

Abstract
Ewing sarcoma is the second most common sarcoma of the bone, afflicting predominantly the pediatric population. While patients with localized disease exhibit favorable survival rates, patients with metastatic disease suffer a dismal 5-year rate of ~25%. Thus, there is a great need to develop treatments to combat disseminated disease. Ubiquitin-specific protease 6 (USP6/TRE17) has been implicated as the key etiological factor in several benign mesenchymal tumors, including nodular fasciitis (NF) and aneurysmal bone cyst (ABC). However, the role of USP6 in the biology of malignant entities remains unexplored. Previously, it was observed that USP6 is sufficient to drive formation of tumors mimicking ABC and NF, and that it functions through JAK1/STAT3 signaling. However, in the context of Ewing sarcoma (ES), USP6 does not enhance transformation, but rather triggers an interferon (IFN) response signature, both in cultured ES cells in vitro and in clinical specimens in vivo. Not only does USP6 independently induce activation of the IFN signaling mediators JAK1 and STAT1, but it also renders ES cells exquisitely responsive to exogenous IFNs, potentiating activation of STAT1 and STAT3. Furthermore, IFNβ (a Type I IFN) induces apoptosis specifically in USP6-positive but not USP6-negative ES cells. Finally, apoptosis is mediated through the pro-apoptotic ligand TRAIL, which is synergistically induced by Type I IFN and USP6.
IMPLICATIONS
These findings provide the first insights into USP6 functions in a clinically relevant malignant entity, and raise the possibility of using IFN for targeting USP6-positive Ewing sarcoma.
Keywords: USP6, TRE17, Interferon, apoptosis, Ewing Sarcoma
INTRODUCTION
Sarcomas are a diverse class of malignancies that represent a significant challenge in oncology. Ewing sarcoma (ES) is the second most common bone sarcoma, and typically affects individuals in the first two decades of life1. While patients with localized disease experience 5-year survival rates of 75%, metastatic patients face a dismal survival probability of ~20%. Thus, there is an urgent need to identify biomarkers that can predict recurrence and response to therapy, and develop strategies to combat metastatic disease.
The key etiologic agent in ES is a translocation product that fuses the EWS RNA-binding protein with an Ets family transcription factor, most commonly FLI12. Sustained EWS-FLI1 activity is required for transformation, and significant efforts have been aimed at identifying its critical targets. Multiple effectors that contribute to pathogenesis have been identified, both in cultured cells in vitro and in murine models. Furthermore, therapeutics have been developed against some of these effectors, including IGF, VEGF, and EWS-FLI1 itself3, 4. However, their clinical efficacy has been limited, underscoring the need to identify novel targets and approaches for ES treatment.
Our research focuses on the ubiquitin-specific protease 6 (USP6) oncogene, which is translocated in multiple benign mesenchymal tumors, including primary aneurysmal bone cyst (ABC), and nodular fasciitis (NF) 5, 6. USP6 translocations were also identified in fibroma of tendon sheath and giant-cell rich granuloma 7, 8. In all cases, translocation resulted in promoter swapping and high level expression of wild type USP6. USP6 expression is normally highly restricted in adult human tissues, with significant levels observed only in testes9. Though USP6 was first cloned in 199210, until recently little was known regarding its molecular functions, either physiologically or during tumorigenesis. We have shown that when ectopically expressed in candidate cells of origin for ABC and NF (i.e. fibroblasts and pre-osteoblasts), USP6 induces formation of tumors that recapitulate key clinical, histological, and molecular features of the human tumors11−13, with its catalytic activity as a de-ubiquitylating enzyme being essential12. Our more recent work revealed that USP6 promotes tumorigenesis through multiple pathways, including Jak1-STAT3, Wnt/β-catenin, and NF-κB11, 14, 15. Within the Jak1-STAT3 pathway, Jak1 itself is the critical target of USP615. De-ubiquitylation of Jak1 by USP6 rescues it from proteasomal degradation, leading to greatly elevated levels of the kinase, and sensitizing cells to Jak1 agonists such as interleukin-615.
While translocation-driven overexpression of USP6 plays a key role in benign neoplasms, its role in malignant entities where it is not the oncogenic driver remains unexplored. It is often incorrectly cited that USP6 is widely expressed in cancer cell lines. However, this erroneous conclusion is based on an early study in which Northern probes cross-reacted with the highly related, widely expressed USP32 gene10. Later reverse transcription-quantitative PCR (RT-qPCR) of primary tumors with USP6-specific primers indicated that its expression is far more restricted: high USP6 expression appears to occur predominantly in tumors of mesenchymal origin16. Yet, to date there have been few publications exploring USP6 functions in malignant cells, with most in HeLa cells14, 17–20
We sought to investigate functions of USP6 in Ewing sarcoma (ES), one of the malignancies shown to express high levels16. We show that USP6 triggers a gene signature reflective of response to interferon (IFN), a Jak1 agonist that functions in immunity. USP6 renders ES cells exquisitely sensitive to exogenous IFNs: not only is STAT1-mediated gene expression dramatically potentiated in USP6-expressing cells by IFN treatment, but Type I IFN is selectively cytotoxic to USP6-positive but not USP6-negative ES cells. IFN-induced death is mediated by TRAIL, a potent pro-apoptotic ligand. This work represents one of the first studies to examine USP6 functions in malignant cells, and suggest that it might serve as a prognostic indicator for response of ES to IFN treatment.
MATERIALS AND METHODS
Cell Lines and CRISPR-mediated gene targeting
RD-ES and TC-71 were from Dr. Frederic Barr and Dr. Lee Helman, respectively. CHLA-10 and SK-N-MC were from Dr. Irfan Asangani. Lines expressing USP6 in a doxycycline-inducible manner were generated as previously described12. Cells were tested for mycoplasma every 3–6 months, and prophylactically maintained in Mycoplasma Removal Agent (MP Biomedicals #09350044) for 2 weeks after thawing. All experiments used cells maintained for fewer than 20 passages after thawing. Cell line identity was confirmed by STR analysis just prior to manuscript submission.
Validated CRISPR target sequences for Jak1, STAT1, and STAT3 were from published sequences21. Target gRNAs (Jak1 (CACCGTCCCATACCTCATCCGGTAG); STAT1 (CACCGTCCCATTACAGGCTCAGTCG); and STAT3 (CACCGAGATTGCCCGGATTGTGGCC)) were subcloned into LentiCRISPRv2 (Addgene #52961) as described21. USP6/RD-ES cells were transfected with CRISPR constructs and subjected to puromycin selection. Clones were screened by immunoblotting.
Reagents
Doxycycline was from ClonTech (#8634–1). Jak Inhibitor I (CAS 457081–03-7; #420099) and PS-1145 (P6624) were from Sigma. Lipofectamine 2000 was from Life Technologies. Interferon α, β, and γ were obtained PBL Assay Science (#11410–2 and #11200–1) and PeproTech (#300–02) respectively. ZVAD (FMK001) and IETD (FMK007) were from R&D Systems. TRAIL (Cat # 752904) and anti-TRAIL (Cat # 308202) were from Biolegend. Caspase-3/7 (#G8090) and Caspase-9 (#G8210) activation kits were purchased from Promega, and assays were performed on Molecular Devices SpectroMax. Annexin V staining kit was from ebioscience (#88–8007-72), and samples analyzed on BD Bioscience Accuri C6 and LSR II machines.
Immunoblotting and RT-qPCR
Cell lysis was performed as previously described12. Jak1 (cs-3332), pSTAT1 (cs-9167), pSTAT3 (cs-9145), TRAIL (cs-3219), PARP (cs-9542), Caspase-8 (cs-9746), and Bid (cs-2002) were from Cell Signaling. HA (sc-805), STAT1 (sc-346), STAT3 (sc-482), and p65 (sc-372) were from Santa Cruz. Erk antibody was from Dr. John Blenis. Quantification was performed using the Image Studio Lite. Trizol was used for RNA isolation, and qPCR was performed using SYBR Green (Cat # 436765, ThermoFisher). Erk, STAT3, and p65 were used as protein loading controls as previously described22–25; their levels were comparable across conditions as shown.
Gene Expression Profiling/Pathway Analysis and DNA Methylation
RNA was isolated from USP6/RD-ES cells treated with or without doxycycline and IFNα for 24h. RNA-sequencing, alignment, processing, and repository deposit was performed by the University of Pennsylvania Next-Generation Sequencing Core (GSE107307). The CDF files for the Affymetrix U133A and U133 Plus 2.0 arrays were edited to remove probes from the USP6 probe set (206405_x_at) that cross-reacted with USP32 or other genes. This refined USP6-specific probe set comprised Probes 4, 8, 9, and 11. Publicly available Ewing sarcoma datasets (GSE700726 and GSE37371 U133A27) were sorted by USP6 expression. Patients with the highest USP6 levels were compared to those with the lowest (5 per group). For the germ cell tumor dataset GSE10615 28, samples were segregated as seminomas (USP6high) vs. yolk sac tumors (USP6low). Gene Set Enrichment Analysis (GSEA) was performed as previously described, using the “Hallmarks” molecular signature database. Gene expression analysis in nodular fasciitis was compared to other USP6-non-expressing mesenchymal tumors, as previously described15.
DNA methylation datasets for five Ewing sarcoma cell lines (CADO-ES1, SK-NMC, A673, RD-ES, and SK-ES-1) was procured from the Cancer Cell Line Encyclopedia (CCLE) (https://portals.broadinstitute.org/ccle) 29, 30, and relative CpG methylation for various genes was plotted using GraphPad. Methylation probe IDs for the USP6 promoter were obtained from MExpress31 and used in conjunction with GSE8904132
RESULTS
USP6 triggers an IFN response in ES in patient samples and cultured cells
Little is known about how USP6 functions in the context of malignant cells where it is not the oncogenic driver, with only a handful of reports largely restricted to HeLa14, 17–20. As mentioned, USP6 expression in neoplasms is far more restricted than initially believed: screening of a broad panel of primary samples demonstrated that high expression was predominantly confined to tumors of mesenchymal origin, including Ewing sarcoma (ES)16
To explore what functions USP6 might have in ES, gene expression patterns were investigated in primary patient samples. Most large ES patient datasets utilize Affymetrix microarrays, which use probesets consisting of 11 distinct probes against a given gene. However, most probes in the USP6 probeset (206405_x_at) cross-reacted with USP32 or other genes. Therefore, GSEA analysis was refined to use only the USP6-specific subset of probes, comparing ES tumors with the highest vs. lowest levels of USP6 expression. From two independent patient datasets, IFNα (Type I) and IFNγ (Type II) responses emerged among the top signatures associated with high USP6 expression (Figure 1A; see Supplemental Table 1 for expanded GSEA results). In addition, the IL6-Jak-STAT3 pathway was potently activated, which we previously showed to be induced by USP6 in our model of ABC
Figure 1. USP6 induces an IFN response in ES cells in vitro and in primary tumors. A).
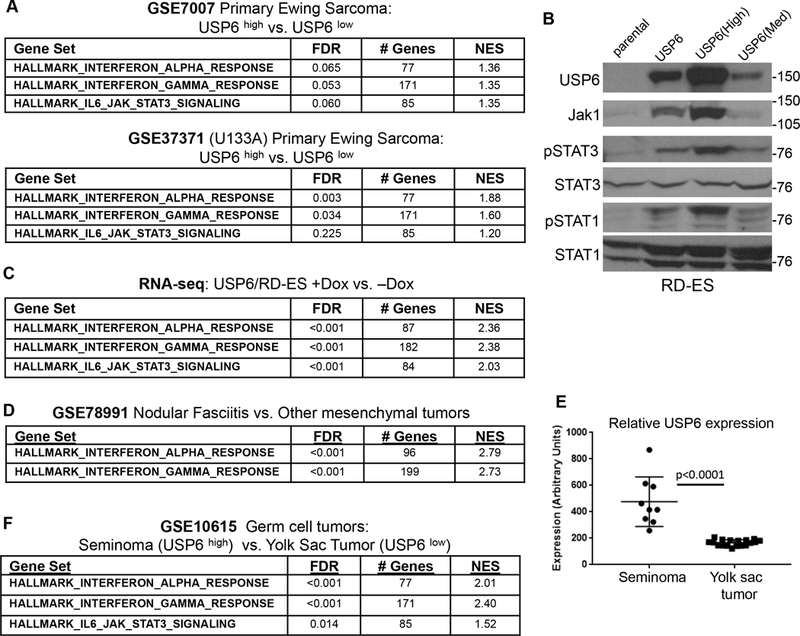
Samples from primary Ewing sarcoma datasets (GSE7007 and GSE37371) were ranked by USP6 expression level, and GSEA was performed comparing the 5 samples with the highest levels to the 5 with the lowest. B) The indicated RD-ES cell lines were grown in the presence of doxycycline (dox) overnight, then blotted as indicated. The USP6 line represents a pooled population, whereas USP6(high) and USP6(Med) are clonal. C) RNA-sequencing was performed on USP6/RD-ES treated with or without dox, followed by GSEA pathway analysis. D) GSEA was performed for nodular fasciitis (NF) dataset 15, which utilized the Illumina Human HT12 v4.0 BeadChip. This platform contains one USP6 probe, which is specific for USP6. E) Relative USP6 levels were evaluated in samples from the germ cell tumor dataset GSE10615 (U133A Microarray). F) GSEA of the germ cell tumor dataset, comparing seminomas to yolk sac tumors.
To determine whether USP6 directly induces these gene signatures, we sought to perform mechanistic studies in immortalized ES cells. However, none of the commonly used ES lines expressed appreciable USP6 levels (data not shown). We found extensive CpG methylation across the USP6 promoter in all immortalized ES cell lines examined, while comparatively low methylation was observed in primary ES tumors (Supplemental Figure 1A). CpG methylation heat maps revealed significant silencing of USP6 in multiple ES cell lines relative to genes known to be highly expressed in ES, such as Myc and EZH2 (Supplemental Figure 1B). How USP6 becomes methylated upon cell immortalization is unknown, but regardless, this necessitated expression of USP6 ectopically. We generated clonal and pooled stable lines expressing varied levels of USP6 in a doxycycline (dox)-inducible manner in the patient-derived ES cell line, RD-ES (Figure 1B). USP6 induced dose dependent upregulation of the Jak1 kinase (which we recently reported to be stabilized by direct de-ubiquitylation by USP6) and phosphorylation of STAT3 (Figure 1B), similar to what we observed in our models of ABC and NF15. In addition, we observed robust phosphorylation of STAT1, the key STAT family member that mediates IFN signaling (Figure 1B). RNA-seq was performed comparing the pooled cell line, USP6/RD-ES, in the presence vs. absence of dox. As in primary ES samples, IFNα and IFNγ responses emerged as the top “hits,” followed by IL6-Jak-STAT3 activation (Figure 1C and Supplemental Table 1). Together, these results demonstrate that not only is USP6 associated with an IFN response in ES in vivo, but that it is sufficient to activate this pathway. They also validate that our RD-ES cell model faithfully reflects physiological USP6 functions in ES patient samples.
We next explored whether USP6 was associated with an IFN response in other tumor types. An IFN signature was also induced in nodular fasciitis (NF), which is driven by translocation-driven overexpression of USP6 (Figure 1D and Supplemental Table 1). In addition, USP6 expression was associated with an IFN response in germ cell tumors. Yolk sac tumors (i.e. germ cell tumors arising from cells lining the yolk sac that are normally destined to become ovaries or testes) uniformly express low USP6 levels, whereas seminomas (i.e. germ cell tumors arising from the germinal epithelium of the testes) exhibit high levels (Figure 1E). GSEA comparing seminomas to yolk sac tumors revealed that high USP6 expression was again correlated with IFN and Jak-STAT3 signatures (Figure 1F). Together, these results indicate that USP6 may be more broadly associated with an IFN response in human tumors.
USP6 sensitizes ES cells to Exogenous IFN treatment
We speculated that in addition to triggering an IFN response by itself, USP6 might render RD-ES hypersensitive to exogenous IFN due to the elevated Jak1 levels. Indeed, dramatic enhancement and prolongation of STAT1/3 activation in USP6/RD-ES cells was observed with Type I and II IFNs (IFNα/IFNβ and IFNγ, respectively) (Figure 2 and Supplemental Figure 2). Treatment of parental RD-ES cells with IFNα or IFNγ induced phosphorylation of STAT1 and STAT3, which peaked within 30 minutes and gradually declined by 8hr. In contrast, STAT1 and STAT3 phosphorylation was augmented and prolonged in USP6/RD-ES, with significant activation persisting at 8hr (Figure 2A,B and Supplemental Figure 2). Interestingly, we also noted that Type I IFNs induced downregulation of USP6 (Figure 2A; discussed below).
Figure 2. USP6 enhances signaling and sensitivity of ES cells to Type I and Type II IFNs. A).
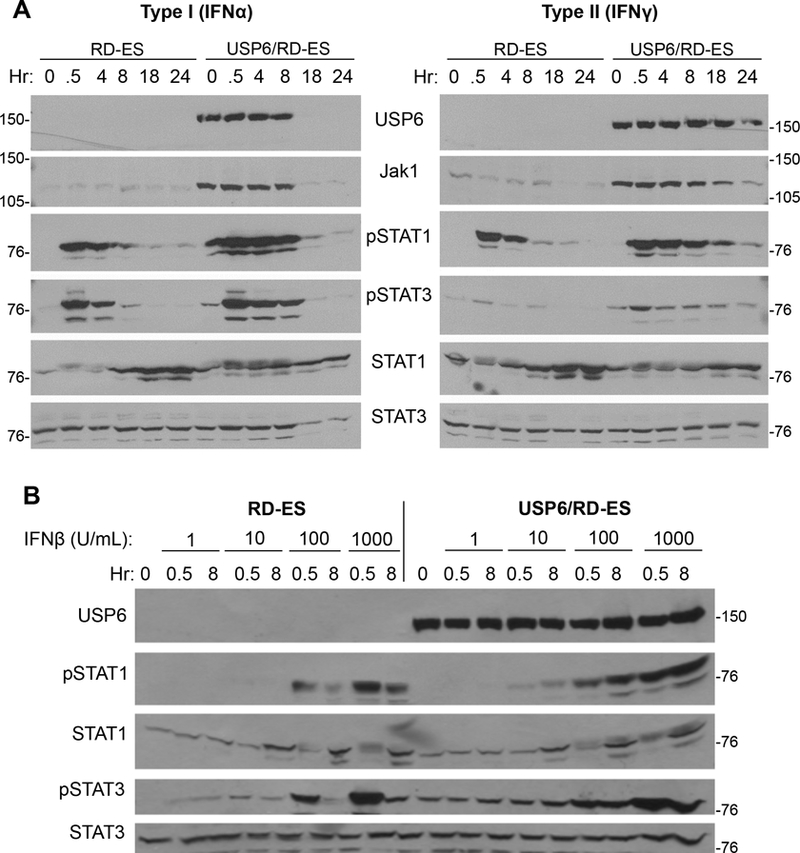
Parental or USP6/RD-ES cells were grown in dox overnight, then treated with IFNα (left) or IFNγ (right) (1000 U/mL) for the indicated times, and blotted. B) Cells were treated with dox overnight, then treated with the indicated dose of IFNβ for 0.5h or 8h. Samples were blotted as indicated; STAT3 was used as a loading control.
In addition to prolonging STAT1/3 activation, USP6 heightened sensitivity to low dose IFN (Figure 2B and Supplemental Figure 2). At doses ranging from 10–1000 U/mL, USP6/RD-ES cells showed elevated STAT1/3 phosphorylation compared to parental RD-ES. The ability of USP6 to enhance and/or prolong STAT activation was confirmed in three additional patient-derived ES lines, TC-71, CHLA-10, and SK-N-MC (Supplemental Figures 3), indicating that its effects are widely observed in ES, and are not a peculiarity of the RD-ES line.
Strikingly, we noticed that with prolonged treatment, Type I IFN was selectively cytotoxic to USP6-expressing but not parental RD-ES cells. IFNβ exhibited the greatest cytotoxicity, followed by IFNα, then IFNγ, as monitored by PARP cleavage and trypan blue exclusion (Figure 3A and3B). Annexin V staining confirmed that IFNβ-induced death occurred through apoptosis (Figure 3C and Supplemental Figure 4A). IFNβ induced apoptosis more effectively than IFNα at doses up to 2500 U/mL (Supplemental Figure 4B), likely due to its greater affinity for the Type I IFN receptor. Furthermore, USP6 conferred sensitivity to IFNβ in a dose-dependent manner, as the extent of death correlated with the level of USP6 expression (Figure 3D). USP6 also sensitized TC-71 cells to IFNβ-induced apoptosis (Figure 3E and3F). However, USP6 minimally enhanced death in CHLA-10 cells and SK-N-MC cells (Supplemental Figure 4C); the mechanism underlying this differential response is discussed below. Notwithstanding, these results indicate that USP6 can dictate the magnitude of response to IFN, and can greatly sensitize ES cells to the apoptotic potential of IFN.
Figure 3. USP6 renders ES cells sensitive to apoptosis by Type I IFN.
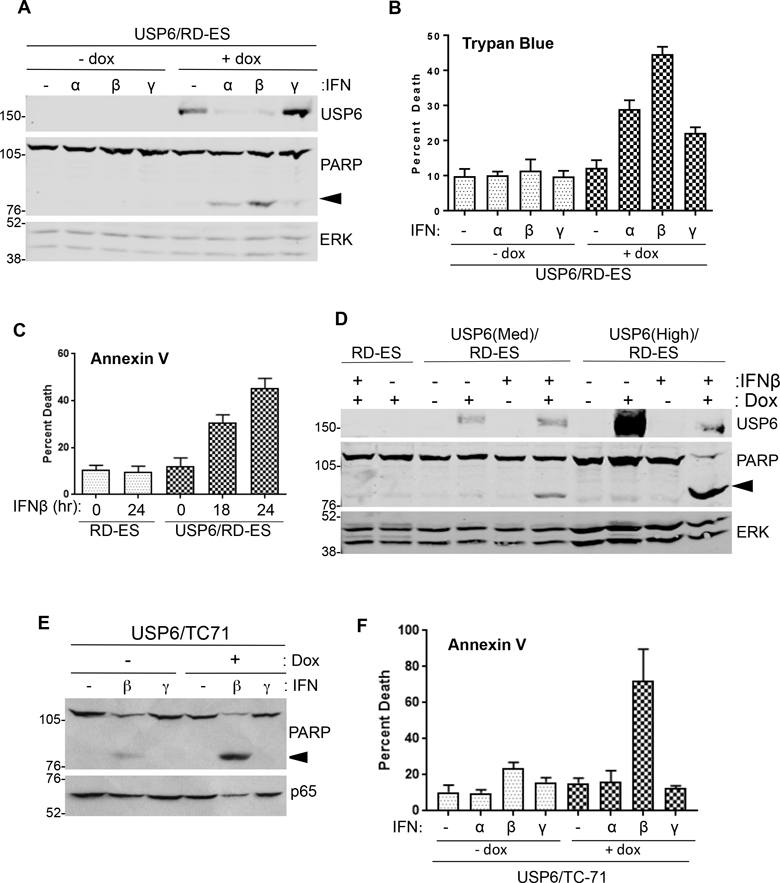
USP6/RD-ES cells were grown in the absence or presence of dox, then treated with the indicated IFN at 1000 U/mL for 24h. Cells were subjected to blotting (A), or trypan blue exclusion assays to monitor viability (n=3) (B). C) Cells were grown in dox, then treated with 1000 U/mL IFNβ for 18h or 24h. Apoptosis was quantified by Annexin V staining (n=4). D) USP6(Med), USP6(High), and parental RD-ES cells were treated with dox and IFNβ overnight, then blotted as indicated. Arrowhead indicates cleaved PARP product. E) USP6/TC-71 cells were grown in the absence or presence of dox, then treated with 100 U/mL of the indicated IFN for 24h. F) Cells were grown in dox, then treated with 100 U/mL of the indicated IFN for 24h. Apoptosis was quantified by Annexin V staining (n=3). ERK or p65 was used as a loading control.
IFN-induced apoptosis involves extrinsic and intrinsic pathways, and requires Jak1-STAT1/3
We next sought to dissect the mechanism of IFN-induced apoptosis. Depending on cell type, IFN can trigger extrinsic apoptosis, which occurs through ligand binding to cell surface receptors, or intrinsic apoptosis, which occurs through mitochondrial dysregulation. These pathways can be distinguished by their requirement for distinct caspase proteases (see Supplemental Figure 5A for pathway summary). Extrinsic apoptosis requires cleavage/activation of caspase-8, followed by caspase-3/7; intrinsic apoptosis entails cleavage of the mitochondrial protein Bid and caspase-9 activation, which also triggers caspase-3/7 activation. However, in some circumstances extrinsic apoptosis induced by IFN can feed into the mitochondrial route, and trigger cleavage/activation of Bid and caspase-9.
IFN−induced death of USP6/RD-ES was blocked by the caspase-8-specific inhibitor IETD (Figure 4A) and was accompanied by caspase-8 cleavage (Figure 4B), implicating the extrinsic pathway. However, IFNβ also induced Bid cleavage (Figure 4B) and caspase-9 activation (Figure 4C), indicating engagement of the mitochondrial route. Activation of Caspase-3/7 was also observed, and could be blocked by caspase-8 inhibitor (Figure 4D). In sum, these data indicate that IFNβ-induced death of USP6/RD-ES cells occurs through a cell surface-mediated, extrinsic route that entails mitochondrial dysregulation.
Figure 4. IFNβ-Induced apoptosis requires Jak1-STAT1/STAT3 and entails extrinsic and intrinsic death pathways. A, B).
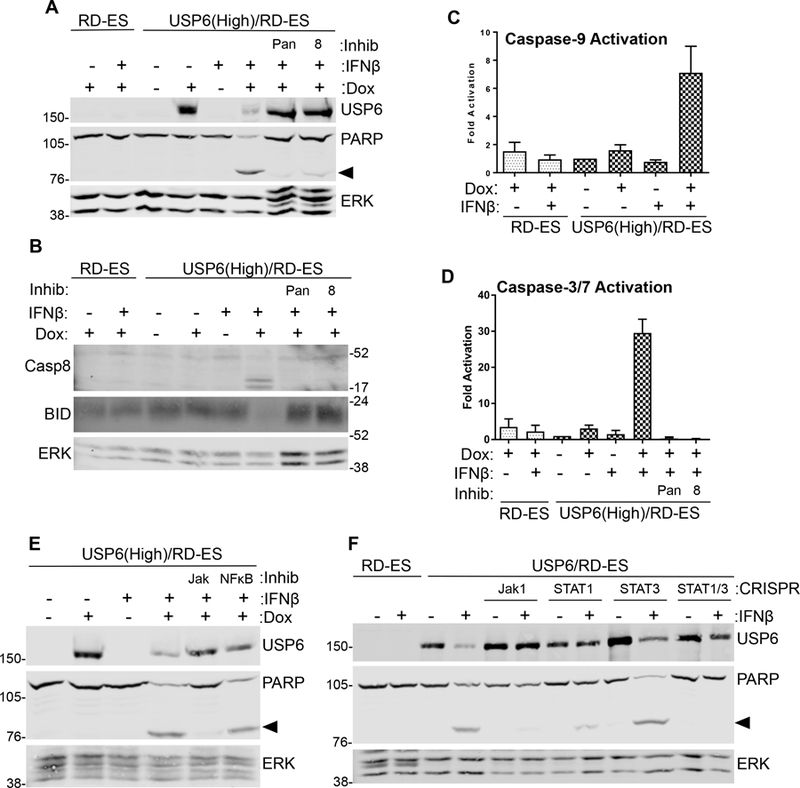
Cells were treated with dox and IFNβ overnight, in the absence or presence of 50 μM pan-caspase inhibitor ZVAD (pan) or caspase-8 inhibitor IETD (8). Lysates were blotted as indicated. C, D) Cells treated with dox and IFNβ overnight, in the presence of caspase inhibitors as indicated, and caspase-9 (n=6) and caspase-3/7 activity was measured (n=3). Activity was calculated as fold relative to that in untreated USP6(High)/RD-ES. E) USP6(High)/RD-ES were treated with dox and IFNβ overnight, in the presence of a pan-Jak inhibitor (1 μM) or NFκB inhibitor PS-1145 (15 μM). F) Jak1, STAT1, or STAT3 were deleted by CRISPR gene editing. Cells were treated with dox and IFNβ overnight, then blotted as shown. Arrowhead indicates cleaved PARP product; ERK was used as a loading control.
To further dissect the signaling mechanisms underlying apoptosis, we examined the roles of Jak1-STAT and NF-κB, both of which have been shown to participate in IFN-mediated death(22,23). A pan-Jak family inhibitor completely blocked apoptosis of USP6/RD-ES, whereas the NF-κB inhibitor was ineffective (Figure 4E). Reporter assays confirmed that the NF-κB inhibitor was functional (Supplemental Figure 5B). To confirm the requirement of the Jak1-STAT pathway, CRISPR-mediated knockouts of Jak1, STAT1, and STAT3 were generated in USP6/RD-ES cells (Figure 4F and Supplemental Figure 5C). Figure 4F shows that Jak1 deletion significantly reduced death. Deletion of both STAT1 and STAT3 was required to obtain robust inhibition of death, indicating that they play distinct roles in the apoptotic response, consistent with their ability to function as homo- and hetero-dimers in response to IFN. These genetic and pharmacological approaches demonstrate that Jak1, STAT1, and STAT3 are required for IFNβ-mediated apoptosis of USP6/RD-ES.
IFNβ-induced apoptosis of USP6/RD-ES cells is mediated by TRAIL pathway
IFN can induce expression of the pro-apoptotic ligands FasL and TRAIL. Our RNA-seq data indicated that TRAIL, but not Fas, was synergistically induced by IFN in USP6/RD-ES relative to parental cells. RT-qPCR confirmed that IFNs had little or no effect on TRAIL expression in RD-ES (Figure 5A). However, TRAIL mRNA levels were dramatically increased in USP6/RD-ES treated with IFNβ. Induction was also observed, but to a much lesser degree, with IFNα and IFNγ (Figure 5A), correlating with the extent of death induced by each (Figure 3). In contrast, FasL expression was not significantly affected by USP6 (Supplemental Figure 6). We also examined expression of the five TRAIL receptors, both the active (DR4/DR5) and inactive decoy (TNFRSF10C/D and OPG) forms, whose balance has been shown to a play an important role in sensitization of cancer cells to TRAIL-induced apoptosis33. USP6 did not alter expression of receptors in a manner consistent with sensitization to death (Supplemental Figure 6).
Figure 5. IFNβ induces apoptosis of USP6-positive ES cells through synergistic production of TRAIL. A,B).
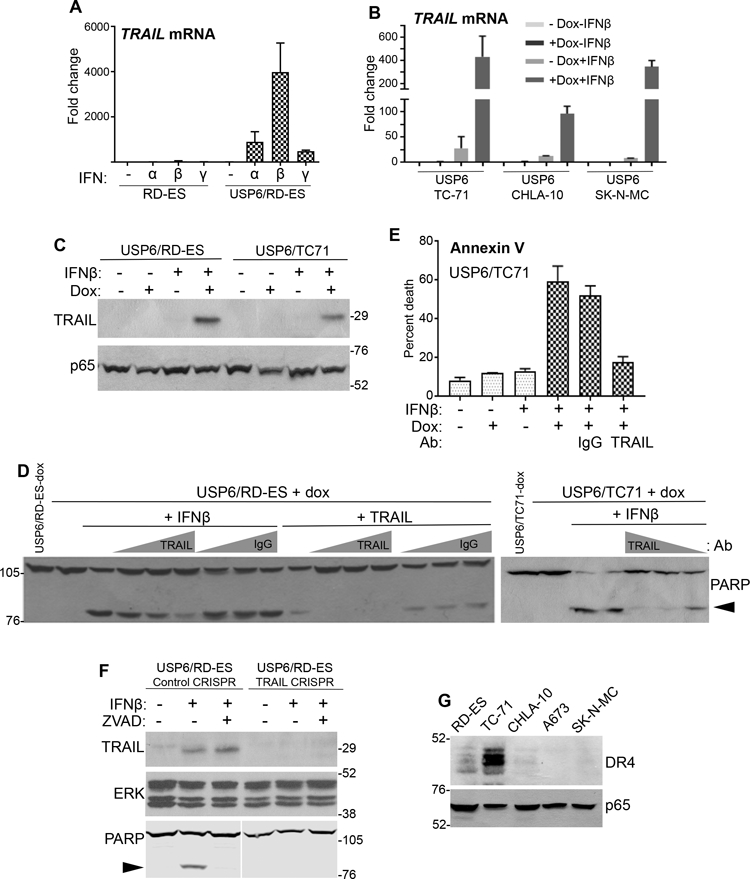
Cells were treated with dox and the indicated IFN (1000 U/mL) for 24h. TRAIL mRNA levels were quantified by RT-qPCR, and fold-induction relative to untreated RD-ES determined. C) The indicated cells were treated with dox and IFNβ (1000 U/mL) for 24h, and blotted as indicated. D,E) The indicated cells were treated overnight with IFNβ (1000 U/mL) or reTRAIL (200 ng/mL), in the presence of increasing amounts (1.0, 1.5, or 2.0 μg) of anti-TRAIL or control IgG. Samples were blotted in D, or subjected to Annexin V staining in E. F) TRAIL was depleted from USP6/RD-ES cells using CRISPR. Cells were treated overnight as indicated, and blotted as shown. G) The indicated ES cell lines were blotted as shown. ERK or p65 was used as a loading control.
Figure 5C confirms that TRAIL protein was strongly induced upon IFNβ treatment in USP6/RD-ES in a dox-dependent manner. Induction of TRAIL transcription and protein was also confirmed in the USP6/TC-71 ES cell line (Figure 5B,C). Neutralizing anti-TRAIL antibody inhibited IFNβ-induced apoptosis of both of USP6/RD-ES and USP6/TC-71 cells, as measured by PARP cleavage and Annexin V staining (Figure 5D and 5E). Furthermore, CRISPR-mediated deletion of TRAIL completely abrogated death of USP6/RD-ES by IFNβ (Figure 5F). These data confirm that TRAIL plays a dominant role in mediating IFNβ-induced apoptosis of USP6-positive ES cells.
As described above, the various ES lines exhibited differential sensitivities to IFNβ-induced apoptosis in the presence of USP6: RD-ES and TC-71 were very sensitive, while CHLA-10 and SK-N-MC were largely unresponsive (Figure 3 and Supplemental Figure 4). To determine whether this was due to disparate induction of TRAIL in these lines, RT-qPCR was performed. However, we found that TRAIL transcription was also synergistically induced in insensitive ES lines (Figure 5B). We then explored whether the differential responsiveness might arise from varied expression of TRAIL receptor. Strikingly, we found sensitivity to IFNβ in the presence of USP6 correlated precisely with expression of the TRAIL receptor DR4: DR4 levels were highest in RD-ES and TC-71, and largely undetectable in the insensitive ES lines (Figure 5G).
IFN triggers USP6 downregulation through TRAIL-dependent caspase activation
As mentioned above, Type I IFNs induce downregulation of USP6 protein (Figure 2,3,4). We noted that TRAIL also triggered USP6 downregulation, in a time- and dose-dependent manner (Figure 6A,B). TRAIL acted more rapidly, with USP6 downregulation observed within 4h, whereas IFNβ required 12–18h (Figures 2A and6B). Since caspases play a key role in TRAIL signaling, we tested whether they mediate USP6 downregulation. Both the pan-caspase inhibitor ZVAD and the caspase-8 inhibitor IETD completely blocked IFNβ- and TRAIL-induced USP6 downregulation (Figures 4A and6C, respectively). Together, these results reveal a negative feedback mechanism whereby USP6 induces TRAIL transcription, which then signals through DR4 to trigger caspase-dependent downregulation of USP6 (see Model Figure 6D). Notably, this identifies Type I IFNs and TRAIL as the first physiological agonists to regulate USP6.
Figure 6. Type I IFN induces USP6 downregulation through a TRAIL- and caspase-mediated mechanism. A-C).
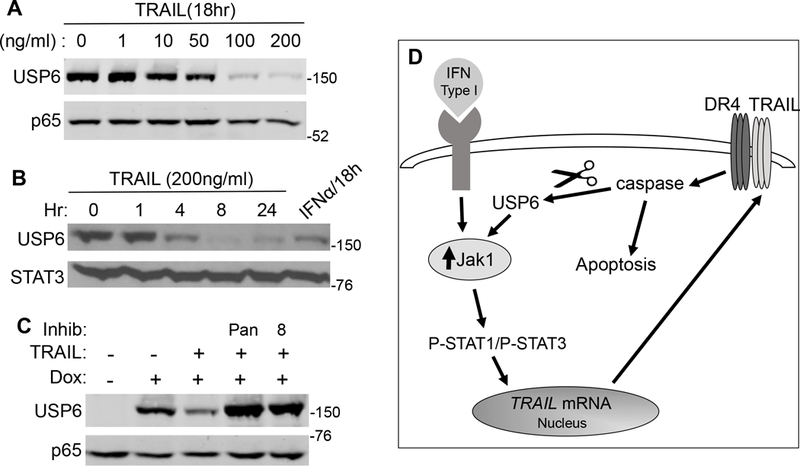
USP6/RD-ES cells were treated with dox and the indicated doses of TRAIL or IFNα (1000 U/mL) for the indicated times. In C, TRAIL was used at 10 ng/mL, and pan-caspase inhibitor ZVAD (pan) or caspase-8 inhibitor (8) was added as shown. STAT3 or p65 was used as a loading control. D) Mechanism of IFN-induced apoptosis of USP6-positive ES cells: Jak1 levels are upregulated by USP6-mediated de-ubiquitination, greatly sensitizing cells to IFNs. Type I IFNs, particularly IFNβ, induces transcription of TRAIL, which induces apoptosis through binding its receptor DR4. TRAIL/DR4 then trigger USP6 downregulation through caspase activation.
DISCUSSION
While it has long been recognized that USP6 plays a key etiologic role in several benign neoplasms, its functions in the biology of malignant entities is poorly understood. Analysis of primary tumor samples by Oliveira et al. revealed that among human malignancies, highest USP6 expression was most commonly observed in mesenchymal cancers, including Ewing sarcoma. The current study is the first to explore functions of USP6 in Ewing sarcoma. We have found that USP6 expression is associated with an IFN signature in primary ES tumors. Furthermore, USP6 is sufficient to trigger this response when inducibly expressed in cultured ES cells. USP6 also confers exquisite sensitivity of ES cells to exogenous IFNs. Strikingly, Type I IFNs (particularly IFNβ) induce TRAIL-mediated apoptosis of USP6-positive but not USP6-negative ES cells, in a DR4-dependent manner (see Figure 6D for results summary).
To date there are notably few studies on USP6, and thus nothing is known of how its expression is regulated, how its activity modulated, or what normal physiological processes it participates in. We identify Type I IFNs and TRAIL as the first physiological agonists to induce post-translational modification of USP6. We show that TRAIL triggers the caspase-dependent processing and downregulation of USP6, and that Type I IFN can also trigger this downregulation through induction of TRAIL signaling. We speculate that this negative feedback loop (wherein USP6 serves to amplify IFN-mediated induction of TRAIL, which then elicits downregulation of USP6) may play an important role during normal physiology to restrict TRAIL-induced functions, which include not only apoptosis but also inflammation34, 35.
Along this vein, a key area for future pursuit is determining the consequences of USP6-mediated IFN signaling in ES pathogenesis. Numerous studies have indicated that IFNs can either promote or antagonize tumor progression across broad tumor types36–38. This complexity can be ascribed to its ability to act not only on tumor cells, but also on immune cells and other cells in the tumor microenvironment. In some scenarios, IFN can promote and inflammatory microenvironment that enhances proliferation and metastasis of tumor cells38. In others, IFN can stimulate immune infiltration and thereby promote tumor cell killing. Thus, future studies will determine whether activation of IFN signaling by USP6 acts in a pro- or anti-tumorigenic manner in ES in vivo. Notably, previous studies have shown that IFNs and TRAIL largely function in an anti-tumorigenic capacity in ES, both in vitro and in murine xenografts39–41. Results have been somewhat variable, with IFN being sufficient to block proliferation and induce death in some studies, but requiring co-treatment with other agents in others. Our work provides a potential mechanism by which ES cells acquire sensitivity to the apoptotic effects of IFNs
Standard of care for Ewing sarcoma patients has progressed minimally over the past two decades. General cytotoxic chemotherapy is typically inefficacious in patients with disseminated or recurrent disease. Therefore, critical goals have been to develop novel therapies to prevent and treat recurrent/disseminated disease, and to identify biomarkers that can predict response to therapy. Type I IFN has previously been explored as a potential therapeutic for several cancers, but its use is currently restricted to advanced cases of melanoma42. However, its broader use has been limited by severe systemic side effects due to its potent immunostimulatory activity42. Our current results may help alleviate this issue, since USP6 greatly sensitizes cells to low-dose IFN. Thus, reduced IFN doses could be utilized that would retain tumoricidal activity while minimizing systemic side effects. Furthermore, since USP6 appears to be associated with an IFN response in other cancers, our findings may be applicable to other malignancies in which USP6 is overexpressed.
Supplementary Material
Acknowledgments
We thank Dr. Gerd A. Blobel for critical reading of the manuscript. This work was funded by NIH/NCI grants CA168452 and CA178601 (M.M.C.) and TG 32GM008076 (I.C.H.).
Footnotes
Conflict of Interest: This manuscript presents original research that has not been or is in the process of being published elsewhere. The authors declare no conflict of interest.
REFERENCES
- 1.Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop 2016;7:527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cidre-Aranaz F, Alonso J. EWS/FLI1 Target Genes and Therapeutic Opportunities in Ewing Sarcoma. Front Oncol 2015;5:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol 2015;33:3036–46. [DOI] [PubMed] [Google Scholar]
- 4.Toomey EC, Schiffman JD, Lessnick SL. Recent advances in the molecular pathogenesis of Ewing’s sarcoma. Oncogene 2010;29:4504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliveira AM, Hsi B, Weremowicz S, Rosenberg AE, Dal Cin P, Joseph N, et al. USP6 (Tre2) Fusion Oncogenes in Aneurysmal Bone Cyst. Cancer Research 2004;64:1920–3. [DOI] [PubMed] [Google Scholar]
- 6.Erickson-Johnson MR, Chou MM, Evers BR, Roth CW, Seys AR, Jin L, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest 2011;91:1427–33. [DOI] [PubMed] [Google Scholar]
- 7.Agaram NP, LeLoarer FV, Zhang L, Hwang S, Athanasian EA, Hameed M, et al. USP6 gene rearrangements occur preferentially in giant cell reparative granulomas of the hands and feet but not in gnathic location. Hum Pathol 2014;45:1147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter JM, Wang X, Dong J, Westendorf J, Chou MM, Oliveira AM. USP6 genetic rearrangements in cellular fibroma of tendon sheath. Mod Pathol 2016;8:865–9. [DOI] [PubMed] [Google Scholar]
- 9.Paulding CA, Ruvolo M, Haber DA. The Tre2 (USP6) oncogene is a hominoid-specific gene. Proc Natl Acad Sci U S A 2003;100:2507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura T, Hillova J M- SR, Onno M HK, Cannizzaro LA, Boghosian-Sell L, Croce CM, et al. A novel transcriptional unit of the tre oncogene widely expressed in human cancer cells. Oncogene 1992;7:733–41. [PubMed] [Google Scholar]
- 11.Pringle LM, Young R, Quick L, Riquelme DN, Oliveira AM, May MJ, et al. Atypical mechanism of NF-kappaB activation by TRE17/ubiquitin-specific protease 6 (USP6) oncogene and its requirement in tumorigenesis. Oncogene 2012;31:3525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye Y, Pringle LM, Lau AW, Riquelme DN, Wang H, Jiang T, et al. TRE17/USP6 oncogene translocated in aneurysmal bone cyst induces matrix metalloproteinase production via activation of NF-kappaB. Oncogene 2010;29:3619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau AW, Pringle LM, Quick L, Riquelme DN, Ye Y, Oliveira AM, et al. TRE17/ubiquitin-specific protease 6 (USP6) oncogene translocated in aneurysmal bone cyst blocks osteoblastic maturation via an autocrine mechanism involving bone morphogenetic protein dysregulation. J Biol Chem 2010;285:37111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Madan B, Walker MP, Young R, Quick L, Orgel KA, Ryan M, et al. USP6 oncogene promotes Wnt signaling by deubiquitylating Frizzleds. Proceedings of the National Academy of Sciences 2016;113:2945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quick L, Young R, Henrich IC, Wang X, Asmann YW, Oliveira AM, et al. Jak1-STAT3 Signals Are Essential Effectors of the USP6/TRE17 Oncogene in Tumorigenesis. Cancer Res 2016;76:5337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliveira AM, Perez-Atayde AR, Dal Cin P, Gebhardt MC, Chen CJ, Neff JR, et al. Aneurysmal bone cyst variant translocations upregulate USP6 transcription by promoter swapping with the ZNF9, COL1A1, TRAP150, and OMD genes. Oncogene 2005;24:3419–26. [DOI] [PubMed] [Google Scholar]
- 17.Masuda-Robens JM, Kutney SN, Qi H, Chou MM. The TRE17 Oncogene Encodes a Component of a Novel Effector Pathway for Rho GTPases Cdc42 and Rac1 and Stimulates Actin Remodeling. Molecular and Cellular Biology 2003;23:2151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rueckert C, Haucke V. The Oncogenic TBC Domain Protein USP6/TRE17 Regulates Cell Migration and Cytokinesis. Biology of the Cell 2012;104:22–33. [DOI] [PubMed] [Google Scholar]
- 19.Li L, Yang H, He Y, Li T, Feng J, Chen W, et al. Ubiquitin-Specific Protease USP6 Regulates the Stability of the c-Jun Protein. Molecular and Cellular Biology 2017;38:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Funakoshi Y, Chou MM, Kanaho Y, Donaldson JG. TRE17/USP6 regulates ubiquitylation and trafficking of cargo proteins that enter cells by clathrin-independent endocytosis. J Cell Sci 2014;127:4750–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang SG, Yu SS, Ryu JH, Jeon HB, Yoo YJ, Eom SH, et al. Regulation of beta-catenin signaling and maintenance of chondrocyte differentiation by ubiquitin-independent proteasomal degradation of alpha-catenin. J Biol Chem 2005;280:12758–65 [DOI] [PubMed] [Google Scholar]
- 23.Huang H, Muddiman DC, Tindall DJ. Androgens negatively regulate forkhead transcription factor FKHR (FOXO1) through a proteolytic mechanism in prostate cancer cells. J Biol Chem 2004;279:13866–77 [DOI] [PubMed] [Google Scholar]
- 24.Banerjee S, Byrd JN, Gianino SM, Harpstrite SE, Rodriguez FJ, Tuskan RG, et al. The neurofibromatosis type 1 tumor suppressor controls cell growth by regulating signal transducer and activator of transcription-3 activity in vitro and in vivo. Cancer Res 2010;70:1356–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 2011;25:2125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007;11:421–9 [DOI] [PubMed] [Google Scholar]
- 27.Martignetti L, Laud-Duval K, Tirode F, Pierron G, Reynaud S, Barillot E, et al. Antagonism pattern detection between microRNA and target expression in Ewing’s sarcoma. PLoS One 2012;7:41770–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmer RD, Barbosa-Morais NL, Gooding EL, Muralidhar B, Thornton CM, Pett MR, et al. Pediatric malignant germ cell tumors show characteristic transcriptome profiles. Cancer Res 2008;68:4239–47. [DOI] [PubMed] [Google Scholar]
- 29.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Cell Line Encyclopedia C, Genomics of Drug Sensitivity in Cancer C. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015;528:84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koch A, De Meyer T, Jeschke J, Van Criekinge W. MEXPRESS: visualizing expression, DNA methylation and clinical TCGA data. BMC Genomics 2015;16:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huertas-Martinez J, Court F, Rello-Varona S, Herrero-Martin D, Almacellas-Rabaiget O, Sainz-Jaspeado M, et al. DNA methylation profiling identifies PTRF/Cavin-1 as a novel tumor suppressor in Ewing sarcoma when co-expressed with caveolin-1. Cancer Lett 2017;386:196–207 [DOI] [PubMed] [Google Scholar]
- 33.von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nature Reviews Cancer 2017;17:352–66. [DOI] [PubMed] [Google Scholar]
- 34.Zoller V, Funcke JB, Roos J, Dahlhaus M, Abd El Hay M, Holzmann K, et al. Trail (TNF-related apoptosis-inducing ligand) induces an inflammatory response in human adipocytes. Sci Rep 2017;7:5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azijli K, Weyhenmeyer B, Peters GJ, de Jong S, Kruyt FA. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell Death Differ 2013;20:858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bekisz J, Sato Y, Johnson C, Husain SR, Puri RK, Zoon KC. Immunomodulatory effects of interferons in malignancies. J Interferon Cytokine Res 2013;33:154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang BX, Platanias LC, Fish EN. STAT activation in malignancies: roles in tumor progression and in the generation of antineoplastic effects of IFNs. J Interferon Cytokine Res 2013;33:181–8. [DOI] [PubMed] [Google Scholar]
- 38.Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res 2011;17:6118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kontny HU, Hammerle K, Klein R,Shayan P,Mackall CL, and Niemeyer CM. Sensitivity of Ewing’s sarcoma to TRAIL-induced apoptosis. Cell Death and Differentiation 2001;8:9. [DOI] [PubMed] [Google Scholar]
- 40.Wietzerbin AAaJ. Involvement of TNF‐Related Apoptosis‐Inducing Ligand (TRAIL) Induction in Interferon γ‐Mediated Apoptosis in Ewing Tumor Cells. Annals of the New York Academy of Sciences 2003;1010:4. [DOI] [PubMed] [Google Scholar]
- 41.Picarda G, Lamoureux F, Geffroy L, Delepine P, Montier T, Laud K, et al. Preclinical evidence that use of TRAIL in Ewing’s sarcoma and osteosarcoma therapy inhibits tumor growth, prevents osteolysis, and increases animal survival. Clin Cancer Res 2010;16:2363–74. [DOI] [PubMed] [Google Scholar]
- 42.Wang BX, Rahbar R, Fish EN. Interferon: current status and future prospects in cancer therapy. J Interferon Cytokine Res 2011;31:545–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.