

Abstract
Purpose:
Prostate cancers show remarkable resistance to emerging immunotherapies, partly due to tolerogenic STAT3 signaling in tumor-associated myeloid cells. Here, we describe a novel strategy combining STAT3 inhibition with Toll-like Receptor-9 (TLR9) stimulation to unleash immune response against prostate cancers regardless of the genetic background.
Experimental Design:
We developed and validated a conjugate of the STAT3 antisense oligonucleotide (ASO) tethered to immunostimulatory TLR9 agonist (CpG oligonucleotide) to improve targeting of human and mouse prostate cancer and myeloid immune cells, such as myeloid-derived suppressor cells (MDSCs).
Results:
CpG-STAT3ASO conjugates showed improved biodistribution and potency of STAT3 knockdown in target cells in vitro and in vivo. Systemic administration of CpG-STAT3ASO (5mg/kg) eradicated bone-localized, Ras-/Myc-driven and Ptenpc−/−Smad4pc−/−Trp53c−/− prostate tumors in the majority of treated mice. These antitumor effects were primarily immune-mediated and correlated with an increased ratio of CD8+ to regulatory T-cells and reduced pSTAT3+/PD-L1+ MDSCs. Both innate and adaptive immunity contributed to systemic antitumor responses as verified by the depletion of Gr1+ myeloid cells and CD8+, CD4+ T-cells, respectively. Importantly, only the bi-functional CpG-STAT3ASO, but not control CpG oligonucleotides, STAT3ASO alone nor the co-injection of both oligonucleotides, succeeded in recruiting neutrophils and CD8+ T-cells into tumors. Thus, the concurrence of TLR9 activation with STAT3 inhibition in the same cellular compartment is indispensable for overcoming tumor immune tolerance and effective antitumor immunity against prostate cancer.
Conclusions:
The bi-functional, immunostimulatory and tolerance-breaking design of CpG-STAT3ASO offers a blueprint for the development of effective and safer oligonucleotide strategies for treatment of immunologically “cold” human cancers.
INTRODUCTION
Emerging immunotherapies exhibit robust clinical activity across a broad spectrum of late-stage tumor types, with notable exception of prostate cancers.(1,2,3) Advanced prostate tumors are dependent on multiple cancer cell intrinsic and extrinsic mechanisms to promote tumorigenesis, escape immunosurveillance and actively block immune responses.(2,4,5) Genetic drivers of carcinogenesis, such as expression of MYC oncogene overexpression or PTEN tumor suppressor deficiency, can differentially shape immunocyte composition of the tumor microenvironment and drive distinct mechanisms of immune evasion in prostate cancer and in other solid tumors.(6–8) Tolerogenic effects of prostate tumors extend beyond the PD-1 immune checkpoint regulation. Therefore, there is a need for combinatorial immunotherapeutic approaches to disrupt complex signaling networks in the tumor microenvironment.(9–13)
The STAT3 transcription factor is a multifaceted oncogene and a master regulator of immunosuppression commonly activated in the majority of human cancers.(14–16) Extensive evidence suggests that tumors, such as advanced prostate cancers, critically depend on STAT3 for their survival, vascularization and metastasis, whereas normal cells do not.(17–19) The role of STAT3 in determining prostate cancer cell fate seems to be context dependent. Constitutive STAT3 activity can result in tumor progression towards hormone-refractory/castration-resistant prostate cancer (CRPC) phenotype and correlate with poor overall survival.(4,20,21) Conversely, in PTEN-null prostate cancer cells STAT3 activation mediates cell senescence and restrict tumor growth.(22) However, STAT3 serves a consistent role as a key mediator of tumor immune evasion regardless of tumor genotype as well documented in prostate cancers, as well as other human malignancies.(21,23–25)
Previous studies demonstrated that stress and inflammation can trigger and/or sustain STAT3 activity in prostate tumors and especially in tumor-associated myeloid cells, such as macrophages (MACs) and myeloid-derived suppressor cells (MDSCs).(26,25,27) Cell death causes the release of Toll-like receptor 9 (TLR9) ligands, such as mitochondrial DNA, and TLR9/NF-κB-induced secretion of IL-6-type cytokines. IL-6 and LIF in turn stimulate STAT3 activity in cancer cells and myeloid cells in the tumor microenvironment.(26,25,27) More recently, we confirmed high TLR9 expression and STAT3 activation in polymorphonuclear MDSCs (PMN-MDSCs), a potently immunosuppressive cells, which accumulate in circulation in prostate cancer patients during disease progression from localized to metastatic/castration-resistant prostate cancer (mCRPC).(25,28) The TLR9+ PMN-MDSCs relied on the STAT3-mediated expression of Arginase-1 (ARG-1) to block T-cell proliferation and activity.(28) These effects underscore well-known pivotal role of MDSCs in prostate cancer progression and poor overall survival.(25,28–32)
As a master regulator operating in both cancer cells and in tumor-associated immune cells, STAT3 is a unique and highly desirable target for cancer therapy.(14,16,29,33) Due to the lack of enzymatic activity, pharmacologic inhibition of STAT3 is challenging.(33,34) Emerging oligonucleotide-based strategies to inhibition of STAT3 signaling, such as decoy and antisense oligonucleotides (ASO), showed promise in phase I clinical trials.(35,36) However, the efficacy of STAT3ASO is limited by the lack of cell-selectivity and targeted delivery, which decrease oligonucleotide penetration into the solid tumor microenvironment and efficacy.(29,36) To improve the pharmacological properties of oligonucleotide therapeutics, we previously developed a strategy for targeted delivery of oligonucleotides, such as siRNA, specifically to tumor-associated TLR9+ immune cells and TLR9+ cancer stem-like cells in prostate tumors.(37,27,28,38,29) Here, we describe the conjugate of CpG oligodeoxynucleotide (ODN), a synthetic TLR9 ligand, with chemically modified STAT3ASO molecules, with improved nuclease-resistance suitable for systemic administration. We characterized effects of CpG-STAT3ASO on both human and mouse cellular targets in vitro and in two syngeneic models of bone-localized prostate tumors. Our studies assessed two-pronged effects of the conjugate, directly on prostate cancer cells and indirectly, through immune-mediated antitumor immune responses.
MATERIALS AND METHODS
Cell lines and uptake studies
Human DU-145 prostate cancer (HTB-81) and mouse RAW264.7 cells (TIB-71) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Mouse DC2.4 cells were originally from Dr. K. Rock (University of Massachusetts Medical School, MA), RM9 cells were kindly provided by Dr. T. Thompson (MD Anderson, TX) and PPS (Ptenpc−/− Trp53pc−/− Smad4pc−/−) cells were recently generated by Drs. R. DePinho and X. Lu.(6) All tested cells were cultured for less than 6 months before experiments and tested bimonthly to be free from mycoplasma infections. For oligonucleotide uptake studies, DU-145 cells were pre-treated for 1 h in the presence of 50 μg/ml dextran sulfate (#42867), 100 ¼g/ml of fucoidan (#F8190), 1 mM of amiloride (#A7410), 150 ¼M of genistein (#G6649), 2 ¼g/ml of filipin (#F4767), 100 ¼M of cadaverine (#33211), 50 μg/ml chondroitin sulfate (#4384, Sigma-Aldrich, St. Louis, MO) or at +4 °C prior to treatment with oligonucleotides followed by flow cytometric analysis.
Oligonucleotide design and synthesis
The CpG-ODN conjugates were synthesized in the DNA/RNA Synthesis Core (COH) by linking CpG-ODNs to STAT3 ASO similarly as previously described (37).The resulting ODN conjugates are shown below (x, indicates a single C3 unit; asterisk, indicates 2’O methylation; underline, indicates phosphorothioation site):
CpG-STAT3ASO - (CpGD19 ODN + human STAT3 ASO targeting sequence):
5’ GGTGCATCGATGCAGGGGGG-xxxxx-C*A*G*C*A*GATCAAGTCCA*G*G*G*A* 3’.
STAT3 ASO (human STAT3 ASO targeting sequence):
5’ C*A*G*C*A*GATCAAGTCCA*G*G*G*A* 3’.
CpG-scrON - (CpGD19 ODN + scrambled ASO sequence):
5’ GGTGCATCGATG CAGGGGGG-xxxxx-A*G*A*G*C*CTAACGGAAGG*C*A*C*T* 3’.
CpG-mSTAT3ASO - (CpG1668 ODN + mouse STAT3 ASO targeting sequence):
5’ TCC ATG ACG TTC CTG ATG CT-xxxxx-G*A*C*T*C*TTGCAGGAATC*G*G*C*T* 3’
mSTAT3ASO - (mouse STAT3 ASO targeting sequence):
5’G*A*C*T*C*TTGCAGGAATC*G*G*C*T* 3’
CpG-scrON (CpGD1668 ODN + scrambled ASO targeting sequence):
5’ TCC ATG ACG TTC CTG ATG CT-xxxxx-A*G*A*G*C*CTAACGGAAGG*C*A*C*T* 3’
For internalization studies, oligonucleotides were labeled on 3’ ends using Cy3 or Alexa488 fluorochromes.
In vivo studies
NOD/SCID/IL-2RγKO (NSG) and C57BL/6 mice, aged between 6–8 weeks, were purchased from the Jackson Laboratory (Bar Harbor, ME). Mouse care and experimental procedures were performed under pathogen-free conditions in accordance with established institutional guidance and approved protocols from Institutional Animal Care and Use Committees. For subcutaneous tumor growth experiments, RM9 (1.5×105) or PPS (8×105) cells were injected subcutaneously (SC) and the tumors size was measured every other day. When tumors reached ~150 mm3 size, mice were injected intratumorally every other day using 5 mg/kg CpG-STAT3ASO, STAT3ASO or CpG-scrON. For the intratibial tumor experiments, the animals were injected intratibially with 1×104 of RM9- or PPS mCherry/luciferase-expressing cells. Tumor engraftment and progression was monitored using bioluminescent imaging (BLI) on the AmiX (Spectral Instruments, Tucson, AZ). After tumor engraftment, mice were injected systemically using 5 mg/kg of CpG-STAT3ASO, STAT3ASO or CpG-scrON, every other day.
Quantitative real-time PCR
Total RNA was extracted from cultured or in vivo grown tumor cells using Maxwell system (#AS1390, Promega, Madison, WI) and then transcribed into cDNAs using iScript cDNA Synthesis kit (#1725064, Bio-Rad, Hercules, CA). The qPCR was carried out using specific primers for STAT3, 18S, actin and TBP, as previously described,(38,39) using CFX96 Real-Time PCR Detection System (Bio-Rad).
Western blotting and immunohistochemical staining
Total cellular lysates were prepared and analyzed using antibodies specific to tyrosine 705-phosphorylated or total STAT3 (#9131, Cell Signaling, Danvers, MA), and β-actin (#A3854, Sigma-Aldrich, St. Louis, MO). Immunohistochemistry using anti-pSTAT3 and anti-Ly6B (clone 7/4, # CL8993AP, Cedarlane, Burlington, Canada) antibodies were performed as previously described.(37) For immunohistochemical staining, the formalin fixed paraffin-embedded tumor sections were stained using primary antibodies and HRP-conjugated secondary antibodies and then analyzed on the Observer II microscope (Zeiss, Oberkochen, Germany).
Flow cytometry
Single-cell suspensions were prepared by mechanic dispersion and enzymatic digestion of tumor tissues. Extracellular staining was performed using fluorochrome-labeled antibodies to major histocompatibility complex (MHC) class II (AF6–120.1, #553552), CD3 (145–2C11, #11–0031-85), CD4 (RM4–5, #12–0042-82), CD8 (53–6.7, #12–0081-83), CD11b (M1/70, #45–0193-82), CD40 (1C10, #12–0401-83), CD80 (16–10A1, #11–0801-85), CD86 (GL1, #25–0862-82), Ly6C (HK1.4, #53–5932-82) or Ly6G (RB6–8C5, #25–5931-82, eBioscience, San Diego, CA) after anti-FcγIII/IIRBlock were used (#558636, BD Biosciences, Franklin, NJ). Human immune cells and PBMCs were analyzed using the following antibodies: HLA-DR (LN3, #12–9956-42), CD1c (L161, #17–0015-14), CD3 (OKT3, #11–0037-42), CD14 (61D3, #11–0149-42), CD15 (MC-480, #12–8813-42), CD16 (CB16, #17–0168-42), CD19 (HIB19, #11–0199-42), CD303 (201A, #11–9818-42). For intracellular staining, cells were fixed/permeabilized and immunostained for pSTAT3 (4/P-STAT3, #562071, BD Biosciences), FoxP3 (236A/E7, #53–4777-41, eBioscience), or Arginase-1 (SL6ARG, #IC5868F, R&D Systems, Minneapolis, MN) as previously described.(28) Fluorescence data were analyzed on BD Fortessa and an AccuriC6 Flow Cytometer (BD Biosciences) using FlowJo software (TreeStar, Ashland, OR).
Confocal microscopy and in situ ligation proximity assays (PLA)
DU-145 cells were cultured in RPMI 1640 medium with 10% FBS using 24-well plates on top of laminin-coated coverslips, and then treated using 250 nM CpG-STAT3ASOCy3. The coverslips were fixed in 2% paraformaldehyde (#15710, EMS, Hatfield, PA), permeabilized with 0.1% Triton X-100 (#X100–500ML Sigma-Aldrich), and stained using primary anti-EEA1 (sc-33585, Santa Cruz Biotechnology, Santa Cruz, CA) or anti-RNase H1 (#NBP2–38501, Novus Biologicals, Littleton, CO) and Alexa488–coupled secondary antibodies (#A32723, Sigma-Aldrich). The proximity ligation assays were performed using Cy3- and RNase H-specific antibodies as reported.(40) Slides mounted in Vectashield Hard-Set medium (#H-1400, Vector Laboratories, Burlingame, CA) were visualized on an LSM510-Axiovert inverted confocal microscope (Zeiss) and analyzed using LSM ImageBrowser (version 4.2.0.121; Zeiss).
T-cell proliferation and activation studies
Studies using human blood samples were performed in accordance with the ethical standards and according to the Declaration of Helsinki under the Institutional Review Board approvals (IRB13141 and IRB 12367) with written, informed consent of all patients. For studies on human PMN-MDSCSs, CD15+ cells were isolated from peripheral blood of patients with metastatic prostate cancers or from healthy subjects and analyzed as previously described (28). Flow cytometric analysis was performed to assess T-cells proliferation using CFSE dilution (#C34554, Sigma-Aldrich), IFNγand granzyme-B production by CD8+ T-cells using specific antibodies to CD3, CD8 (RPA-T8, #25–0088-42), IFNγ (#13–1191-82, eBioscience), and granzyme-B (GB11, #561998, BD Biosciences).
Statistics
Unpaired t test was used to calculate two-tailed P value to estimate statistical significance of differences between two treatment groups. One- or two-way ANOVA plus Bonferroni post-test were applied to assess differences between multiple groups or in tumor growth kinetics experiments. Statistically significant P values were indicated in figures as follows: ***, P <0.001; **, P <0.01 and *, P < 0.05. Data were analyzed using Prism software v. 6.01 (GraphPad).
RESULTS
CpG-STAT3ASO design and cell-selective internalization in vitro
To improve the efficiency and selectivity of STAT3 inhibition in the microenvironment of prostate tumors, we conjugated a STAT3 antisense oligonucleotide (ASO) with a TLR9 agonist, single-stranded CpG(D19) ODN, using a synthetic carbon linker (Fig. 1A).(37) The STAT3ASO has a standard gapmer design, with targeting sequence flanked by 2’-O-methyl-modified nucleotides, to induce RNase H1-dependent knockdown as previously characterized.(41) For enhanced nuclease-resistance the sugar backbone of the conjugate was also partly phosphorothioated. The half-life of CpG-STAT3ASO in 50% human serum exceeded 4 days (T1/2 = 106 h; Supplemental Fig.S1). The conjugation of STAT3ASO to CpG moiety improved oligonucleotide delivery to target primary human immune cells compared to the antisense molecule alone (Fig. 1B, left). When incubated for 1 h with peripheral blood mononuclear cells (PBMCs), fluorescently-labeled CpG-STAT3ASOAlexa488 was internalized by dendritic cells (DCs) and B cells more effectively than STAT3ASOAlexa488 alone at 500 nM concentration. The uptake of CpG-STAT3ASO was dose-dependent and detectable even at 50 nM in human and mouse myeloid cells and B cells, but not in T-cells (Fig. 1B, right; Supplemental Fig.S2). For testing on mouse target cells, we used conjugates with sequences optimized for immunostimulation and targeting STAT3 in mice (CpG1668-mSTAT3ASO) but these modifications did not affect the pattern of conjugate uptake (Fig. 1C and Supplemental Fig.S2). The CpG-STAT3ASO was also effectively internalized by TLR9+ prostate cancer cells such as human DU145 or mouse RM9 cells (Fig. 1C).(27) Cultured prostate cancer cells also internalized STAT3ASO alone but required significantly higher concentrations (500 nM) compared to CpG-STAT3ASO conjugates.
Figure 1. CpG-STAT3ASO conjugate design and cell-selective uptake.
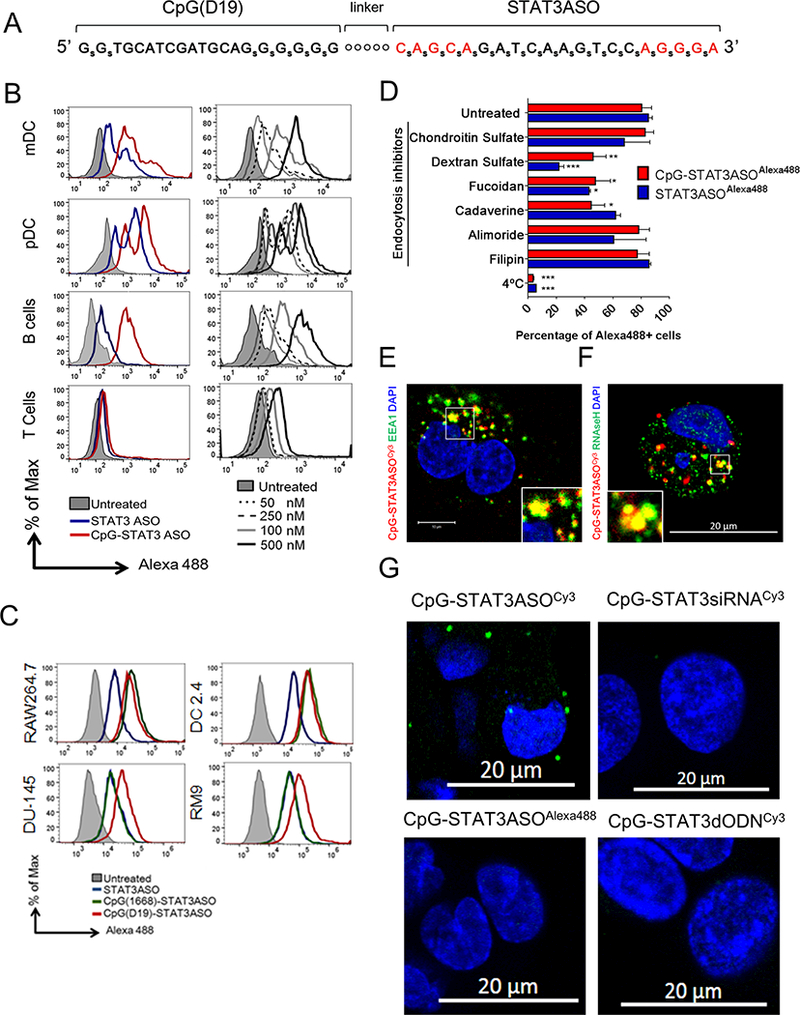
(A) Single-stranded CpG-STAT3ASO design; subscript “S” = phosphothioated nucleotides; “o” = C3 units of the carbon linker; red = 2’-O-methyl-modified nucleotides. (B, C) The in vitro uptake of CpG-STAT3ASOAlexa488 compared to STAT3ASOAlexa488 by: (B) primary human immune cells (pDC: CD303+, mDCs: CD1c+, B cells: CD19+, and T-cells: CD3+); (C) mouse dendritic (DC2.4) and macrophage (RAW264.7) cells, and prostate cancer cells (DU145 and RM9). Cells were incubated for 1 h with 500 nM (B-left panel, C) or with various concentrations (B-right panel) of CpG-STAT3ASOAlexa488 and STAT3ASOAlexa488 without any transfection reagents. Oligonucleotide uptake was measured cytofluorimetrically. (D) CpG-STAT3ASO is internalized by prostate cancer cells via scavenger receptor- and clathrin-dependent endocytosis. DU145 cells were pretreated using various endocytosis inhibitors or placed in 4°C for 1 h before incubation with CpG-STAT3ASOAlexa488 (250 nM) or STAT3ASOAlexa488 (750 nM) for another hour. The percentage of Alexa488-positive cells was assessed by flow cytometry; shown are means+SEM from three independent experiments. (E, F) Partial colocalization of CpG-STAT3ASO with early endosomes and with RNase H1 after cellular uptake. The confocal microscopy to visualize Cy3-labeled oligonucleotides and (E) early endosomal antigen 1 (EEA1) or (F) RNase H1 in prostate cancer cells (DU145) after 15 min and 4 h of incubation with 250 nM CpG-STAT3ASOCy3, respectively. (G) The direct interaction of CpG-STAT3ASOCy3 with RNAse H1 as measured by in situ proximity ligation assay and confocal microscopy. Cells were incubated with 250 nM CpG-STAT3ASOCy3 or other labeled control oligonucleotides for 4 h before the analysis; shown are representative images from one of three independent experiments.
Uptake of antisense oligonucleotides can occur through various surface receptors and endocytic mechanisms but some of these internalization pathways are “non-productive” as they do not result in target gene knockdown.(42) To identify these mechanisms, we compared the effect of endocytosis inhibitors on the uptake of fluorescently-labeled CpG-STAT3ASO vs. STAT3ASO by DU145 cells, at 250 nM and 750 nM, respectively, for comparable baseline level of uptake. Target cells were pre-incubated at reduced temperature (4°C) or in the presence of dextran sulfate, a general inhibitor of scavenger receptor (SR) mediated internalization, or with other more specific inhibitors of endocytosis, such as fucoidan, amiloride, cadaverine and filipin. We observed that both oligonucleotides underwent active and mainly SR-A-mediated internalization (Fig. 1D and Supplemental Fig.S3). However, the presence of CpG moiety enhanced clathrin-mediated uptake as indicated by the increased sensitivity to cadaverine, which is one of the “productive” uptake pathways for ASO molecules.(42) The improved internalization of CpG-STAT3ASO could be facilitated by the increased size and the additional phosphorothioate modifications of the conjugate.(43) When assessed using confocal microscopy, most of CpG-STAT3ASOCy3 was found located in early endosomes within 30 min of incubation (Fig. 1E). After 1 h, the endosomal signal of CpG-STAT3ASOCy3 decreased, which likely indicates cytoplasmic release of the conjugate (Supplemental Fig.S4).(40) At 4 h, CpG-STAT3ASOCy3 partially colocalized and directly interacted with RNase H1 in the cytoplasm as detected using confocal microscopy (Fig. 1F) and in situ proximity ligation assay (PLA) using antibodies specific to Cy3 and RNase H1 (Fig. 1G). In contrast, we did not detect interaction of the RNase H1 with control Cy3-labeled conjugates (CpG-STAT3siRNA or CpG-STAT3dODN), or with the Alexa488-labeled CpG-STAT3ASO (Fig. 1G).
CpG-STAT3ASO leads to accelerated STAT3 knockdown in target cells
The antisense oligonucleotides induces of RNase H1-dependent cleavage of the specific target mRNA, which is then degraded by cytoplasmic nucleases (44,45). To verify whether a CpG-STAT3ASO conjugate retains similar activity as the STAT3ASO alone, we tested STAT3 knockdown in several immune and prostate cancer cells (Fig. 2). Consistent with the pattern of uptake (Fig. 1C), both CpG-STAT3ASO and STAT3ASO induced similar levels of STAT3 knockdown in mouse immune cells, such as DCs and macrophages (Fig. 2A). Both oligonucleotides had similar activity also in mouse RM1/9 prostate cancer cells and to a lesser extent in PPS (Ptenpc−/− Trp53pc−/− Smad4pc−/−) cells,(6) likely due to poor internalization of both molecules (Fig. 2B and Supplemental Fig.S5A). At lower concentrations, CpG-STAT3ASO was clearly more effective in reducing STAT3 protein levels in mouse immune cells (Fig. 2C). The improved inhibitory effect of CpG-STAT3ASO was more pronounced in human prostate cancer cells such as LAPC4, CWR-22rv1 (Fig. 2D) and DU145 cells (Fig. 2E). In DU145 cells, CpG-STAT3ASO showed accelerated kinetics of STAT3 knockdown, which was already detectable at 8–12 h compared to 24 h required for STAT3ASO (Fig. 2E). At the protein level, CpG-STAT3ASO reduced STAT3 activation (Y705-phosphorylation) after 24 h and the total protein levels were significantly reduced after 48 h (Fig. 2F). Correspondingly, CpG-STAT3ASO showed increased cytotoxicity against STAT3-dependent prostate cancer cells such as DU145 and RM9 compared to STAT3ASO (Supplemental Fig.S6). Together, these results show that in vitro CpG-STAT3ASO conjugate shows better or at least comparable potency against human and mouse target cells compared to STAT3ASO.
Figure 2. STAT3 knockdown in human and mouse target cells in vitro after treatment with CpG-STAT3ASO or STAT3ASO alone.
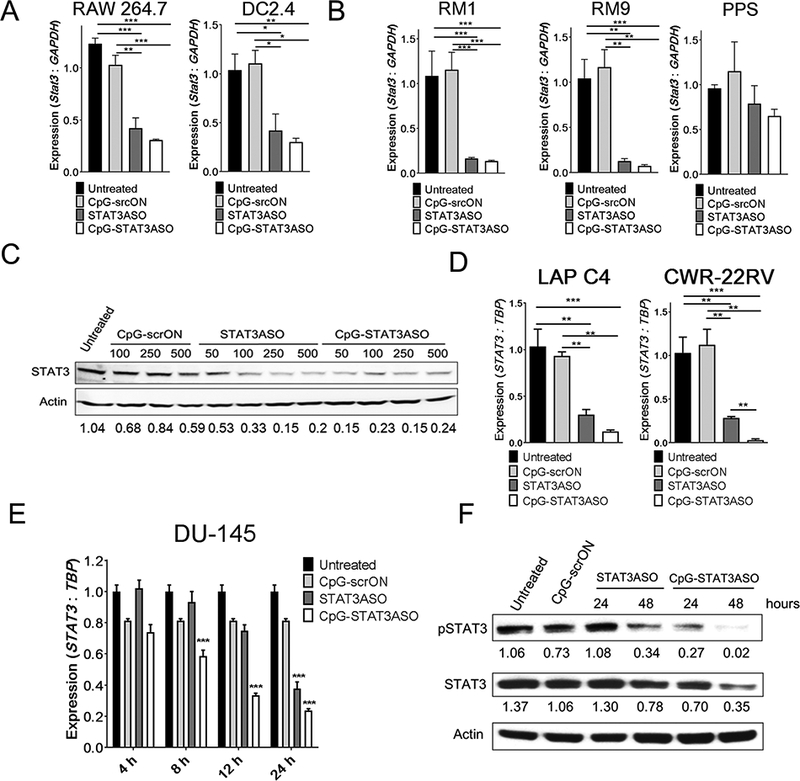
(A-B) CpG-STAT3ASO induces STAT3 knockdown in mouse myeloid (A) and prostate cancer (B) cells. Cells were treated using 500 nM CpG-STAT3ASO, STAT3ASO or control CpG-scrON for 18 h. The STAT3 mRNA levels were assessed using qPCR; means+SEM from one of three independent experiments performed in triplicates. (C) Dose-dependent STAT3 inhibition in mouse splenocytes derived from RM9-tumor bearing mice. Splenocytes were treated ex vivo with the indicated dose of CpG-STAT3ASO, STAT3ASO or CpG-scrON for 48 h and evaluated by Western blotting, with normalization to β-actin. Shown are the representative results from one of three independent experiments. (D) CpG-STAT3ASO reduces STAT3 expression in human CRW-22rv1 and LAPC4 prostate cancer cells. Cells were treated using 500 nM CpG-STAT3ASO, STAT3ASO or control CpG-scrON for 18 h. The STAT3 mRNA levels were assessed using qPCR; shown are means+SEM from one of three independent experiments performed in triplicates. (E, F) Time-dependent STAT3 knockdown by CpG-STAT3ASO vs. STAT3ASO in human DU145 cells at mRNA (E) or protein levels (F) as assessed using qPCR or Western blot, respectively; shown are means+SEM from one of three independent experiments. The relative STAT3 band intensities normalized to β-actin are indicated.
Local administration of CpG-STAT3ASO triggers systemic antitumor effects in two genetically distinct mouse prostate cancer models
As noted, STAT3 serves multiple context-specific roles in tumor biology.(22) We first assessed local and systemic antitumor effects of CpG-STAT3ASO vs. STAT3ASO or CpG immunostimulation alone, using the syngeneic Ras/Myc-driven (Pten intact) RM9 prostate cancer model, which generates potently immunosuppressive tumor microenvironment.(46) In vitro, RM9 cancer cells showed sensitivity to STAT3 inhibition (Supplemental Fig.S6B). Mice with RM9 tumors engrafted subcutaneously (SC) into opposite flanks were treated using intratumoral (IT) injections of either CpG-STAT3ASO, CpG-scrambled conjugate (CpG-scrON), STAT3ASO alone (5 mg/kg each) or vehicle only into only one tumor site. As shown in Figure 3A, both CpG-STAT3ASO and STAT3ASO induced comparable STAT3 knockdown. Both treatments inhibited growth of RM9 tumors in the locally treated site but the antitumor effect of STAT3ASO was transient and followed by tumor regrowth (Fig. 3B). The control CpG-srcON conjugate, which is immunostimulatory but non-targeting, did not affect growth of the potently immunosuppressive RM9 tumors (Fig. 2B).(25) Only CpG-STAT3ASO reduced tumor progression in the distant/non-injected site, which suggests generation of systemic antitumor effects (Fig. 3C).
Figure 3. Local administration of CpG-STAT3ASO triggers systemic antitumor immunity against two genetically distinct mouse prostate cancer models.
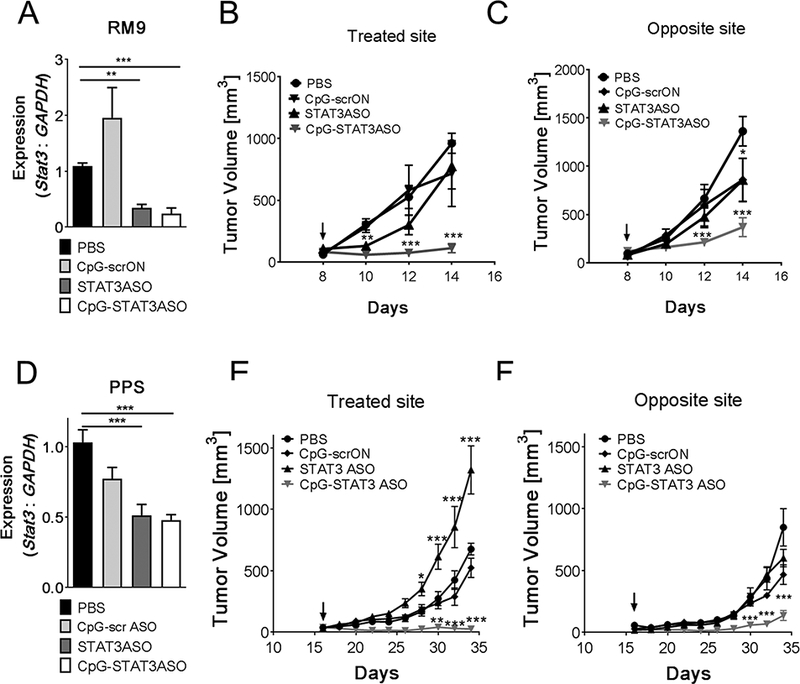
C57BL/6 mice were injected subcutaneously at two sites with mouse syngeneic RM9 (A-C) or PPS (D-F) prostate cancer cells to generate dual tumor models. After tumors were established, one site was injected every other day intratumorally using 5 mg/kg of indicated oligonucleotides; the arrows indicate treatment initiation. (A, D) STAT3 mRNA levels were assessed using real-time qPCR at the end of the experiment; means+SEM (n = 6). RM9 and PPS tumor growth kinetics measured at the treated (B, E) and at the distant (C, F) tumor sites; means±SEM (n = 12). Shown are results combined from three independent experiments.
Next, we compared antitumor efficacy of these oligonucleotides in the genetically distinct, PPS (Ptenpc−/−Trp53c−/−Smad4pc−/−) metastatic prostate cancer model, which is engineered with mutations commonly found in human tumors.(6) In contrast to RM9, our preliminary in vitro studies found that PPS cells were resistant to STAT3 knockdown and did not reduce their proliferation and viability (Supplemental Fig.S5B and S5C). As before, PPS tumors were engrafted SC in mice on both flanks but only tumors in the left site were treated. Local injections of CpG-STAT3ASO and STAT3ASO alone reduced STAT3 expression in whole tumors although less efficiently than in RM9 tumors (Fig. 3A and Fig. 3D). In the PPS model, only CpG-STAT3ASO abrogated tumor growth in both treated and distant locations, with complete rejection of 7 out of 10 in primary and 3 out of 10 in distant site. Moreover, in the treated site, STAT3ASO injections seemed to stimulate tumor progression (Fig. 3E-F). Tumor-promoting effect of STAT3ASO at high local oligonucleotide concentrations may indicate the role of STAT3 in regulating cancer cell senescence in PTEN-deficient prostate tumors by yet unclear p53-independent molecular mechanism.(22) The inhibitory effect of control CpG-scrON was minimal, indicating failure of TLR9 stimulation alone to overcome tolerogenic effects of the tumor microenvironment. Importantly, our results suggest that the concomitant STAT3 inhibition and TLR9 stimulation are necessary for the generation of systemic antitumor immune effects against prostate tumors, independently from the intrinsic sensitivity of prostate cancer cells to STAT3 inhibition.
Generation of immune responses against prostate tumors requires combination of STAT3 inhibition with TLR9 stimulation
Prostate tumors are known for harboring potently tolerogenic microenvironments, which thwarts antitumor immunity. Thus, we assessed whether CpG-STAT3ASO administration was capable of alleviating tumor immunosuppression and stimulating systemic T-cell activity. Previous studies from our group and others documented the critical role of tumor-associated myeloid cells, and specifically polymorphonuclear-MDSCs (PMN-MDSCs), in sustaining prostate cancer immune evasion.(6,25,27–29,47) We used flow cytometry to characterize changes in immune cell populations infiltrating RM9 or PPS tumors after local IT administration of either 5 mg/kg CpG-STAT3ASO conjugates, STAT3ASO alone, control CpG-scrON or vehicle as before. The immunophenotypic analysis revealed treatment-dependent differences in the composition of the tumor microenvironment in the two tumor models (Fig. 4 and Supplemental Fig.S7). The untreated RM9 tumors showed low level of infiltration by immune cells, mainly immature myeloid cells with almost undetectable levels of T-cells compared to PPS tumors (Fig. 4A and Fig. 4B). Only CpG-STAT3ASO treatment, but not CpG-scrON or STAT3ASO alone, significantly altered the RM9 microenvironment by inducing recruitment of CD11b+Ly6G+ myeloid cells into primary tumors and CD8 T-cells into the distant tumor site (Fig. 4A). Immunohistochemical analysis indicated that these CD11b+Ly6G+ cells represented activated Ly6B.2+ neutrophils rather than PMN-MDSCs (Fig. 4C). This observation corresponds to our original findings of increased neutrophil activity in mice with hematopoietic cell-selective Stat3-ablation, especially in response to local TLR9 stimulation.(48,49) In fact, the CD11b+Ly6G+ myeloid cells infiltrating RM9 tumors in CpG-STAT3ASO-treated mice showed reduced phospho-STAT3 (pSTAT3) levels compared to mice from other treatment groups as assessed by intracellular staining and flow cytometric analysis (Fig. 4D). Although STAT3ASO decreased STAT3 activity in the treated tumor site as assessed by immunohistochemistry (Fig. 4C), it did not significantly inhibit STAT3 in CD11b+Ly6G+ myeloid cells recruited into tumors (Fig. 4D). STAT3 is known to be a direct upstream regulator of PDL1 transcription in human or mouse monocytes and cancer cells.(50,51) Consistent with the level of STAT3 inhibition, injections of CpG-STAT3ASO, but not STAT3ASO, reduced PD-L1 levels by ~80% in RM9 tumor-associated CD11b+Ly6G+ cells (Fig. 4E).
Figure 4. STAT3-inhibition combined with TLR9-stimulation is crucial for disrupting tolerogenic prostate tumor microenvironment and for immune cell recruitment.
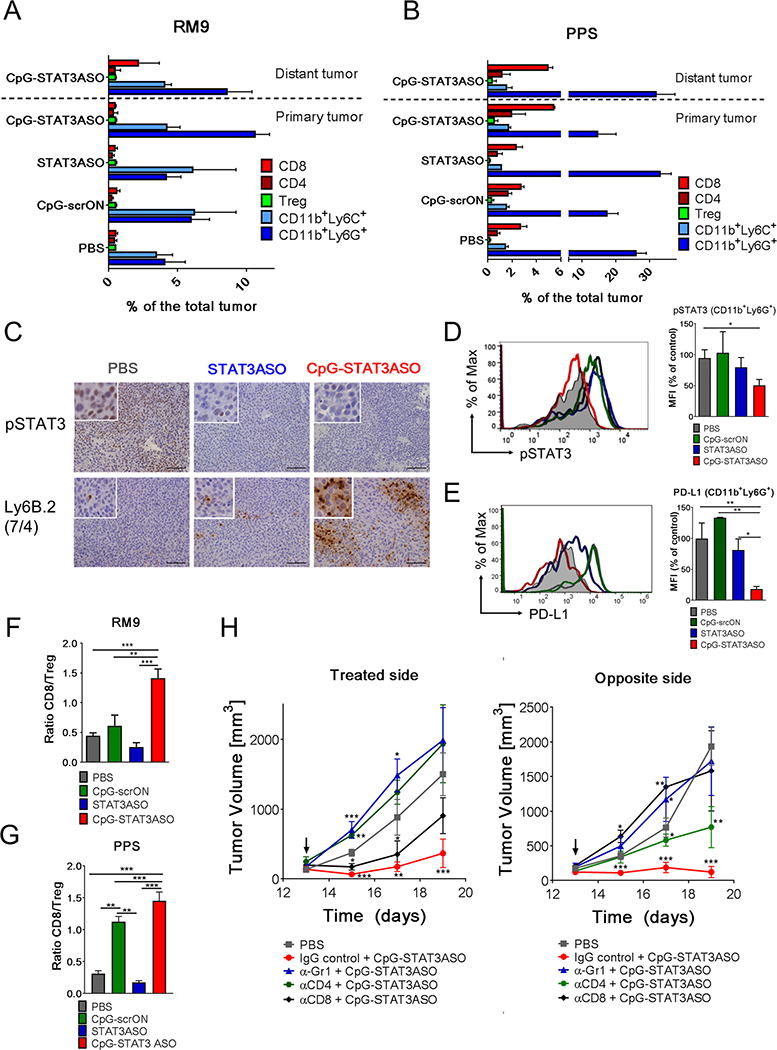
Dual tumor models were established as described in Fig. 3. The left tumor site was injected intratumorally using 5 mg/kg of CpG-STAT3ASO, STAT3ASO or CpG-srcON every other day. (A, B) Immunophenotypic analysis showing differences in composition of the tumor microenvironment in RM9 (A) and PPS (B) tumors in both locations during the experiment. The percentages of immune cell populations, such as granulocytic (CD11b+Ly6G+Ly6CLO) and monocytic (CD11b+Ly6G–Ly6C+) myeloid cells, CD3+CD8+ T-cells, CD3+CD4+FoxP3– T-cells or CD3+CD4+FoxP3+ Tregs infiltrating tumors were measured using flow cytometry; means+SEM (n = 6/each treatment group). The detailed gating strategy is presented in the Supplemental Fig.S7. (C) STAT3 inhibition (top row) and recruitment of activated neutrophils (Ly6B.2+; clone 7/4) (bottom row) were assessed using immunohistochemical staining in treated tumors. (D) Activation of pSTAT3 and expression of PD-L1 (E) in CD11b+Ly6G+Ly6CLO cells isolated from RM9 tumors after oligonucleotide treatments. pSTAT3 and PD-L1 expression levels in the tumor and in the tumor-associated CD11b+Ly6G+Ly6CLO were assessed using flow cytometry; means+SEM. (n = 6). (F, G) Ratio of CD8 T-cell (CD3+CD8+) to Tregs (CD3+CD4+FOXP3+) in treated RM9 (F) and PPS (G) tumors as assessed using flow cytometry. Shown are ratios of CD8+ T-cells to Tregs; means+SEM (n = 6).
We next compared effects of locally administered (IT) oligonucleotides on the PPS tumor microenvironment, which is potently tolerogenic due to high percentage of tumor-associated PMN-MDSCs.(6,47) Local IT injections of CpG-STAT3ASO reduced the population of putative PMN-MDSCs, CD11b+Ly6G+Ly6CLO granulocytic myeloid cells, in treated tumors from 26% to 14% on average, while stimulating recruitment of CD8+ T-cells to both tumor sites (Fig. 4B). Unexpectedly, treatment with STAT3ASO alone had the opposite effect. It increased the percentage of CD11b+Ly6G+Ly6CLO myeloid cells in treated site but failed to recruit CD8+ T-cells into tumors (Fig. 4B).(6,47) The expansion of tumor-associated myeloid cells by the local STAT3ASO treatment could support the unexpected effect of the accelerated progression of primary PPS tumors (Fig. 3E). The effect of CpG-scrON on myeloid and T-cell populations was negligible (Fig. 4B), indicating failure of TLR9 stimulation alone to counteract the immunosuppressive effect of the tumor microenvironment. While inhibiting tolerogenic CD11b+Ly6G+ cells, CpG-STAT3ASO also augmented activation of dendritic cells (DCs) in tumor-draining lymph nodes as detected by the elevated levels of major histocompatibility complex (MHC) class II, as well as CD40 and CD80 co-stimulatory molecules and decreased percentage of regulatory T-cells (Treg) (Supplemental Fig.S8AB). Further flow cytometric analysis confirmed that that CpG-STAT3ASO and to a lesser extent CpG-scrON, but not STAT3ASO, increased percentage of CD8+ T-cells while reducing Tregs in distant tumors in both RM9 and PPS models. The significant rise of CD8:Treg ratio is a critical indicator of successful generation of systemic T-cell-mediated response (Fig. 4F and Fig. 4G).
To further assess the contribution of granulocytic myeloid cells and T-cell lymphocyte populations in the antitumoral immune responses induced by CpG-STAT3ASO, we individually depleted Gr1+, CD4+ or CD8+ cells in RM9 tumor-bearing mice. Following the antibody-mediated neutralization, we treated the mice using 5 mg/kg of CpG-STAT3ASO at the left tumor site, as before. The CD8-depletion partly impaired antitumor CpG-STAT3ASO activity against treated tumors, while completely eliminating any effects against distant tumors (Fig. 4H). The depletion of Gr1+ and CD4+ cells had a strong negative impact on the antitumor effects of CpG-STAT3ASO against tumors in both treated and the distant sites (Fig. 4H). These results suggest that only the combination of STAT3 inhibition with TLR9 stimulation had potential to disrupt tumor immune evasion while effectively engaging multilayered cellular immune network, thereby combining both innate, neutrophil-dependent, and adaptive, T-cell-mediated antitumor immunity.
Intravenous injections of CpG-STAT3ASO can eradicate bone-localized prostate tumors
The potential to induce systemic antitumor immune responses using CpG-STAT3ASO conjugates, prompted us to test the feasibility of using this strategy to treat metastatic disease. First, we compared biodistribution of CpG-STAT3ASO vs. STAT3ASO alone after systemic administration into tumor-bearing mice. Mice with established intratibial RM9 tumors were injected intravenously (IV) using 2.5 mg/kg of fluorescently Cy3-labeled oligonucleotides. Cellular biodistribution of both oligonucleotides was measured using flow cytometry in various immune cell populations, such as DCs, MACs, total MDSCs (CD11b+Gr1+) and T-cells in bone marrow and spleen after 3 h. As shown in Figure 5A, CpG-STAT3ASO was internalized by >80% of MDSCs and ~45% of DCs in the bone marrow and at a lower percentage (~30%) in the spleen but not by T-cells in any of the tested organs. While STAT3ASO showed similar biodistribution pattern, it penetrated a significantly lower percentage of MDSCs (~65%) and DCs (~30%) in the bone marrow. Given the superior uptake of ASOs by bone marrow-localized myeloid cells, we decided to test their efficacy against prostate tumors in the clinically relevant localization.(52) Mice were injected intratibially using luciferase-expressing RM9-luc cancer cells. After tumors were established, as verified using bioluminescent imaging (BLI), mice were IV injected with every other day using 5 mg/kg CpG-STAT3ASO, STAT3ASO, CpG-scrON or PBS. The levels of STAT3 activity were assessed in tumor sections using immunohistochemistry and flow cytometry one day after the third injection. Repeated systemic administration of CpG-STAT3ASO strongly reduced the overall pSTAT3 levels in bone-localized RM9 tumors in contrast to both control treatments (Fig. 5B). Flow cytometric analysis indicated reduction of pSTAT3 levels in various tumor-infiltrating immune cell populations, such as DCs and MDSCs after IV treatment using CpG-STAT3ASO and STAT3ASO (Fig. 5C). Consistent with the lack of oligonucleotide uptake, STAT3 activity was not reduced but slightly elevated in tumor-resident T-cells after CpG ODN and CpG-STAT3ASO treatments (Fig. 5C). It remains to be tested whether such STAT3 upregulation could indicate CD8 T-cells activation and expansion.(53)
Figure 5. Systemic administration of CpG-STAT3ASO induces regression of bone-localized mouse prostate tumors in immunocompetent mice.
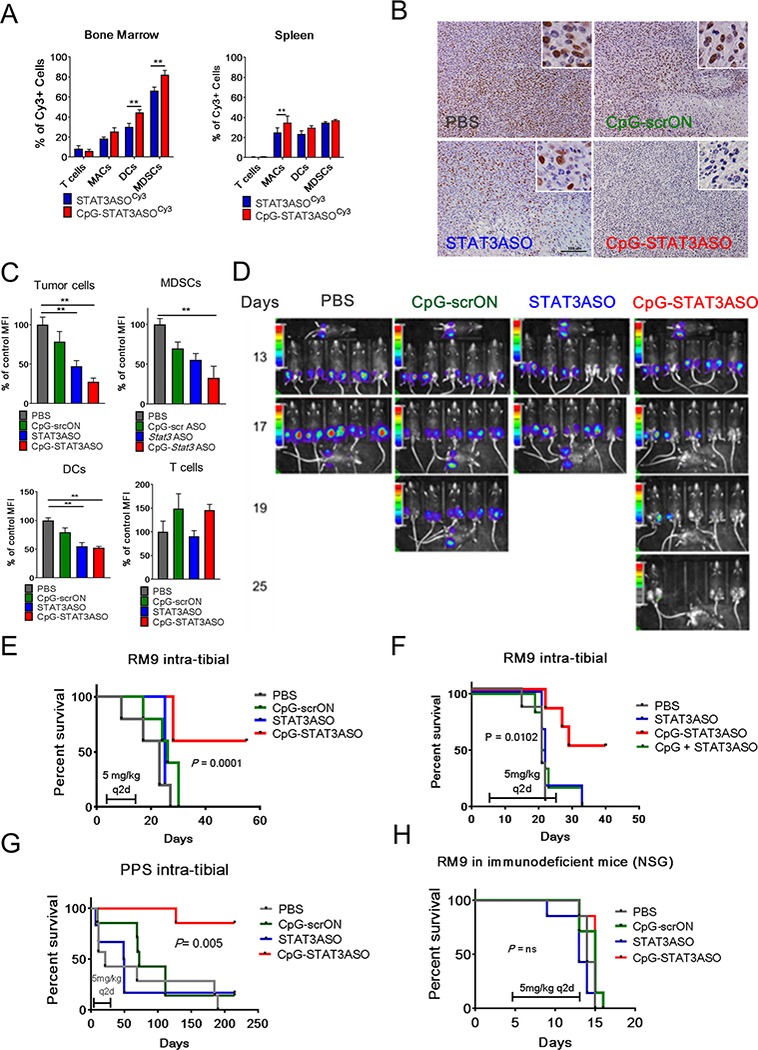
C57BL/6 mice were injected intratibially using RM9 or PPS prostate cancer cells. (A) Biodistribution of systemically injected CpG-STAT3ASOCy3 and STAT3ASOCy3 in RM9 tumor-bearing mice. Mice were injected IV using 2.5 mg/kg of either oligonucleotide and euthanized 3 h later. Percentages of Cy3+ T-cells (CD3+), macrophages (CD11b+F4/80+), DCs (CD11b+CD11c+) and MDSCs (CD11b+/Gr1+) were assessed using flow cytometry in single-cell suspensions of bone marrow or spleen. Results of two independent experiments using a total of 6 mice analyzed individually; means+SEM. (B-C) Systemic administration of CpG-STAT3ASO reduces STAT3 activation in bone-localized prostate tumors and in the tumor-associated immune cells. After tumors were established, mice were treated using IV injections (q2d) of 5 mg/kg of indicated oligonucleotides. After the third treatment, mice were euthanized and pSTAT3 activation was assessed in the tumors using immunohistochemistry (B) and flow cytometry (C) in tumor cells (LSCHISCCHICD11b-CD3-), MDSCs (CD11b+/Gr1+), DCs (CD11b+CD11c+), and T-cells (CD3+). C57BL/6 (D-G) or NSG (H) mice were intratibially injected using RM9-Luc or PPS-Luc prostate cancer cells. After tumors were established mice were treated using IV injections (q2d) of 5 mg/kg of indicated oligonucleotides. (D) Tumor progression was monitored using bioluminescent imaging on the AmiX (Spectral Instruments). (E) Repeated systemic administration of CpG-STAT3ASO induces regression of bone-localized tumors and increases the overall survival of mice. Shown are combined results from two independent experiments (n = 12 mice/each group). (F) Co-injection of CpG ODN and STAT3ASO fails to reproduce the efficacy of the bi-functional CpG-STAT3ASO conjugate against bone-localized RM9-Luc tumors (n = 6 mice/each group). (G) Systemic administration of CpG-STAT3ASO induced tumor regression in the bone-localized Pten-deficient tumor model (PPS-Luc). Results were combined from two independent experiments (n = 12 mice/each group). (H) The antitumor effect of CpG-STAT3ASO depended on the presence of an intact immune system and cannot be achieved in immunodeficient NSG mice (n = 6 mice/each group).
Based on these results, we decided to compare the efficacy of CpG-STAT3ASO, STAT3ASO alone and control CpG-scrON against bone-localized RM9 prostate tumors. Mice with established (day 13) intratibial RM9-luc tumors were treated every other day using IV injections of 5 mg/kg of above mentioned oligonucleotides or PBS for a total of six injections. As shown in Figure 5D, tumors progressed rapidly in all control experimental groups except for CpG-STAT3ASO. Already after the second injection (day 15), CpG-STAT3ASO treatment halted tumor progression and within the following week tumors regressed in the majority of treated mice (Fig. 5D). The surviving CpG-STAT3ASO-treated mice remained tumor free until the end of experiment at 58 days, while survival of mice in other treatment groups did not exceed 30 days (Fig. 5E). Importantly, the coinjection of unconjugated CpG ODN together with STAT3ASO failed to significantly extend animal survival (Fig. 5E and Fig. 5F).
Our earlier results (Fig. 5B) indicated that the efficacy of systemic administration of CpG-STAT3ASO could depend on direct STAT3 targeting in RM9 prostate cancer cells as well as in myeloid cells in the tumor microenvironment. In contrast, the PPS tumor model allows for more selective analysis of cancer cell-extrinsic effects of tested oligonucleotides due to the resistance of cancer cells to direct cytotoxic effect of the conjugate (Supplemental Fig.S5), Mice with established intratibial, luciferase expression PPS tumors (PPS-luc) were injected IV every other day using CpG-STAT3ASO, STAT3ASO alone and CpG-scrON (5 mg/kg) or PBS and monitored using BLI as before. Treatment with CpG-STAT3ASO led to complete regression of PPS tumors and resulted in tumor-free survival of the 90% of treated mice for up to 230 days after engraftment (Fig. 5G). None of other treatments resulted in statistically significant extended animal survival compared to PBS group. Finally, to assess the contribution of immune responses to antitumor effects of CpG-STAT3ASO, we engrafted RM9-luc tumors into the tibia of immunodeficient NOD/SCID/Il2rγKO (NSG) mice. Similarly as before, after tumors were established as verified by BLI, mice were treated every other day using IV injections of 5 mg/kg of CpG-STAT3ASO, STAT3ASO alone, CpG-scrON or PBS. As shown in the Fig. 5H, in the absence of functional immune cells CpG-STAT3ASO injections failed to control RM9 tumor progression and did not extend mice survival. These results suggested that presence of functional immune cells is critical for the antitumor efficacy of CpG-STAT3ASO. In agreement with these results, flow cytometric analysis confirmed that IV injections of CpG-STAT3ASO, but not control oligonucleotides, improve about four-fold the ratio of effector to regulatory T-cell populations in the RM9 prostate tumor microenvironment (Supplemental Fig.S8C-E). These data further underscore therapeutic potential of combining systemic disruption of STAT3-mediated prostate tumor immune evasion with concomitant CpG immunostimulation in order to unleash antitumor immunity against bone-localized prostate cancers.
STAT3-inhibition combined with TLR9-stimultation alleviates tolerogenic activity of PMN-MDSCs from human prostate cancers
We previously described accumulation of potently immunosuppressive population of TLR9+ PMN-MDSCs in prostate cancer patients during disease progression.(28) Since human PMN-MDSCs rely on STAT3 signaling for their tolerogenic effects on T-cells, we tested whether CpG-STAT3ASO can target human PMN-MDSCs. We first assessed the spontaneous uptake of CpG-STAT3ASO and unconjugated STAT3ASO by primary PBMCs from patients with advanced prostate cancers. Within 1 h of incubation with fluorescently-labeled oligonucleotides, 80–90% of PMN-MDSCs (Lin-HLA-DR-CD15+) internalized both CpG-STAT3ASOAlexa488 and STAT3ASOAlexa488, although the intracellular levels of the CpG-STAT3ASO conjugate were significantly higher as indicated by mean fluorescent intensity (Fig. 6A). Subsequently the level of target gene silencing was assessed in CD15+ PMN-MDSCs after 72 h incubation with CpG-STAT3ASO, STAT3ASO or CpG-scrON using real-time PCR. The results showed >90% reduction in STAT3 mRNA for both STAT3 targeting oligonucleotides compared to controls (Fig. 6B). Given the comparable effect of CpG-STAT3ASO and STAT3ASO oligonucleotides on STAT3 knockdown ex vivo, we assessed the effect of both treatments and control CpG-scrON on the immunosuppressive properties of PMN-MDSCs. CD15+ PMN-MDSCs enriched from blood of advanced prostate cancer patients were pre-incubated for three days in the presence of 500 nM of CpG-STAT3ASO, STAT3ASO or CpG-scrONs. Viable PMN-MDSCs were then co-cultured (3:1) with allogeneic CD3+ T-cells and CD3/CD28 co-stimulation for additional 3 days. As indicated in Figure 6C, only the CpG-STAT3ASO conjugate was able to alleviate the multiple immunosuppressive effects of PMN-MDSCs on CD3+ T-cells proliferation The CpG-STAT3ASO conjugate also stimulated the production of granzyme B and IFN-γ by CD8+ T-cells (Fig. 6DE). Neither STAT3ASO alone nor immunostimulatory CpG-scrON resulted in significant improvement of CD8+ T-cells activity. Therefore, we conclude that, as in mouse tumor models, overcoming the MDSC-dependent immunosuppression in the prostate cancer microenvironment requires the concomitant STAT3 inhibition and TLR9 stimulation.
Figure 6. TLR9-stimulation combined with STAT3-inhibition alleviates tolerogenic activity of PMN-MDSCs from prostate cancer patients.
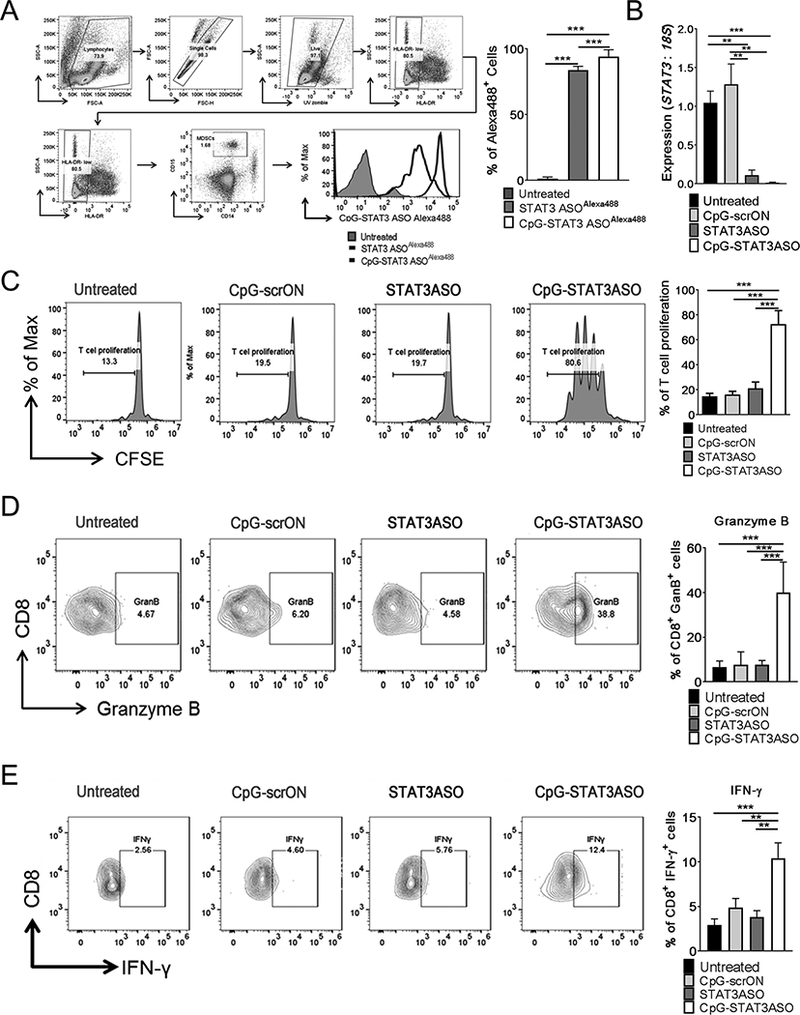
(A) CpG(D19)-STAT3ASO is efficiently internalized by human HLA-DR–CD16–CD15+ prostate cancer-associated PMN-MDSCs. Peripheral blood monocytes isolated from patients with advanced prostate cancers were incubated with 250 nM of fluorescently-labeled CpG(D19)-STAT3ASOAlexa488 conjugate or unconjugated STAT3ASOAlexa488 for 1 h without any transfection reagents. Percentages of Alexa488+ PMN-MDSCs were assessed using flow cytometry. The gating strategy (left panels) and bar graph (right) combining results from 6 individual patient’s samples; means+SD. (B) STAT3 knockdown in PMN-MDSCs treated using CpG(D19)-STAT3ASO or STAT3ASO alone. The CD15+ PMN-MDSCs enriched from prostate cancer patients’ PBMCs were treated using 500 nM of CpG-STAT3ASO, STAT3ASO or CpG-scrON. The levels of STAT3 mRNA were assessed at 18 h using real-time qPCR; shown are means+SD (n = 6). (C-E) Prostate cancer-associated PMN-MDSCs were treated with 500 nM of CpG-STAT3ASO, STAT3ASO or control CpG-scrON for 72 h and then co-cultured with allogeneic CD3+ T-cells at 3:1 ratio with anti-CD3/CD28 co-stimulation. Flow cytometry was used to determine T-cell proliferation using CFSE dilution assay (C), and percentages of IFNγ- (E) or granzyme B-producing (F) CD8+ T-cells. Dot plots from a representative sample (left) and bar graphs with combined results from all tested patients’ samples (right) are shown as means+SD (n = 12).
DISCUSSION
Here, we demonstrate the feasibility of using bi-functional oligonucleotide strategy to overcome de novo therapeutic resistance of potently immunosuppressive and immunologically “cold” prostate tumors. The combination of targeted STAT3 inhibition and TLR9 immunostimulation was required to disrupt myeloid cell-dependent immunosuppression and to unleash antitumor immune responses, eradicating aggressive, bone-localized prostate tumors. Importantly, we showed that neither treatment with STAT3ASO or CpG ODN as single treatments, nor their co-injection were sufficient for tumor eradication. The combination of STAT3 inhibitor and TLR9 agonist in a single molecule maximized treatment efficacy by several mechanisms. First, it enhanced and accelerated cell-selective uptake of systemically administered oligonucleotide, thereby improving STAT3 knockdown. Secondly, TLR9 immunostimulation combined with the STAT3 inhibition resulted in the “push & release” effect, essential for jump-starting antitumor immunity. As shown by our studies comparing the intratumoral injections of CpG-STAT3ASO vs. STAT3ASO alone, even when STAT3 knockdown levels were comparable, only the CpG-STAT3ASO conjugate had potential to overcome tumor immune tolerance and induce systemic antitumor immune responses. This is consistent with our previous report, which demonstrated that combined STAT3-inhibition/TLR9-stimulation specifically in myeloid cells and not in cancer cells is sufficient for eradication of solid tumors through efficient recruitment of innate and adaptive effector cells, such as neutrophils and CD8/CD4 T-cells.(49)
CpG-STAT3ASO strategy provides an opportunity for the development of immunotherapies broadly applicable to therapy of genetically diverse prostate cancers. As recently described, Myc oncogene expression as well as combined deletion of Pten/Smad4 or Pten/Tp53, cause expansion of tumor-associated macrophages and MDSCs, and thereby promote tumor immune tolerance and vascularization.(6,8,47) Both Ras/Myc-driven as well as Pten deletion-dependent prostate tumors were sensitive to CpG-STAT3ASO-induced immune responses, even though cancer cells in both models differed dramatically in their intrinsic sensitivity to STAT3 inhibition. As discussed above, due to pharmacokinetic properties, systemic administration of CpG-STAT3ASO had limited direct effect on cancer cells, as indicated by the lack of significant antitumor activity against STAT3-dependent RM9 tumors in immunodeficient mice. This is of benefit considering that STAT3 may gain tumor suppressor function in Pten-loss prostate tumors.(15,22) STAT3 inhibition in Pten-deficient cancer cells interfered with the induction of p53-dependent cell senescence, promoting tumor growth in vivo. The transplantable PPS (Ptenpc−/−Trp53c−/−Smad4pc−/−) model of castration-resistant prostate tumors used in our studies lacks p53. Therefore, the accelerated PPS tumor progression after local STAT3ASO treatment suggests that STAT3 may have additional p53-independent tumor suppressor functions in Pten-deficient prostate cancers.(6) Our future studies will interrogate molecular mechanisms underlying these effects, which can be of concern for a number of patients with PTEN-deficient prostate cancers. Importantly, in contrast to STAT3ASO alone, CpG-STAT3ASO showed consistent if not higher antitumor activity against Pten-deficient prostate tumors, as observed in Ras/Myc-driven tumors. These results underscore broad therapeutic potential of targeting myeloid cells in the prostate tumor microenvironment, which likely reflects the fundamental role of myeloid cell-dependent immune evasion for tumor progression.(31)
To date, prostate cancers have proven to be difficult targets for emerging immunotherapies, including cancer vaccines and immune checkpoint blockade.(5,9) Recent studies highlighted the role of the tumor microenvironment, especially MDSCs, in shielding prostate cancers from antitumor immunity.(25,31,47) Lu et al. demonstrated in mouse tumor models that targeting MDSCs, predominantly of the PMN-MDSCs subtype, can augment the modest efficacy of immune checkpoint inhibitors against mCRPC.(47) We recently found that potently tolerogenic TLR9+ PMN-MDSCs, with high levels of STAT3 activity, accumulate in blood of prostate cancer patients with progression of the disease.(28) STAT3 activation in PMN-MDSCs correlated with elevated plasma levels of IL-6-type cytokines, such as LIF, suggesting a potential cross-talk mechanism promoting tumor immune evasion.(25) IL-23, another cytokine and STAT3-regulated target in tumor-associated myeloid cells,(54) was recently identified as a potential driver of castration-resistant prostate cancers.(55) Both RM9 and PPS prostate tumor models replicate well these clinical findings and show high percentage of pSTAT3+ tumor-infiltrating PMN-MDSCs.(6,25,47) STAT3 activity in these tumor-infiltrating MDSCs correlated with elevated levels of PD-L1, a known a STAT3 target gene (50,51) and a key immune checkpoint regulator.(56,57) However, targeting MDSCs in clinical setting remains a challenge. The lack of distinct surface markers for these immature myeloid cells complicates their neutralization using antibodies. Recent studies focused on small molecule drugs inhibiting immunosuppressive functions of MDSCs. Blocking Jak/STAT3 signaling using broadly specific tyrosine kinase inhibitors, such as sunitinib or cucurbitacin B, or in vitro inhibition of STAT3 was shown to alleviate tolerogenic MDSCs activity.(30,58) PI3K inhibitors, such as BEZ235 and cabozantinib (HGF/PI3K), can also overcome MDSC-mediated immunosuppression, while promoting innate immune responses against prostate tumors.(47,59) At the same time, targeting signaling molecules across the immune network can result in contradictory outcomes in various immune cell populations. This is of concern for both PI3K/mTOR and Jak/STAT3 inhibitors due to complex role of these target pathways in T-lymphocytes. For example, STAT3 seems to reduce antitumor activity of CD8 T-cells and expand tumor-promoting Th17 lymphocytes but it is also indispensable for the generation of memory T-cells and long-term antitumor immunity.(60–62) Targeting Jak1/2 kinases upstream from STAT3 was shown to reduce numbers of MDSCs, while paradoxically increasing their immunosuppressive activity and blocking T-cells proliferation.(63) Similarly, PI3K and PI3K/mTOR inhibitors can interfere with T-cell activation and induce tolerance.(64) Therefore, targeting tolerogenic signaling in tumor-associated myeloid cells demands cell-selective strategies to avoid potential adverse effects and toxicities, while maximizing therapeutic efficacy.
Oligonucleotide therapeutics (ONTs) provide an alternative and clinically relevant strategy for targeting tumor-associated myeloid cells.(29) The sensitivity of innate immune cells to ONTs has long been a major obstacle to the clinical development of these reagents beyond oncology. Paradoxically, the myeloid cells may be essential cellular targets for cancer immunotherapy. This is best exemplified by the recently reported results from the phase I clinical trial on STAT3ASO molecule (AZD9150) in patients with various relapsed/refractory tumors.(36,65) Similar as in preclinical models, IV injected STAT3ASO was found to primarily target non-malignant, stromal cells rather than cancer cells. Importantly, non-autonomous effects of STAT3ASO in tumor cells were likely essential for the observed antitumor effects.(65,66) Consistent with our preclinical data, partial clinical responses to STAT3ASO alone occurred in patients with B-cell lymphoma but not with the advanced prostate cancer.(36) The limited penetration of the STAT3ASO into tumors was likely a result of the slow uptake kinetic and the limited circulatory half-life of ASOs due to renal clearance rather than degradation. Targeted and rapid cellular internalization of CpG-STAT3ASO can improve therapeutic efficacy in vivo compared to the unconjugated STAT3ASO even at significantly lower dosing.(36) The new types of chemical modification of ASO molecules, such as locked nucleic acids, are likely to further improve the potency, while reducing potential toxicities of these ONTs. Both CpG ODNs and STAT3ASO molecules were well tolerated by patients, when tested as single agents in clinical trials.(36,67) The most common adverse effects for both reagents were flu-like symptoms likely related to production of interferons and proinflammatory cytokines. In addition, the whole class of PS-modified ONTs can potentially trigger platelet activation and thrombocytopenia at high dosing. In case of CpG-STAT3ASO, such risks are mitigated by the significantly lower effective dosing and reduced extent of PS-modifications compared to unconjugated and fully phosphorothioated ASOs. The preclinical studies to fully assess safety and pharmacokinetic/pharmacodynamic properties of CpG-STAT3ASO are ongoing. We believe that STAT3-inhibition/TLR9-stimulation targeted to myeloid-cells using a single oligonucleotide agent can provide broadly applicable, yet effective and safe strategy to overcome resistance of metastatic prostate cancers to immunotherapy.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Prostate cancers have proven resilient to emerging clinical immunotherapies, including cancer vaccines and immune checkpoint blockade. Growing evidence emphasizes the role of the tolerogenic tumor microenvironment, with essential role of STAT3 signaling in myeloid cells, in shielding prostate cancers from antitumor immunity. Here, we describe new bi-functional CpG-STAT3ASO molecules, which trigger TLR9 immunostimulation while eliminating negative effect of STAT3. Despite known challenges in penetrating solid tumors by intravenously injected oligonucleotides, CpG-STAT3ASO effectively targeted TLR9+ myeloid cells in bone-localized prostate tumors, resulting in immune-mediated eradication of tumors regardless of their genetic background. Our study highlights the potential of using CpG-ASO strategy to target other “undruggable” master regulators of tumorigenic functions of myeloid cells. We believe that these findings, underscored by the ongoing IND-enabling studies of first generation CpG-STAT3 inhibitors, have broad implications for the design of oligonucleotide strategies for prostate cancer immunotherapy.
ACKNOWLEDGEMENTS
We would like to thank Dr. S. Vonderfecht (Veterinary Pathology) for his assistance and acknowledge the dedication of staff members at the Analytical Cytometry, Analytical Pharmacology, Light Microscopy, Pathology Cores and Animal Resources Center (COH).
This work was supported by the Department of Defense (Prostate Cancer Research Program) grant number W81XWH-16–1-0499, Prostate Cancer Foundation, Israel Cancer Research Fund “Jacki and Bruce Barron Cancer Research Scholars Program” (M. Kortylewski) and by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572 (City of Hope). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations list:
- STAT3
signal transducer and activator of transcription 3
- TLR9
Toll-like receptor 9
- ASO
antisense oligonucleotide
- MDSC
myeloid-derived suppressor cells
Footnotes
DISCLOSURE OF POTENTIAL CONFLICT OF INTEREST
M.K., P.S., D.M. and S.K.P. are inventors on patent application U.S. Provisional Application No.: 62/264,026 submitted by COH that covers the design of oligonucleotides presented in this report. All other authors declare no potential conflict of interest.
REFERENCES
- 1.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schweizer MT, Drake CG. Immunotherapy for prostate cancer: recent developments and future challenges. Cancer Metastasis Rev. 2014;33:641–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McNeel DG, Bander NH, Beer TM, Drake CG, Fong L, Harrelson S, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of prostate carcinoma. J Immunother Cancer. 2016;4:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Marzo AM, Platz EA, Sutcliffe S, Xu J, Grönberg H, Drake CG, et al. Inflammation in prostate carcinogenesis. Nat Rev Cancer. 2007;7:256–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyahira AK, Kissick HT, Bishop JL, Takeda DY, Barbieri CE, Simons JW, et al. Beyond immune checkpoint blockade: new approaches to targeting host-tumor interactions in prostate cancer: report from the 2014 Coffey-Holden prostate cancer academy meeting. Prostate. 2015;75:337–47. [DOI] [PubMed] [Google Scholar]
- 6.Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016;6:80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bezzi M, Seitzer N, Ishikawa T, Reschke M, Chen M, Wang G, et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat Med. 2018 [DOI] [PubMed] [Google Scholar]
- 8.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell. 2017;171:1301–15.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alme AKB, Karir BS, Faltas BM, Drake CG. Blocking immune checkpoints in prostate, kidney, and urothelial cancer: An overview. Urol Oncol. 2016;34:171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM, van Bokhoven A, et al. Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 2015;18:325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Shea JJ, Kanno Y, Chan AC. In search of magic bullets: the golden age of immunotherapeutics. Cell. 2014;157:227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. 2010;10:580–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. [DOI] [PubMed] [Google Scholar]
- 15.Avalle L, Camporeale A, Camperi A, Poli V. STAT3 in cancer: A double edged sword. Cytokine. 2017 [DOI] [PubMed] [Google Scholar]
- 16.Califano A, Alvarez MJ. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer. 2017;17:116–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhir R, Ni Z, Lou W, DeMiguel F, Grandis JR, Gao AC. Stat3 activation in prostatic carcinomas. Prostate. 2002;51:241–6. [DOI] [PubMed] [Google Scholar]
- 18.Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002;62:6659–66. [PubMed] [Google Scholar]
- 19.Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, et al. Stat3 promotes metastatic progression of prostate cancer. Am J Pathol. 2008;172:1717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Culig Z, Puhr M. Interleukin-6 and prostate cancer: Current developments and unsolved questions. Mol Cell Endocrinol. 2017 [DOI] [PubMed] [Google Scholar]
- 21.Bishop JL, Thaper D, Zoubeidi A. The Multifaceted Roles of STAT3 Signaling in the Progression of Prostate Cancer. Cancers (Basel). 2014;6:829–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pencik J, Wiebringhaus R, Susani M, Culig Z, Kenner L. IL-6/STAT3/ARF: the guardians of senescence, cancer progression and metastasis in prostate cancer. Swiss Med Wkly. 2015;145:w14215. [DOI] [PubMed] [Google Scholar]
- 23.Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9:75–89. [DOI] [PubMed] [Google Scholar]
- 24.Hossain DMS, Moreira D, Zhang Q, Nechaev S, Swiderski P, Kortylewski M. TLR9-Targeted SiRNA Delivery In Vivo. Methods Mol Biol. 2016;1364:183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Won H, Moreira D, Gao C, Duttagupta P, Zhao X, Manuel E, et al. TLR9 expression and secretion of LIF by prostate cancer cells stimulates accumulation and activity of polymorphonuclear MDSCs. J Leukoc Biol. 2017;102:423–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao C, Kozlowska A, Nechaev S, Li H, Zhang Q, Hossain DMS, et al. TLR9 signaling in the tumor microenvironment initiates cancer recurrence after radiotherapy. Cancer Res. 2013;73:7211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreira D, Zhang Q, Hossain DMS, Nechaev S, Li H, Kowolik CM, et al. TLR9 signaling through NF-κB/RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget. 2015;6:17302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hossain DMS, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, et al. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin Cancer Res. 2015;21:3771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kortylewski M, Moreira D. Myeloid cells as a target for oligonucleotide therapeutics: turning obstacles into opportunities. Cancer Immunol Immunother. 2017;66:979–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. 2013;123:1580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopez-Bujanda Z, Drake CG. Myeloid-derived cells in prostate cancer progression: phenotype and prospective therapies. J Leukoc Biol. 2017;102:393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sen M, Grandis JR. Nucleic acid-based approaches to STAT inhibition. JAKSTAT. 2012;1:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh JE, Toniolo PA, Frank DA. Targeting transcription factors: promising new strategies for cancer therapy. Curr Opin Oncol. 2013;25:652–8. [DOI] [PubMed] [Google Scholar]
- 35.Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2:694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7:314ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27:925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Q, Hossain DMS, Nechaev S, Kozlowska A, Zhang W, Liu Y, et al. TLR9-mediated siRNA delivery for targeting of normal and malignant human hematopoietic cells in vivo. Blood. 2013;121:1304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hossain DMS, Dos Santos C, Zhang Q, Kozlowska A, Liu H, Gao C, et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood. 2014;123:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nechaev S, Gao C, Moreira D, Swiderski P, Jozwiak A, Kowolik CM, et al. Intracellular processing of immunostimulatory CpG-siRNA: Toll-like receptor 9 facilitates siRNA dicing and endosomal escape. J Control Release. 2013;170:307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward AJ, Norrbom M, Chun S, Bennett CF, Rigo F. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014;42:5871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crooke ST, Wang S, Vickers TA, Shen W, Liang X-H. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 2017;35:230–7. [DOI] [PubMed] [Google Scholar]
- 43.Phosphorothioates Eckstein F., essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014;24:374–87. [DOI] [PubMed] [Google Scholar]
- 44.Gleave ME, Monia BP. Antisense therapy for cancer. Nat Rev Cancer. 2005;5:468–79. [DOI] [PubMed] [Google Scholar]
- 45.Castanotto D, Lin M, Kowolik C, Wang L, Ren X-Q, Soifer HS, et al. A cytoplasmic pathway for gapmer antisense oligonucleotide-mediated gene silencing in mammalian cells. Nucleic Acids Res. 2015;43:9350–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson TC, Southgate J, Kitchener G, Land H. Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell. 1989;56:917–30. [DOI] [PubMed] [Google Scholar]
- 47.Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543:728–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–21. [DOI] [PubMed] [Google Scholar]
- 49.Kortylewski M, Kujawski M, Herrmann A, Yang C, Wang L, Liu Y, et al. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009;69:2497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105:20852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wölfle SJ, Strebovsky J, Bartz H, Sähr A, Arnold C, Kaiser C, et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol. 2011;41:413–24. [DOI] [PubMed] [Google Scholar]
- 52.Gartrell BA, Saad F. Managing bone metastases and reducing skeletal related events in prostate cancer. Nat Rev Clin Oncol. 2014;11:335–45. [DOI] [PubMed] [Google Scholar]
- 53.Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, et al. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012;72:3570–81. [DOI] [PubMed] [Google Scholar]
- 54.Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calcinotto A, Spataro C, Zagato E, Di Mitri D, Gil V, Crespo M, et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells-An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology. 2015;4:e954829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patnaik A, Swanson KD, Csizmadia E, Solanki A, Landon-Brace N, Gehring MP, et al. Cabozantinib eradicates advanced murine prostate cancer by activating antitumor innate immunity. Cancer Discov. 2017;7:750–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kujawski M, Zhang C, Herrmann A, Reckamp K, Scuto A, Jensen M, et al. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010;70:9599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016;31:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maenhout SK, Du Four S, Corthals J, Neyns B, Thielemans K, Aerts JL. AZD1480 delays tumor growth in a melanoma model while enhancing the suppressive activity of myeloid-derived suppressor cells. Oncotarget. 2014;5:6801–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herrero-Sánchez MC, Rodríguez-Serrano C, Almeida J, San Segundo L, Inogés S, Santos-Briz Á, et al. Targeting of PI3K/AKT/mTOR pathway to inhibit T cell activation and prevent graft-versus-host disease development. J Hematol Oncol. 2016;9:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kroemer G, Galluzzi L, Zitvogel L. STAT3 inhibition for cancer therapy: Cell-autonomous effects only? Oncoimmunology. 2016;5:e1126063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Woessner RD, McCoon P, Grosskurth S, Lyne P, Bell K, Collins M, et al. Abstract A94: STAT3 antisense treatment decreases M2 macrophage infiltration and enhances the activity of checkpoint inhibitors in preclinical tumor models. Mol Cancer Ther. 2015;14:A94–A94. [Google Scholar]
- 67.Krieg AM. CpG still rocks! Update on an accidental drug. Nucleic Acid Ther. 2012;22:77–89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.