Table 4).
Alternatively, only 12 genes were significantly expressed at higher levels in airway AMs during infection, when compared with non-airway AMs isolated from Mtb infected mice (Fig. 4e and Supplementary Table 5), including inhibitor of DNA binding 1 (Id1; expressed by tissue-resident macrophages28) and growth differentiation factor-15 (Gdf15), which has been reported to drive expression of CCR2 in macrophages29. Further, significantly higher gene expression in airway AMs was observed for Toll like receptor (TLR)-induced inflammatory responses such as Polo-like kinase 3 (Plk3)30 and Pleckstrin homology like domain, family A, member 1 (Phlda1)31 and the cholesterol–trafficking START domain containing 9 (Stard9) genes. Genes significantly higher in airway AMs from infected mice compared to uninfected mice demonstrated significant enrichment for pathways associated with metabolic pathways, antigen processing and phagocytosis (Supplementary Table 6). In addition, genes associated with several key signaling pathways associated with cytoskeletal rearrangement and diapedesis were downregulated during infection in airway AMs (Supplementary Table 7). Together, our results indicate that airways AMs during infection upregulate a unique transcriptional signature associated with phagocytosis and antigen- presentation, when compared to non-airway AMs, who are more classically activated for intracellular killing and T cell activation.
To mechanistically examine the ability of CCR2+ AMs to egress from the airways and localize within the TB granulomas during HN878 infection, we used an adoptive transfer model with CCR2-GFP-expressing AMs. We purified CD11c+ lung cells (Supplementary Fig. 5b) from HN878-infected CCR2-GFP(+/KI) mice32 or CCR2-GFP(KI/KI) mice. CCR2-GFP(+/KI) mice have one functional allele for CCR2, but also express GFP, while CCR2-GFP(KI/KI) mice have both nonfunctional, GFP+ alleles. We adoptively transferred the CD11c+ cells into the airways of HN878-infected B6 mice, and using SiglecF to further identify AMs, CD11c+ SiglecF+ CCR2-GFP+ AMs were tracked within TB granulomas (Fig. 5a). We observed that CD11c+ SiglecF+ CCR2-GFP(+/KI) AMs adoptively transferred into the airways localized within the TB granulomas (Fig. 5b). In contrast, adoptive transfer of CCR2-GFP(KI/KI) AMs into the airways resulted in notably reduced accumulation within the TB granulomas (Fig. 5c). Furthermore, we observed increased migration of Mtb-stimulated CCR2-GFP(+/KI) macrophages in response to HN878-infected but not H37Rv-infected epithelial cell supernatant. However, Mtb-stimulated CCR2-GFP(KI/KI) macrophages did not migrate in response to Mtb-infected epithelial cell supernatants, suggesting that the macrophage migration in response to infection is CCR2-dependent (Fig. 5d). Furthermore, localization of Ccl2 mRNA is within TB granulomas in HN878-infected B6 lungs, while Ccl2 mRNA expression was localized outside of cavitary TB granulomas in CCR2−/− HN878-infected lungs (Fig. 5e). Additionally, a higher percentage of AMs were found in the airways of CCR2−/− HN878-infected lungs when compared to B6 HN878-infected mice (Fig. 5f). These data suggest that CCR2 plays a role in AM localization within the TB granuloma, and that without a functional CCR2, AMs less efficiently migrate from the airways to localize within the TB granulomas.
Figure 5: CCR2 is required for AM localization within TB granulomas.
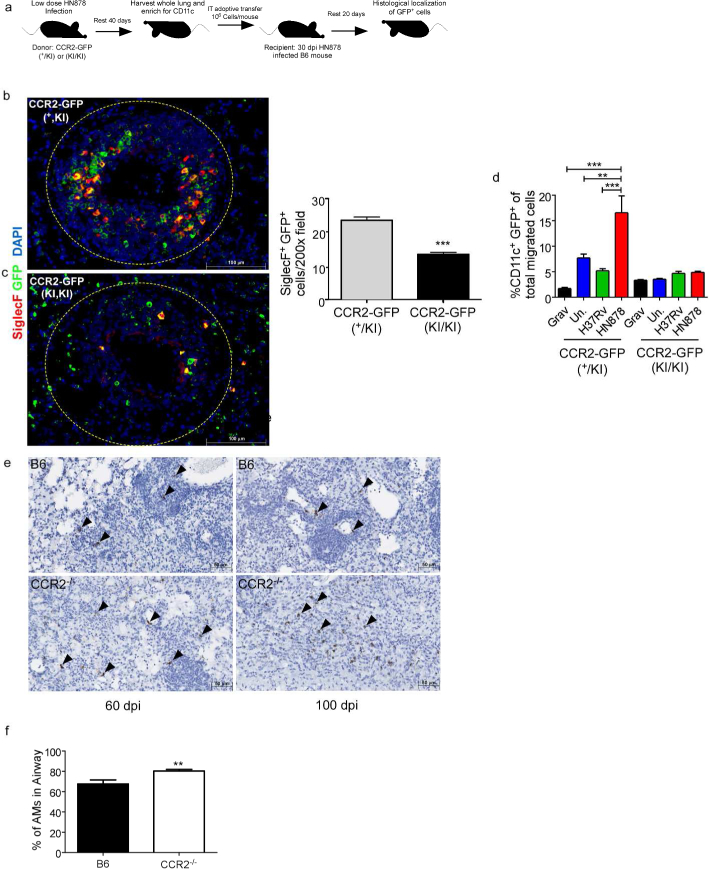
(a) CD11c+ cells were purified from lungs of 30 dpi HN878-infected CCR2-GFP(+/KI) or CCR2-GFP(KI/KI) mice and 106 cells were IT transferred into B6 HN878-infected mice (n=5) at 30 dpi.(a-c) Lungs were harvested at 50 dpi and examined for localization of SiglecF+ GFP+ cells within TB granulomas using the morphometric tool of the Zeiss Axioplan microscope. (d) CCR2-GFP(+/KI) or CCR2-GFP(KI/KI) BMDMs were stimulated in vitro with 20 μg/mL irradiated Mtb HN878 for 24 hours. Migration towards uninfected, H37Rv- or HN878-infected epithelial cell supernatants was analyzed via transwell chemotaxis assays and flow cytometry (n=3). (e) Ccl2 mRNA localization was determined within FFPE lung sections from B6 and CCR2−/− HN878-infected using RNAScope in situ hybridization (ISH). Arrows point to Ccl2 mRNA localization(brown). (f) B6 and CCR2−/− mice (n=5) were infected with HN878 and percentage of AMs with airway label CD45.2 delivered IT was calculated on 14 dpi by flow cytometry. Grav=Gravity control, Un.=uninfected, AMs=Alveolar Macrophages. n=5 (b,c) Student’s t-test. n=3, (d) 2-Way ANOVA with Bonferroni posttest. (f) Student’s t-test.
CCR2 expression at the time of airway AM egress is critical for control of Mtb HN878 infection
CCR2-DTR mice have diphtheria toxin receptor (DTR) inserted between the first and second codon of CCR233, and treatment of CCR2-DTR mice with diphtheria toxin (Dtx) resulted in depletion of all CCR2-expressing cells. Previously, depletion of CCR2-expressing cells following Mtb Erdman infection reduced monocyte and monocyte-derived populations, but did not result in increased Mtb susceptibility2. To address the timing of the CCR2 requirement for protection following HN878 infection, we similarly used CCR2-DTR mice2. CCR2-DTR mice were infected with HN878, and CCR2-expressing cells were depleted either early at the time of infection (−1 to 5 dpi) or later at the time of AM egress from the airways (12–18 dpi) (Fig. 4d). Transient depletion of all CCR2-expressing cells around 12–18 dpi but not at the time of infection resulted in a significant increase in Mtb burden (Fig. 6a-d). Depletion of CCR2-expressing cells at 12–18 dpi also resulted in increased accumulation of neutrophils, monocytes and RMs in the lung and sharply decreased number of AMs, when compared to PBS treated Mtb-infected mice (Fig. 6b). Additionally, CCR2-DTR that received Dtx between 12–18 dpi also showed increased inflammation, when compared to control CCR2-DTR mice that received PBS (Fig. 6c). In contrast, CCR2-DTR mice that received Dtx around the time of infection did not show any differences in AM numbers, but showed decreased accumulation of neutrophils ,monocytes and RMs and coincident decreased inflammation (Fig. 6e,f). Together, these results demonstrate that transient depletion of CCR2-expressing cells coincident with the time of AM egress from the airways resulted in decreased AM accumulation and increased susceptibility to HN878 infection. In contrast, depletion of CCR2-expressing cells around the time of infection,while dampening RM and monocyte accumulation, did not impact AM accumulation and only minimally increased Mtb control.
Figure 6: Depletion of CCR2+ cells at the time of AM egress from airways increases susceptibility to HN878 infection.

CCR2-DTR mice (n=4) were infected with HN878 and administered Dtx (a-c) IP at 12, 14, and 16 dpi or (d-f) at −1, 1, and 3 dpi. (a, d) Lung bacterial burden was determined by plating on 30 dpi. (b, e) Neutrophil, AM, monocyte and RM numbers were determined in PBS treated and Dtx treated CCR2-DTR mice at 30 dpi. (c, f) Pulmonary histology was assessed on FFPE lung sections stained with H&E, and inflammatory area was quantified using the morphometric tool of the Zeiss Axioplan microscope. (g) B6 (n=5) and CCR2−/− mice (n=8 per group) were infected with HN878, and CCR2−/− mice received either PBS or HN878-stimulated BMDMs delivered IT on 15 and 21 dpi. Lungs were harvested at 30 dpi and bacterial burden was determined by plating. Neuts=Neutrophils, AMs=Alveolar Macrophages, Monos=Monocytes, RMs=Recruited Macrophages. (a-f) Student’s t-test. (g) 1-way ANOVA with Tukey’s posttest.
To further confirm the role of CCR2+ AMs in protection against HN878 Mtb infection, we delivered B6 macrophages stimulated in vitro with irradiated Mtb into the airways of CCR2−/−HN878-infected mice. Adoptive transfer of B6 macrophages rescued the increased susceptibility observed in CCR2−/− Mtb-infected mice, yielding decreased lung bacterial burden equivalent to B6 Mtb-infected mice (Fig. 6g). These data suggest that the susceptibility associated with CCR2−/− mice coincides with lack of macrophage accumulation in the lung, and that adoptive transfer of B6 macrophages into airways is sufficient to reverse susceptibility Mtb in CCR2−/−Mtb-infected mice.
Dependence on CCR2 for protective immunity to Mtb HN878 is not driven solely by PGL expression
Recently, M. marinum expressing PGL has been shown to be the involved in induction of CCL2 and recruitment of CCR2-expressing permissive macrophages in zebrafish mycobacterial infection model7,8. Thus, we next addressed if absence of PGL in Mtb HN878 would result in lack of a role for CCR2 in controlling Mtb HN878 infection in mice. Thus, B6 and CCR2−/− mice were infected with a recombinant Mtb HN878 mutant harboring a mutation within pks1-15(HN878 pks1-15::hygB) rendering Mtb PGL deficient 5. Upon infection with Mtb HN878 pks1–15::hygB, CCR2−/− mice were still more susceptible to infection when compared with B6 infected mice (Fig. 7a). This coincided with decreased accumulation of AMs, RMs and monocytes ,increased neutrophil accumulation, (Fig. 7b) and severe pulmonary disease (Fig. 7c) in CCR2−/−13infected mice when compared with B6 infected mice. Additionally, while in vitro infection of macrophages and DCs with HN878 pks1–15::hygB resulted in decreased CCL2 when compared to HN878 infection5 (Fig. 7d), comparable induction of CCL2 was observed upon infection of lung epithelial cells with HN878 and HN878 pks1–15::hygB (Fig. 7e). These data are also supported by localized expression of Ccl2 mRNA within TB granulomas in lungs of B6 and CCR2−/− mice infected with HN878 pks1–15:hygB (Fig. 7f). These data suggest that PGL expression is not the only key determinant for a protective role for CCR2 in Mtb HN878 infection, as Mtb H37Rv and HN878 also differ in expression of several other key components in their cell walls34.
Figure 7: Dependence on CCR2 for protective immunity to Mtb HN878 is not driven by PGL expression.
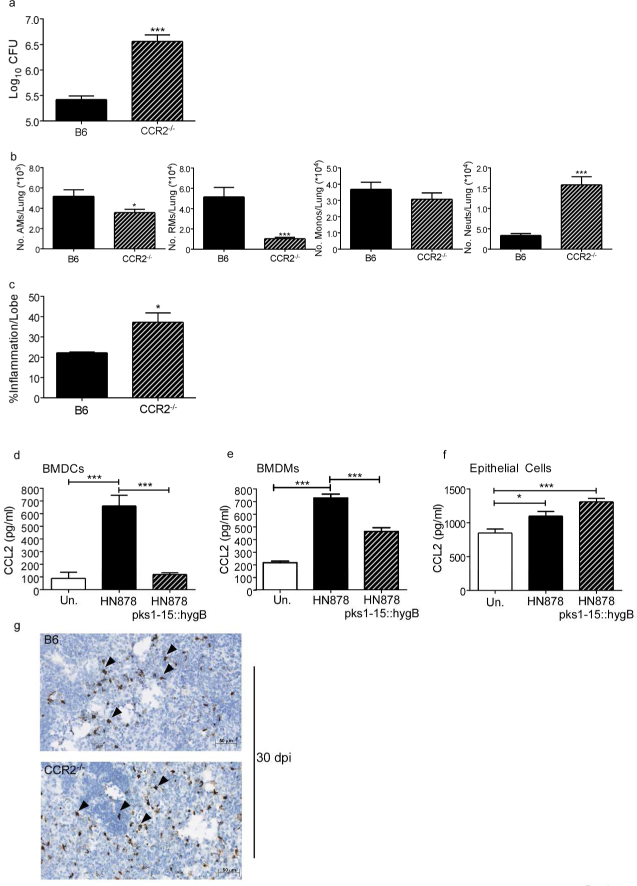
B6 and CCR2−/− mice (n=5) were aerosol-infected with ~100 CFU HN878 pks1–15::hygB. (a) Lung bacterial burden was determined by plating on 30 dpi. (b) Lung myeloid cell populations of AMs, RMs, monocytes and neutrophils were enumerated in B6 and CCR2 −/− HN878 pks1–15:hygB infected mice using flow cytometry. (c) Pulmonary histology was assessed on FFPE lung sections stained with H&E, and inflammatory area was quantified using the morphometric tool of the Zeiss Axioplan microscope. (d-f) BMDCs, BMDMs, and C10 epithelial cells were cultured and infected with indicated Mtb strains at an MOI of 1 for 48 hours (n=5). Supernatants 34 were analyzed by multiplex or ELISA assay for CCL2. (g) Ccl2 mRNA localization was determined within FFPE lung sections from B6 and CCR2−/− HN878-infected using RNA Scope in situ hybridization (ISH). Arrows point to Ccl2 mRNA localization (brown). AMs=Alveolar Macrophages, RMs=Recruited Macrophages, Monos=Monocytes, Neuts=Neutrophils. (a-c) Student’s t-test. (d-f) 1-way ANOVA with Tukey’s posttest.
Discussion
In humans, polymorphisms in CCL2 have been associated with pulmonary TB4, while mouse infection studies have shown that CCR2 expression is dispensable for protective immunity to infection with aerosolized Euro-American lineage 4 Mtb strains1. Zebrafish modeling of granulomas using M. marinum has proposed that the CCR2-CCL2 axis through interactions with virulence factors drives the generation of permissive macrophages and promotes mycobacterial replication and pathogenesis7. In the current study, using murine models, we provide novel evidence that the CCR2 axis is critical for protective immunity against infection with emerging Mtb lineages, such as the W-Beijing Mtb strains. Our data demonstrate that AMs express CCR2 that is required for localization of AMs within the TB granulomas. In the absence of CCR2, AMs fail to localize within TB granulomas, resulting instead in accumulation of neutrophils and development of necrotic TB lesions, a characteristic of failed immunity in TB. Additionally, we show that the requirement for CCR2 for protective immunity to Mtb HN878 is not solely dependent on expression of PGL and PGL-mediated modulation of this chemokine axis. Thus, our study provides novel insights by projecting that CCR2 expression, especially on AMs, is required for protective immunity against emerging Mtb strains.
AMs are a tissue resident macrophage lineage that under homeostatic conditions are believed to mature during neonatal development35. However, under conditions of inflammation inflammatory monocytes, perhaps through a transient lung-resident macrophage population may also give rise to AMs35. During Mtb infection, although it is widely believed that tissueresident AMs are the primary innate cell type that are infected36, the functional relevance of AMs during Mtb infection is unclear. Utilizing the zebrafish granuloma model, it has been shown thatM. marinum can be transported across neuroepithelial barriers by macrophages and results inCCL2-mediated recruitment of permissive monocyte-derived macrophages that mediate formation of the granuloma to enhance disease pathogenesis7,8. Using a murine model, our studies here show that following infection with Mtb HN878, bonafide AMs increase in the lung following Mtb infection, and they localize within the airway. As infection progresses and TBranulomas form, our results show that AMs respond to CCL2-dependent chemokine signals likely from lung epithelial cells to egress from the airways. Furthermore, adoptive transfer ofCD11c+ SiglecF+ AMs expressing CCR2 localized within granulomas, and AMs not expressingCCR2 did not localize within granulomas, suggesting that AMs may require CCR2 expression torespond to epithelial signals and localize into granulomas. Additionally, we show that HN878pks1–15::hygB infection still results in increased susceptibility in CCR2−/− mice. Thus, the protective versus disease-promoting features of the CCR2 axis and AMs in mycobacterial infections in mouse versus zebrafish model may be due to 1.) differences in the animal models used, 2.) a specific feature of AM trafficking from airways that are not observed in zebrafish macrophages, or 3.) due to the fact that M. marinum PGLs contain a monosaccharide while MtbPGL contain a trisaccharide domain37.
Ligands for CCR2, namely CCL2, CCL7 and CCL12 are induced in response to Mtb infection in mice, non-human primates and humans1,38,39. Despite this, studies in the last decade have shown that CCR2−/− mice display negligible or low susceptibility to low dose aerosolized Mtb infection belonging to the Euro-American lineage 4, such as H37Rv and Erdman1,11. Our data show that when W-Beijing family lineage 2 clinical Mtb strains are used, absence of CCR2 in the CCR2−/− or deletion of CCR2 in CCR2-DTR mice render mice more susceptible to infection, with coincident defects in macrophage accumulation, instead promoting neutrophil accumulation, an immune cell type associated with failed immunity15–18. The influx of neutrophils may be a direct response to the tissue damage and inflammatory environment brought on by the lack of apoptotic clearance and anti-inflammatory signaling of AMs40. This increase in susceptibility in CCR2−/− mice is not just limited to lineage 2 but also to clinical isolates within the Euro-American lineage and Indo-Oceanic lineages, albeit not to the same extent as in W-Beijing Mtb infection. The observation that infection with the HN878 mutant lacking PGLs still requires CCR2 for AM accumulation and protective immunity supports the idea that expression of other factors by emerging Mtb strains are likely involved. It is possible that the observed Mtb strain-dependent response is due to distinct cellular responses brought on by pattern recognition and phagocytic receptor interactions with Mtb strain-specific membrane lipids and virulence factors34. This is supported by published data that Mtb-associated membrane lipids and other virulence factors lead to differential transcriptional and metabolic profiles in macrophages41–43.
Upon infection with Streptococcus, AMs can leave the airway to migrate to lymph nodes to transport bacteria44. However, whether AMs can egress from the airway and participate in immune responses locally within the lung is unknown. In our study we find that airway AMs apart from expressing the core macrophage signature genes, also express a unique transcriptional signature. Furthermore, airway AMs upon Mtb infection upregulate transcriptional path ways associated with antigen presentation and phagosome maturation. Using a novel I Tlabeling technique, we show that CCR2+ AMs are present within airways during early stages ofMtb infection. As disease progresses, airway AMs can likely respond to chemokine signals from lung epithelial cells (and other cells such as DCs) and egress from the airway and localize within TB granulomas. Upon localization within granulomas, non-airway AMs can undergo classical activation including production of iNOS, upregulation of genes associated with inflammation, pattern recognition receptors, and T cell co stimulation thereby providing optimal Mtb control. Whether the activation of non-airway AMs is due to localization within the inflammatory granulomas and dependent on signals within the granuloma is yet to be studied.
This airway labeling technique we have developed can be useful in the study of airway localized immune cell types in the lung, including the localization of AMs, monocytes, DCs and neutrophils in the airways versus lung tissue. While this labeling technique can be useful for tracking early cellular changes in the airways as in this study, upon formation of granuloma and disease progression to chronic stages of infection, the structural integrity of the lung airway epithelium can be compromised and may be a caveat as airway administered antibody may leak, thus also labeling non-airway cells. Furthermore, with time, fluid accumulation and scarring may occur in the airway, leading to less effective labeling of airway cells. These caveats should be carefully considered in the use of this labeling technique during chronic stages of infection, or under conditions of loss of structural integrity or tissue scarring.
In summary, we show that CCR2 expression, while dispensable for protection against Mtb lineage 4 infections, is critical for protection against emerging Mtb strains. Together our data show that during early HN878 infection, AMs upregulate CCR2 and accumulate in the airways. As infection progresses, some AMs remain in the airway where they likely continue to phagocytose and process TB antigen. Other CCR2+ AMs egress in response to production ofCCL2 by epithelial cells or DCs around forming granulomas, and interact with other inflammatory cells to become classically activated and potentially localize within organized granulomas to mediate Mtb control. Thus, our results provide novel insights into the role of AMs and the CCR2 axis in macrophage anti-microbial mechanisms of protection against Mtb infection.
Materials and Methods
Mice
C57BL/6J (B6), CCR2−/−12 and CCL2−/−45 mice on the B6 background were purchased from The Jackson Laboratory (Bar Harbor, ME). CCR2-DTR33 and CCR2-GFP32 were a kind gift from Drs.Robyn Klein and Marco Colonna at Washington University School of Medicine and bred in house. IKK2fl/fl Sftpc-cre20 were a kind gift from Dr. Pasparakis (University of Cologne). CCR2-DTR mice were administered sterile PBS or 20ng Dtx/g body weight (VWR, Radnor, PA)approximately 500 ng/mouse in 2000µL sterile PBS, intraperitoneally (IP), 3 times as indicated. All mice were maintained in the animal facility at Washington University in St. Louis and bred in house. Experimental mice were age and sex matched and infected between the ages of 6 and 8 weeks.
All mice were maintained and used in accordance with the approved Washington University in St. Louis Institutional Animal Care and Use Committee guidelines. Both male and female mice were used and to our knowledge no sex-based differences were observed.
Mtb strains and experimental infections
Mtb strains H37Rv (Trudeau Institute), CDC1551, HN878, and HN563 were obtained from BEI resources (Manassas, VA) under National Institutes of Health contract AI-75320. HN878 pks1-15::hygB was used as previously published5. H37Rv-GFP and HN878-GFP was transformed with the integrating EGFP expression vector pMV261.kan (provided by Dr. Christina Stallings ,Washington University in St. Louis) to generate GFP-expressing Mtb. Indo-Oceanic T17x strain was acquired from Dr. Karen M. Dobos, Colorado State University. All Mtb strains were cultured in Proskauer Beck medium supplemented with 0.05% Tween 80 and frozen at −80°C while in mid-log phase. Mice were aerosol infected with low doses (~100 CFU) of indicated Mtb strains in sterile PBS using a Glass-col nebulizer46. Mice were monitored and weighed as needed and lungs and spleens were harvested at described time points. Mtb CFU/organ was quantitated by plating serial dilutions of homogenized lung or spleen tissue on 7H11 agar plates (BD Biosciences, Franklin Lakes, NJ). Plates were incubated for 2–3 weeks at 37°C and colonies were counted visually.
In vitro culture of cells
Mouse epithelial C10 cells were cultured in complete Dulbecco’s modified eagle’s medium (cDMEM) to confluence for approximately 2 days. Counts from a representative well were used to calculate the concentration of C10 cells/well, performed visually by hemocytometer. Bone marrow-derived macrophages (BMDMs) and bone marrow-derived DCs (BMDCs) were cultured from bone marrow cells as previously described47. Briefly, bone marrow cells from the femur and tibia of B6 and gene deficient mice were extracted, and 1×107 cells were plated in 10mL of complete Dulbecco’s modified eagle’s medium (cDMEM) supplemented with 20ng/mL mouse recombinant (rm) granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech, Rocky Hill, NJ)46. Cells were then cultured at 37°C in 5% CO2. On day 3, 10mL of cDMEM containing 20ng/mL rmGM-CSF was added. On day 7, adherent cells were collected asmacrophages and non-adherent cells were collected as dendritic cells (DCs).
Chemotaxis Assay
BMDMs were grown as above, and plated at 2×106/mL and stimulated with 200µg/mL irradiated HN878 (BEI) for 24 hours in cDMEM at 37 °C. BMDMs were then added to the upper chamber of the 24 well trans well plate (Costar, Cambridge, CA), 1×105 cells/well in 100µl of Hank’s balanced salt solution containing 1% fetal bovine serum with 600µL of indicated conditioned media beneath the trans well in a 24 well plate. These were incubated for 90 minutes at 37 °C, then transmigrated cells were collected from the lower chamber, stained and analyzed by flow cytometry.
In vitro infections
BMDMs, BMDCs, and C10 epithelial cells were infected with Mtb (multiplicity of infection,MOI:1) in antibiotic-free cDMEM. Cell culture supernatants were collected for analysis of cytokines 48 hours post infection.
Adoptive Transfer
Lung cell suspensions were prepared as before from the lungs of donor CCR2-GFP (+/KI orKI/KI) mice on 40 dpi following HN878 infection. CD11c+ cells were enriched from the lung suspension using magnetic selection with CD11c microbeads (Miltenyi Biotec, Auburn, CA) per manufacturer’s instructions, yielding a population of (>85%) CD11c+ cells. These cells were resuspended at 1×107 cells in 500µL sterile PBS. 50µL (1×106 cells) of this suspension was administered IT to HN878-infected mice at 30 dpi. These mice were harvested on 50 dpi as indicated.
BMDMs were grown as above, and plated at 2×106 cells/mL and stimulated with 20µg/mL irradiated HN878 (BEI) for 24 hours in cDMEM at 37 °C. These cells were then harvested and resuspended at 1×107 cells in 500µL sterile PBS. 50µL (1×106 cells) of this suspension was administered IT to HN878-infected mice at 15 and 22 or 30 dpi as indicated.
Flow Cytometry
For distinguishing between airway and non-airway cells, mice received 0.7µg of V500 or PE conjugated CD45.2 mAb (Clone 104, BD Biosciences and Bio legend, San Diego, CA)),administered IT in 50µL sterile PBS per mouse. Mice were rested for 15 minutes following which lungs were harvested. To validate the technique some mice received 50 uL of HCl in sterile water (pH 1.5) intratracheally and rested for 24 hrs prior to instillation of CD45.2 antibody48. Lung single cell suspensions were prepared as before 46, treated with Fc Block (CD16/CD32,2.4G2, Tonbo Biosciences, San Diego, CA), and stained with appropriate fluorochrome-labeled specific antibodies or isotype control antibodies:
CD11c (HL3, BD Biosciences), CD11b (M1/70, BD Biosciences and Tonbo Biosciences), SiglecF (E50–2440, BD Biosciences), CD64(X54–5/7.1, Bio legend), Gr-1 (RB6–8C5, BD Biosciences),CCR2 (FAB5538P, R&D Systems), CX3CR1 (SA011F11, Bio legend), Ly6C (AL21, BD Biosciences), Ly6G (1A8, BD Biosciences), CD3 (145–2C11, Tonbo Biosciences), CD4 (GK1.5,BD Biosciences), CD44 (IM7, eBioscience), IFN-γ (XMG1.2, BD Biosciences), and rat IgG1 (BD Biosciences). Cells were processed using a Becton Dickinson FACS LSR Fortessa flow cytometer using FACSDiva software, or sorted on a Becton Dickinson FACS Jazz cell sorter using BD FACS sortware sorting software. Cells were gated based on their forward and side scatter characteristics and the frequency of specific cell types was calculated using Flow Jo (Flow Jo, LLC, Ashland, OR). Neutrophils were defined as CD11b+CD11c−Gr-1hi cells, monocytes were defined as CD11b+CD11c−Gr-1lo cells, and recruited macrophages were defined as CD11b+CD11c−Gr-1− cells. mDCs were defined as CD11b+CD11c+ cells. AMs were defined as CD11bloCD11c+ cells (Fig. 1) and subsequently as CD11bloCD11c+Siglec F+ cells. The flow cytometry gating strategy used for lung subset analysis was an approach using our IT labeling method to select for airway populations and a combined gating strategy from several previous publications35,49–52.
For uptake experiments, lung cell suspensions were prepared from H37Rv-GFP- or HN878-GFP-infected B6 mice and stained with appropriate fluorochrome-labeled specific antibodies. Uptake was determined by the co-localization of GFP with known fluorophore-conjugated cell subset markers as measured by flow cytometry.
Broncho-Alveolar Lavage (BAL)
Mice were administered IT CD45.2 Ab as above and ~10 min later euthanized. BAL was then performed as previously described 53,54. Briefly, the chest cavity was opened and the sternum/ribcage was resected. The trachea was isolated and a blunt tipped needle was gently inserted into the trachea. The lungs were lavaged with ~5 mLs (5 × 1 mL washes) with sterile 0.2 mM EDTA (Sigma-Aldrich) in PBS. Cells were collected from these lavages and analyzed by flow cytometry as above.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Lung tissue was homogenized and snap-frozen in RLT buffer (Qiagen, Valencia, CA). Total RNA was extracted from lung tissue using the Qiagen RNeasy Mini kit (Qiagen). RNA was converted to cDNA using ABI reverse transcription reagents (ABI/Thermo Fisher Scientific, Carlsbad, CA) using a Bio Rad DNA Engine Thermal Cycler (Bio Rad, Hercules, CA). cDNA was then amplified using TaqMan reagents on the ABI Viia7 Real-Time PCR detection system (Thermo Fisher Scientific). The primer and probe sequences for murine glyceraldehyde 3-phosphate dehydrogenase (Gapdh) and Tnfa were previously published55. The primers and probes for murine Arg1, Ccl2, Ccl7, and Ccl12 were purchased commercially (ABI Biosystems). The primer and probe sequences for Tgfb1 are forward 5′-TGACGTCACTGGAGTTGTACGG-3′, reverse 5′- GGTTCATGTCATGGATGGTGC-3′, probe 5′ - /56-FAM/TTC AGC GCT CACTGC TCT TGT GAC AG/3BHQ_1/ - 3’. Fold increase was determined over uninfected controls relative to Gapdh expression using the ∆∆CT calculation as previous described55.
RNA-Sequencing of AM populations
AMs were sorted and RNA was extracted as described above. RNA sequencing libraries were generated using Clontech SMART-Seq v4 Ultra Low Input RNA Kit for sequencing and Illumina Nextera XT DNA Library preparation kit following the manufacturer’s protocol. The cDNA libraries were validated using KAPA Biosystems primer premix kit with Illumina-compatible DNA primers and quality was examined using Agilent Tapestation 2200. The cDNA libraries were pooled at a final concentration of 1.8pM. Cluster generation and 75 bp Paired-read dual-indexed sequencing was performed on Illumina Next Seq 500. Single-ended 75bp reads were cleaned using Trimmomatic (version 0.36) to remove adapters and quality trimmed to filter out reads <60bp in length (after trimming). Cleaned sequence data was mapped against the mouse reference genome build GRCh38.90 (Ensembl) using the HISAT256 aligner (version 2.1.0) with default parameters, generating sam format alignment files. These sam files were then used as input for feature Counts (version 1.5.1), requiring a minimum mapping quality score of 10. Raw read counts were used as input for DESeq225 (version 1.16.1) differential expression analysis ,using default settings and an FDR-adjusted P value threshold of 0.05 for significant differential expression. Lists of differentially expressed genes were used to test for significant enrichment among KEGG pathways57 and Mammalian Phenotype Ontology58, using WebGestalt59 (default settings, adjusted P = 0.05 threshold for enrichment). Heatmap clustering was performed using hclust” in R, using Pearson distances and complete linkage, based on Z-scores calculated perrow using Microsoft Excel.
Histology
Lung lobes were perfused with 10% neutral buffered formalin and embedded in paraffin. For immunofluorescent staining, formalin-fixed paraffin-embedded (FFPE) lung sections were cut, immersed in xylene to remove paraffin, and then sequentially hydrated in absolute ethanol, 95% ethanol, 70% ethanol, and water. Antigens were unmasked with a DakoCytomation Target Retrieval Solution (Dako, Carpinteria, CA) and nonspecific binding was blocked with 5% (v/v) normal donkey serum and Fc Block (BD Pharmingen, San Jose, CA). Endogenous biotin (Sigma-Aldrich) was neutralized by adding avidin followed by incubation with biotin.
Antibodies used for identification of lung cells as previously described:
Rabbit anti-Mycobacterium tuberculosis antibody, (MBS534825, MyBiosource.com)
Rabbit anti MCP-1/CCL2 (GTX37379, GeneTex ,Irvine, CA) Goat anti-human/mouse E-Cadherin (AF748, R & D systems)
Monoclonal hamster anti-ITGAX/CD11c (N418, Lifespan Biosciences, Inc., Seattle, WA)
Polyclonal rabbit anti-Siglec 5/CD170 (LS-C97825, Lifespan Biosciences, Inc.). Fluorescein(FITC) affinipure F(ab’)2 fragment donkey anti-rabbit IgG (H+L) (711–096-152, Jackson Immuno research, West Grove, PA).
Biotin-SP (long spacer) affinipure F(ab’)2 fragment rabbit anti-Syrian hamster IgG (H+L) (307–066-003, Jackson Immuno research).
Streptavidin, Alexa fluor® 488 conjugate (S-11223, Thermo Fisher Scientific).
Biotinylated goat anti-GFP (GTX26658, Genetex, Irvine, CA) was visualized with Alexa fluor 568 donkey anti-goat IgG and Alexa fluor 555 streptavidin. GFP stain was pseudo colored to green with the Axiovision software. Images were collected using an inverted Observer.Z1 Axio plan Zeiss Microscope with Colibri system (Zeiss, Thornwood, NY) and the Axiovision Rel 4.8.Software.
FFPE lung sections were stained with hematoxylin and eosin (H&E) and inflammatory features were evaluated by light microscopy. Inflammatory lesions were outlined with the automated tool of the Zeiss Axioplan 2 microscope (Carl Zeiss) and percentage of inflammation was calculated by dividing the inflammatory area by the total area of individual lung lobes. FFPE lung sections were stained for collagen and muscle using Masson’s Trichrome 2000 stain procedure kit(#KTMTR2, American Master tech, Lodi, CA) per the manufacturer’s instructions. Trichrome stained slide images were acquired using a Hamamatsu Nanozoomer 2.0 HT system with NDP scan image acquisition software. Trichrome staining was quantified using Visiomorph image processing software (Visiopharm, Broomfield, CO). FFPE lung sections were subjected to in situ hybridization (ISH) with the mouse-Ccl2 probe using the RNAscope 2.5HD Detection Kit (Brown staining) as per the manufacturer’s recommendations (Advanced Cell Diagnostics, Newark,CA). The representative pictures were taken with the Hamamatsu Nanozoomer 2.0 HT system with NDP scan image acquisition software.
Cytokine production quantification
Cytokine levels within cell culture supernatants were analyzed using the BD OptEIA Mouse CCL2 ELISA kit (BD Biosciences) or using Milliplex Multiplex Assays (Millipore, Billerica, MA),as per standard protocol.
Statistical Analyses
Statistical Analyses were performed using GraphPad Prism 5 and Microsoft Excel. Specific analysis techniques and post tests are mentioned in figure legends. SEM error bars displayed. *p≤0.05, **p≤0.01, ***p≤0.001, n.s.-not significant.
Supplementary Material
Acknowledgements:
This work was supported by Washington University in St. Louis, NIH grant HL105427 to S.A.K., AI111914, AI134236 to S.A.K, and D.K., NIH/NHLBI T32 AI007172 to MDD, NIH/NHLBI T32 HL007317–37 to N.H., and NIH Shared instrumentation Grant S10RR0227552. J.R.M. was supported by funds of the Department of Medicine, University of Rochester, and U19 AI91036. The authors thank Drs. Robyn Klein (Washington University in St.Louis) and Marco Colonna (Washington University in St. Louis) for generously providing mice. We thank Dr. Kimberly Thomas for critical reading of the manuscript, Sarah Squires and Lan Lu(Washington University in St. Louis) for technical support, William Horne (University of Pittsburgh) for carrying out RNA sequencing.
Footnotes
Conflict of Interest:
The authors declare no conflicts of interest.
The authors declare no competing financial interests.
References
- 1.Scott HM & Flynn JL Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: influence of dose on disease progression. Infect. Immun 70, 5946–5954 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samstein M et al. Essential yet limited role for CCR2(+) inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. Elife 2, e01086, 10.7554/eLife.01086 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Domingo-Gonzalez R, Prince O, Cooper A & Khader SA Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol Spectr 4, 10.1128/microbiolspec.TBTB2-0018-2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian G, Li X, Li H, Wang X & Cheng B Systematic meta-analysis of the association between monocyte chemoattractant protein-1 −2518A/G polymorphism and risk of tuberculosis. Genet. Mol. Res 14, 5501–5510, 10.4238/2015.May.25.1 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Reed MB et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431, 84–87, 10.1038/nature02837 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Gopal R et al. Unexpected role for IL-17 in protective immunity against hyper virulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog 10, e1004099, 10.1371/journal.ppat.1004099 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cambier CJ et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 505, 218–222, 10.1038/nature12799 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cambier CJ, O’Leary SM, O’Sullivan MP, Keane J & Ramakrishnan L Phenolic Glycolipid Facilitates Mycobacterial Escape from Microbicidal Tissue-Resident Macrophages. Immunity, 10.1016/j.immuni.2017.08.003 (2017). [DOI] [PMC free article] [PubMed]
- 9.Armstrong JA & Hart PD Phagosome-lysosome interactions in cultured macrophages infected with virulent tubercle bacilli. Reversal of the usual nonfusion pattern and observations on bacterial survival. J. Exp. Med 142, 1–16 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava S, Ernst JD & Desvignes L Beyond macrophages: the diversity of mononuclear cells in tuberculosis. Immunol. Rev 262, 179–192, 10.1111/imr.12217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antonelli LR et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Invest 120, 1674–1682, 10.1172/jci40817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters W et al. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 98, 7958–7963,10.1073/pnas.131207398 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gagneux S & Small PM Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. The Lancet Infectious Diseases 7,328–337, http://dx.doi.org/10.1016/S1473-3099(07)70108-1 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Serbina NV, Shi C & Pamer EG Monocyte-mediated immune defense against murine Listeria monocytogenes infection. Adv. Immunol 113, 119–134,10.1016/b978-0-12-394590-7.00003-8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eruslanov EB et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect. Immun 73, 1744–1753,10.1128/iai.73.3.1744-1753.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lowe DM et al. Neutrophilia independently predicts death in tuberculosis. Eur. Respir.J 42, 1752–1757, 10.1183/09031936.00140913 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lowe DM, Redford PS, Wilkinson RJ, O’Garra A & Martineau AR Neutrophils in tuberculosis: friend or foe? Trends Immunol 33, 14–25, 10.1016/j.it.2011.10.003 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Yeremeev V, Linge I, Kondratieva T & Apt A Neutrophils exacerbate tuberculosis infection in genetically susceptible mice. Tuberculosis (Edinb) 95, 447–451,10.1016/j.tube.2015.03.007 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Vasquez-Loarte T, Trubnykova M & Guio H Genetic association meta-analysis: a new classification to assess ethnicity using the association of MCP-1 −2518 polymorphism and tuberculosis susceptibility as a model. BMC Genet 16, 128, 10.1186/s12863-015-0280-2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pasparakis M et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 417, 861–866, 10.1038/nature00820 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Gigliotti F, Bhagwat SP, Maggirwar SB & Wright TW Pneumocystis stimulates MCP-1 production by alveolar epithelial cells through a JNK-dependent mechanism. Am. J. Physiol. Lung Cell Mol. Physiol 292, L1495–1505,10.1152/ajplung.00452.2006 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Misharin AV et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med 214, 2387–2404,10.1084/jem.20162152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautier EL et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J. Immunol 189, 2614–2624,10.4049/jimmunol.1200495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang L, Nazarova EV, Tan S, Liu Y & Russell DG Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med 215, 1135–1152, 10.1084/jem.20172020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy S et al. Batf2/Irf1 induces inflammatory responses in classically activated macrophages, lipopolysaccharides, and mycobacterial infection. J. Immunol 194, 6035–6044, 10.4049/jimmunol.1402521 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Xu H et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat. Immunol 13, 642–650, 10.1038/ni.2304(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mass E et al. Specification of tissue-resident macrophages during organogenesis. Science 353, 10.1126/science.aaf4238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jager SC et al. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J. Exp. Med 208, 217–225, 10.1084/jem.20100370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu J et al. Polo-like kinase 1 (PLK1) is involved in toll-like receptor (TLR)-mediated TNF-alpha production in monocytic THP-1 cells. PLoS One 8, e78832, 10.1371/journal.pone.0078832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyu JH, Huang B, Park DW & Baek SH Regulation of PHLDA1 Expression by JAK2-ERK1/2-STAT3 Signaling Pathway. J. Cell. Biochem 117, 483–490,10.1002/jcb.25296 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Satpathy AT et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat. Immunol 14, 937–948,10.1038/ni.2679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hohl TM et al. Inflammatory Monocytes Facilitate Adaptive CD4 T Cell Responses during Respiratory Fungal Infection. Cell Host Microbe 6, 470–481,10.1016/j.chom.2009.10.007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Domingo-Gonzalez R et al. Interleukin-17 limits hypoxia-inducible factor 1alpha and development of hypoxic granulomas during tuberculosis. JCI Insight 2,10.1172/jci.insight.92973 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davies LC, Jenkins SJ, Allen JE & Taylor PR Tissue-resident macrophages. Nat. Immunol 14, 986–995, 10.1038/ni.2705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schorey JS & Schlesinger LS Innate Immune Responses to Tuberculosis. Microbiol Spectr 4, 10.1128/microbiolspec.TBTB2-0010-2016 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Arbues A, Lugo-Villarino G, Neyrolles O, Guilhot C & Astarie-Dequeker C Playing hide-and-seek with host macrophages through the use of mycobacterial cell envelope phthiocerol dimycocerosates and phenolic glycolipids. Front Cell Infect Microbiol 4, 173, 10.3389/fcimb.2014.00173 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dutta NK et al. The stress-response factor SigH modulates the interaction between Mycobacterium tuberculosis and host phagocytes. PLoS One 7, e28958, 10.1371/journal.pone.0028958 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flores-Villanueva PO et al. A functional promoter polymorphism in monocyte chemoattractant protein-1 is associated with increased susceptibility to pulmonary tuberculosis. J. Exp. Med 202, 1649–1658, 10.1084/jem.20050126 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hussell T & Bell TJ Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol 14, 81–93, 10.1038/nri3600 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Brace PT et al. Mycobacterium tuberculosis subverts negative regulatory pathways in human macrophages to drive immunopathology. PLoS Pathog 13, e1006367, 10.1371/journal.ppat.1006367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cumming BM et al. Mycobacterium tuberculosis arrests host cycle at the G(1)/S transition to establish long term infection. PLoS Pathog 13, e1006389, 10.1371/journal.ppat.1006389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koo M-S, Subbian S & Kaplan G Strain specific transcriptional response in Mycobacterium tuberculosis infected macrophages. Cell Communication and Signaling : CCS 10, 2–2, 10.1186/1478-811X-10-2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirby AC, Coles MC & Kaye PM Alveolar macrophages transport pathogens to lung draining lymph nodes. J. Immunol 183, 1983–1989, 10.4049/jimmunol.0901089 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu B et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med 187, 601–608 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khader SA et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol 8, 369–377, 10.1038/ni1449 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Lin Y et al. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity 31, 799–810, 10.1016/j.immuni.2009.08.025 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eickmeier O et al. Altered mucosal immune response after acute lung injury in a murine model of Ataxia Telangiectasia. BMC Pulm. Med 14, 93, 10.1186/1471-2466-14-93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Desvignes L, Wolf AJ & Ernst JD Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J. Immunol 188, 6205–6215, 10.4049/jimmunol.1200255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR & Perlman H Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol 49, 503–510, 10.1165/rcmb.2013-0086MA (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bedoret D et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Invest 119, 3723–3738, 10.1172/JCI39717 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jakubzick C et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 39, 599–610, 10.1016/j.immuni.2013.08.007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gopal R et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am. J. Respir. Crit. Care Med 188, 1137–1146,10.1164/rccm.201304-0803OC (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slight SR et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J. Clin. Invest 123, 712–726, 10.1172/jci65728 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khader SA et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen- specific IFN-gamma responses if IL-12p70 is available. J. Immunol 175, 788–795 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357–360, 10.1038/nmeth.3317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanehisa M, Sato Y, Kawashima M, Furumichi M & Tanabe M KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–462, 10.1093/nar/gkv1070 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith CL & Eppig JT The Mammalian Phenotype Ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm. Genome 23, 653–668, 10.1007/s00335-012-9421-3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang B, Kirov S & Snoddy J WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33, W741–748,10.1093/nar/gki475 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.