

Abstract
Patients with pancreatic neuroendocrine tumors (PNETs) commonly develop advanced disease and require systemic therapy. However, treatment options remain limited, in part because experimental models that reliably emulate PNET disease are lacking. We therefore developed a patient-derived xenograft model of PNET (PDX-PNET) which we then used to evaluate two mTOR inhibitor drugs: FDA-approved everolimus and the investigational new drug sapanisertib. PDX-PNETs maintained a PNET morphology and PNET-specific gene expression signature with serial passage. PDX-PNETs also harbored mutations in genes previously associated with PNETs (such as MEN1 and PTEN), displayed activation of the mTOR pathway and could be detected by Gallium-68 DOTATATE PET-CT. Treatment of PDX-PNETs with either everolimus or sapanisertib strongly inhibited growth. As seen in patients, some PDX-PNETs developed resistance to everolimus. However, sapanisertib, a more potent inhibitor of the mTOR pathway, caused tumor shrinkage in most everolimus-resistant tumors. Our PDX-PNET model is the first available, validated PDX model for PNET, and preclinical data from the use of this model suggests that sapanisertib may be an effective new treatment option for patients with PNET or everolimus-resistant PNET.
Introduction
Pancreatic neuroendocrine tumors (PNETs) are a form of pancreatic neoplasm with neuroendocrine features whose precise cell of origin is unknown. PNETs comprise ~10% of all pancreatic malignancies (1), can cause diverse clinical syndromes (2), and have a 5-year mortality rate of approximately 60% (1, 3). PNETs can be either sporadic or the consequence of a hereditary cancer syndrome, such as multiple endocrine neoplasia type 1 (MEN1). Two-thirds of patients diagnosed with PNET have unresectable metastatic disease and therefore require systemic therapy.
Activated mTOR signaling is present in about 14% of PNETs and is associated with a poor prognosis (4). mTOR is a serine-threonine kinase that serves as the catalytic subunit of two distinct signaling complexes, mTORC1 and mTORC2. mTORC1 promotes cell growth through two key effectors: p70S6 Kinase 1 (S6K1) and eIF4E-binding protein 1 (4EBP1). mTORC1 directly phosphorylates S6K1, which can then phosphorylate several downstream substrates such as ribosomal protein S6 (RPS6). 4EBP1 is inactivated by mTORC1 phosphorylation, initiating the translation of proteins. mTORC2 promotes cell proliferation and survival through activation of AKT by phosphorylation (5).
Activation of the mTOR pathway in PNETs results from mutations in genes that encode negative regulators of the pathway, such as PTEN (4, 6, 7). The best known mTOR inhibitor drug is rapamycin, which forms a complex with 12-kD FK506-binding protein (FKBP12) to inhibit mTORC1. Because of mTOR’s central importance in many human diseases, there are many efforts to develop drugs that more effectively inhibit its activity (8). The mTOR inhibitor drug everolimus, a derivative of rapamycin (8), has been approved by the FDA for the treatment of advanced PNETs and other tumors (9, 10). Everolimus, like rapamycin and other homologous agents (rapalogs), allosterically inhibits mTORC1 but does not affect mTORC2. It can also fail to block the phosphorylation of mTORC1 target 4EBP1, and paradoxically increase the activation of AKT due to suppression of a negative-feedback loop (11). Clinically, everolimus typically delays human PNET progression by several months, but significant tumor shrinkage is unlikely and eventual resistance is the rule. Sapanisertib (INK128), is a second generation mTOR inhibitor drug that directly binds to the ATP-binding site of mTOR, thereby potently inhibiting both mTORC1 and mTORC2 (12), and overcoming the everolimus-resistant phosphorylation of 4EBP1 and AKT (13, 14). Also in contrast to rapalogs, sapanisertib has strong cytotoxic activity towards tumors (15–17).
Patient-derived xenografts (PDXs) have emerged as an important platform to elucidate new treatments and biomarkers for cancer (18–21). Human tumor samples, typically derived from biopsy or surgical samples, are implanted and propagated in nude mice in an effort to generate PDX lines that retain signaling pathways (22) and the idiosyncratic intra-tumor heterogeneity that typifies human cancer (23–25). PDX models have proven to be particularly useful tools for predicting the effectiveness of new drug therapies (26–29), and modeling drug resistance (30, 31). Although multiple PDX models have been successfully developed for pancreatic ductal adenocarcinoma (32, 33) and other cancers (20, 21), a PDX model for PNETs has not been described.
Here we report the establishment of the first PDX model for PNETs, and show using this model that the second generation mTOR inhibitor drug sapanisertib may serve as an effective new treatment option for patients with PNET and everolimus-resistant PNET.
Materials and methods
PNET xenografts.
Patient written informed consent was obtained, and the research protocol was approved by the institutional review board of the University of California, San Francisco. All animal studies were conducted under an animal use protocol approved by the University of California San Francisco animal care and use committee. A patient with an advanced PNET producing insulin underwent palliative debulking of liver metastases to ameliorate symptomatic hypoglycemia. To establish xenografts, non-diagnostic portions of tumors removed during the hepatic resection were minced under aseptic conditions and approximately 20 μL of tumor:matrigel (1:1) implanted subcutaneously into female athymic nude mice (Envigo, Indianapolis, IN). Non-implanted fragments were flash-frozen in liquid nitrogen and banked. For the initial implantation, bilateral injections were made into the flanks of mice to establish the xenograft passage 0 (P0). When tumor burden reached ~700–1000 mm3, mice were sacrificed; tumors were then resected, processed and injected subcutaneously into new recipient animals. To establish cohorts of mice for drug trials, individual tumors were implanted into 4–5 mice and allowed to reach maximum protocol size. Tumors were then minced and implanted into 30 mice and allowed to grow. When the average tumor size of the cohort reached 150 mm3, animals with high and low tumor volumes were discarded from the study and the remaining animals randomized into control and treatment groups based on tumor volume. Tumor volume was measured every 3–7 days during drug treatment. For single-arm drug studies, waterfall plots were used to summarize the distribution of tumor growth responses to treatments, as previously described (34). Briefly, the tumor size at the start of treatment was defined as the baseline volume, and the smallest tumor volume recorded during treatment was defined as the best response. If the tumor did not respond and its size did not decrease below baseline throughout treatment, then the largest change in tumor size up to 100% was used instead.
Drug treatment.
Everolimus was purchased commercially and sapanisertib was synthesized by KS and GD. The drugs were prepared as 2.5 mg/mL suspensions in a vehicle of 5% 1-methyl-2 pyrrolidinone (NMP), 80% polyvinyl pyrralidone (PVP) and 15% water for initial pharmacodynamic studies and then in 3.1% NMP, 81.6% PVP, 15.3% water for tumor regression studies to reduce toxicity. Single tumor-bearing mice were dosed once daily with 10 mg/kg body weight (BW) everolimus, 1 mg/kg BW sapanisertib, or vehicle by oral gavage (OG). Mice were sacrificed, and tumors harvested and split and paraffin embedded for histological analysis or flash frozen for whole tissue western blotting and molecular interrogation at experimental endpoints.
MicroPET Imaging.
68Ga-DOTATATE was prepared and purified in the UCSF radiochemistry laboratory. Briefly, 68GaCl3 was eluted from a 68Ge/68Ga generator (Eckert&Ziegler, Valencia CA) and reacted with DOTATATE (DOTA0, Tyr3, Thr8]octreotide) in sodium acetate buffer. Solid phase extraction purification gave 68Ga-DOTATATE in injectable ethanol/saline solution. PNET-PDX mice were injected with 120–180 μCi (4.4–6.7 MBq) of 68Ga-DOTATATE through a tail vein catheter. Static PET/CT images were taken on a Siemens Inveon microPET/CT scanner for 30 minutes at 2.75h post tracer injection. Images were reconstructed and viewed using AMIDE software (freeware, amide.sourceforge.net).
Western blot analysis.
All antibodies for western blot analysis were purchased from Cell Signaling Technology (Danvers, MA). Blots were visualized by using either HRP conjugated secondary antibodies and chemiluminescent reagent from Pierce Protein Research Products (Rockford, IL) or by fluorescent secondary antibodies. GAPDH was used as an internal control and each experiment was done in independent biological duplicates.
Immunohistochemistry.
Tissues were fixed in Z-fix (Anatech, Battle Creek, MI) and processed for paraffin or frozen sections using standard methods. The following primary antibodies were used: rabbit anti-CHGA (Immunostar, Hudson, MA); rabbit anti-5-HT (Immunostar); mouse monoclonal anti-5-HT (Dako, Carpinteria, CA), guinea pig polyclonal anti-INS (Dako), mouse monoclonal anti-Ki-67 (clone MIB-1, Dako). Secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were FITC-conjugated goat anti-mouse and anti-rabbit; FITC-conjugated donkey anti-goat and anti-mouse; Cy3-conjugated donkey anti-goat, goat anti-mouse, and anti-rabbit; HRP-conjugated goat anti-mouse and anti-rabbit. Slides were imaged on an Axioskop 2 microscope (Zeiss, Thornwood, NY) or on an LSM510 META confocal microscope (Zeiss). Ki-67 was used to assess proliferation. The mitotic index was calculated on the H&E sections for the histology samples according to the 2010 WHO guidelines. Ki-67 indices for the cell block sections were calculated as the total number of tumor cells with positive nuclear staining divided by the total number of tumor cells present.
Whole-exome sequencing.
‘Genomic DNA was extracted using standard procedures. Targeted capture and massive parallel sequencing were performed at the UCSF Institute for Human Genetics. Briefly, genomic DNA was sheared by Covaris S2 (Woburn, MA) to a target size of 200–300bp and assembled into a library with TruSeq adapters containing indexes that differentiates different libraries in a capture reaction as well as a sequencing run (Kapa Biosystems, Wilmington, MA). Libraries were pooled into a capture reaction that contains biotinylated DNA oligonucleotides (called ‘baits’) from Roche-Nimblegen SeqCap EZ Human Exome Library v3.0 (Madison, WI) for 72 hours. The DNA bait-DNA hybrids were then pulled out of the complex mixture by incubation with streptavidin-labeled magnetic beads and captured onto a strong magnet. After washing, the targeted DNA of interest was eluted and subjected to 18 cycles of DNA amplification. Captured DNA libraries were sequenced with the Illumina HiSeq 2500 (Hayward, CA), yielding 150 (2X75) base pairs from the final library fragments. Sequence data were mapped to the GRCh37 reference genome and processed using the Bina Genomic Analysis Platform (Belmont, CA). Somatic mutation caller tools included JointSNVMix (35), MuTect (35), Somatic Sniper (36), VarDict (37) and VarScan (37). Somatic mutations were also filtered based on read quality, allele frequency (dbSNP (38), 1000 Genomes (39), ExAC (40), COSMIC (41)), and functional effect (SIFT (42), Polyphen (43), SnpEff (44)). Sequencing data can be found at the Sequence Read Archive (SRA Study Accession Number: SRP162135).
Results
Development and characterization of a PDX model of PNET
To establish xenografts, non-diagnostic portions of PNET liver metastasis tissue were removed during partial hepatic resection surgery in a patient with an advanced, well-differentiated PNET producing insulin, processed as described and subcutaneously implanted into female athymic nude mice (Fig. 1A). The successfully engrafted tumor maintained the characteristics of the patient tumor tissue through multiple rounds of serial transplantation in athymic mice to successfully establish this PDX-PNET.
Figure 1.

Generation and characterization of the patient-derived xenograft model of PNET (PDX-PNET). A, non-diagnostic portion of metastasized PNETs were removed during the hepatic resection and expanded in a cohort of female athymic nude mice. B and C, Comparison of neuroendocrine biomarkers in original patient tumor and passage-6 PDX-PNET tumor using RNA sequencing (B) and immunofluorescence (C). In B, a “+” means a gene is expressed. In C, DNA is in blue while CHGA, INS and 5-HT are in green.
We compared gene expression between the original patient PNET liver metastasis tissue (Liv-Met) and PDX-PNET tissue harvested at passage 6 (Fig. 1B). Using RNA sequencing (RNA-seq), we detected the expression of neuroendocrine tumor genes CHGA, INS and TPH1 (2). We also detected NKX2–2, ASCL1, and FEV, which encode developmental transcription factors critical to the differentiation and hormone expression of neuroendocrine tumors (45, 46). Consistent with this, immunofluorescence analysis of tissue sections revealed strong expression of CHGA, INS and 5-HT, the metabolic product of TPH1, in both Liv-Met and PDX-PNET tissues (Fig. 1C). PDX-PNETs also retained a well-differentiated neuroendocrine tumor morphology with a Ki-67 index of 6–8% (G2).
PNETs express somatostatin receptors (47); consequently, the radiolabeled somatostatin analog Gallium-68 DOTATATE is used as a tracer for positron emission tomography-computed tomography (PET-CT) imaging of PNETs in the clinic (48). We therefore evaluated whether Gallium-68 DOTATATE could be used to perform functional PET-CT imaging of PDX-PNETs in vivo. At 3 hours post-injection, Gallium-68 DOTATATE PET-CT clearly colocalized with the subcutaneous PDX-PNETs (Fig. 2), showing that Gallium-68 DOTATATE can be used to perform functional PET-CT imaging in this model. Importantly, a linear relationship was observed between tumor volume and normalized Gallium-68 DOTATATE counts over a large range of tumor size (Fig. 2C).
Figure 2.
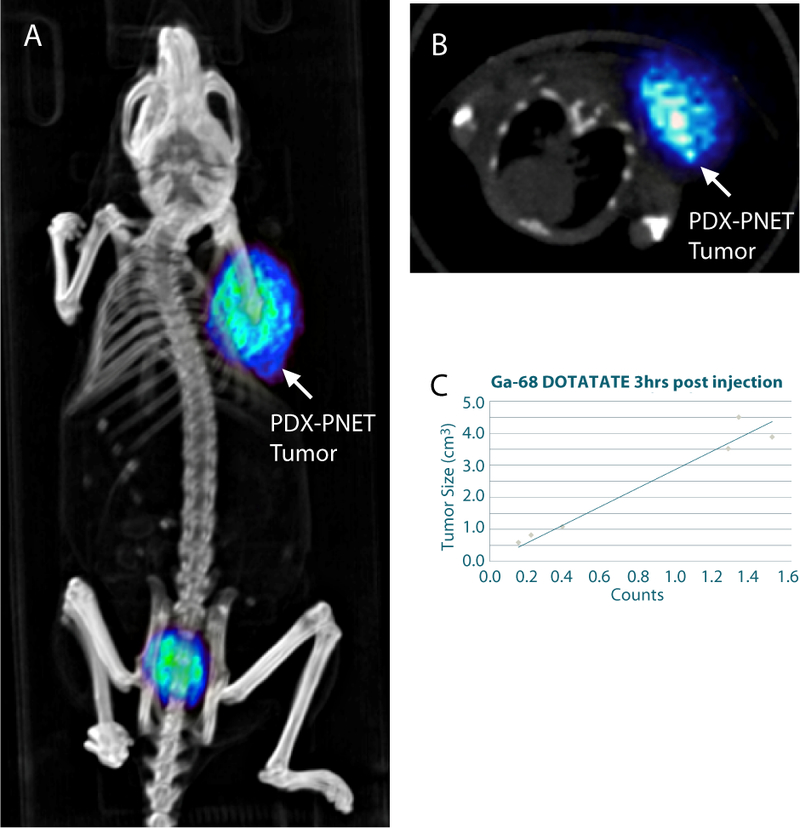
68Ga-DOTATATE PET-CT imaging of the PDX-PNET model. A, 3D rendered PET/CT image showing flank tumor tracer uptake. B, axial view at the level of the tumor. C, relationship between tumor size and normalized 68Ga-DOTATATE PET/CT counts.
Using whole-exome sequencing to identify genetic mutations harbored in the PDX-PNET model, we found that PDX-PNETs contained mutations in known PNET-associated genes (4, 6), such as MEN1, BRCA2, PTEN, and SETD2 (Fig. 3A). We did not observe mutations in genes commonly associated with pancreatic ductal adenocarcinoma, such as KRAS, TP53, CDKN2A, SMAD3, SMAD4, TGFBR1 (49, 50). Together, these findings provide strong evidence that the PDX-PNET model is a bona fide PNET model.
Figure 3.
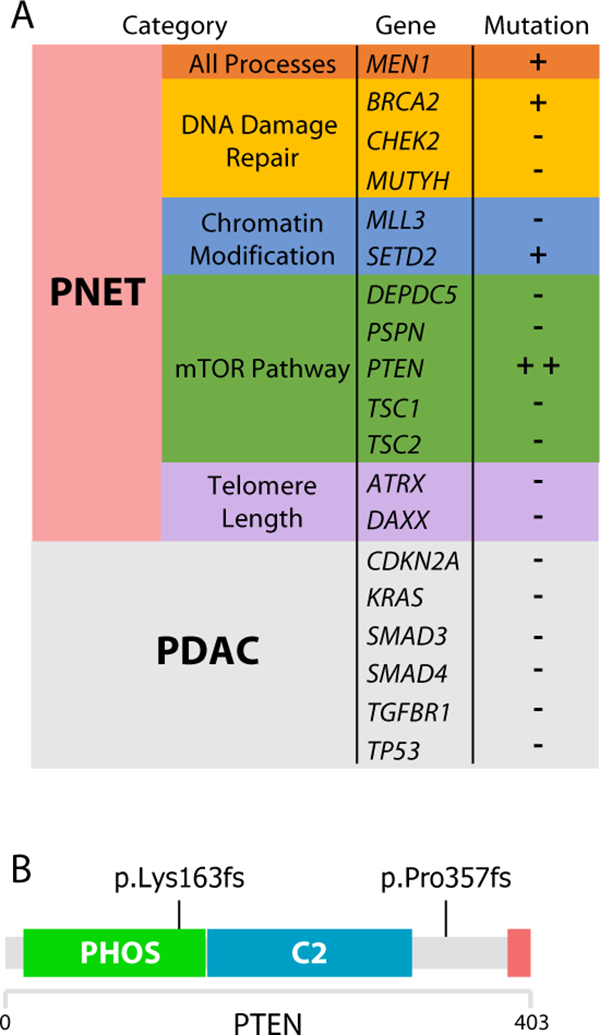
PDX-PNETs harbor mutations in genes commonly associated with PNETs and mTOR pathway activation. A, whole-exome sequencing of PDX-PNET reveals mutations in genes commonly found in PNET. B, two frameshift mutations were identified in PTEN. Green: Phosphatase domain; Blue: C2 domain; Red: PDZ binding domain.
Evaluating mTOR inhibitor drugs everolimus and sapanisertib in the PDX-PNET model
PTEN functions as an inhibitor of the mTOR pathway (51). Mutations in PTEN cause mTOR pathway activation (51), and can predict tumor response to mTOR inhibitor drugs (52). We found two deleterious frameshift mutations in PTEN (Fig. 3B), suggesting mTOR pathway activation in the PDX-PNET model. Consistent with this, western blot analysis revealed phosphorylation of the mTORC1 downstream targets 4EBP1 and RPS6 (Fig. 4B, lanes 1–3). We also detected the phosphorylation of mTORC2 target AKT. Phosphorylation of S6K1, a substrate of mTORC1 that phosphorylates RPS6, was barely detectable in PDX-PNETs.
Figure 4.
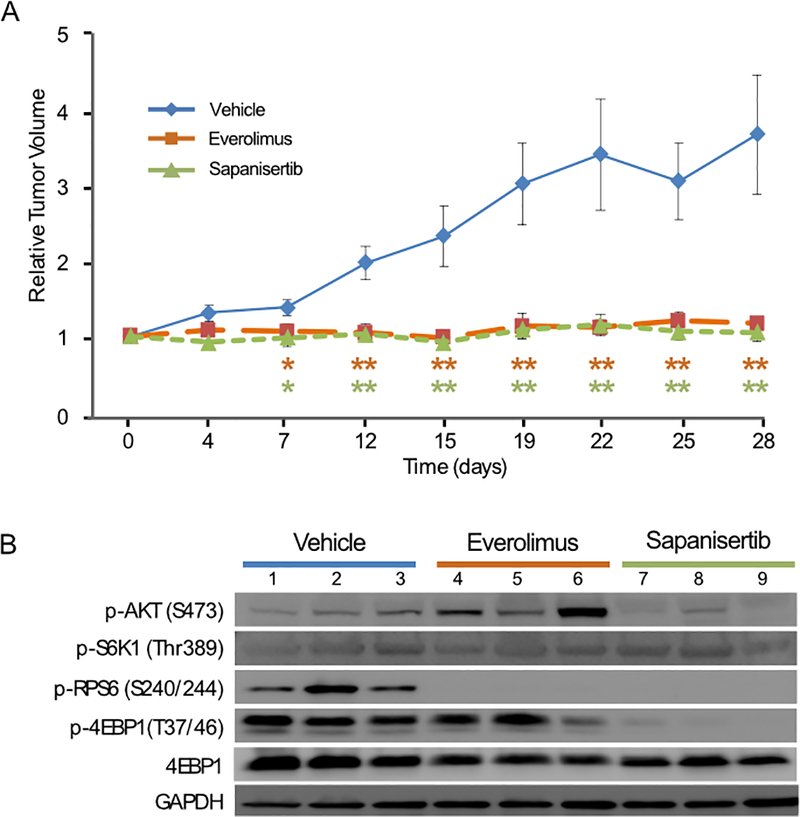
Response of PDX-PNETs to the mTOR inhibitor drugs everolimus and sapanisertib. A, growth chart of PDX-PNETs treated with vehicle control, everolimus or sapanisertib for 28 days (vehicle control n=5, everolimus n=6, sapanisertib n=6). The p-values were calculated by 2-tailed Student’s t test (*p<0.05, **p<0.01). Error bars indicate the standard deviation from the mean. B, Western blot analysis of mTOR pathway targets in PDX-PNETs harvested after 28 days of treatment with either vehicle control, everolimus, or sapanisertib. Each lane represents a single xenograft harvested from a unique mouse.
Everolimus, an mTOR inhibitor drug that inhibits mTORC1 but not mTORC2, demonstrated unequivocal anti-tumor activity in a phase III study leading to its approval for the treatment of advanced PNETs (9). We therefore evaluated the response of PDX-PNETs to treatment with everolimus. We also evaluated sapanisertib, a second-generation mTOR inhibitor that directly binds the ATP binding site of mTOR and inhibits both mTORC1 and mTORC2 (12). The optimal everolimus and sapanisertib dose was determined by pharmacodynamic studies (Supplementary Fig. 1). Single tumor-bearing mice were treated once daily with everolimus (10 mg/kg BW), sapanisertib (1 mg/kg BW), or vehicle by OG for 28-days. Treatment was well tolerated in all groups. We found that vehicle treated control PDX-PNETs grew several-fold in size (n=5, Fig. 4A). In contrast, the growth of PDX-PNETs treated with either everolimus (n=6) or sapanisertib (n=6) was significantly blocked (Fig. 4A).
To evaluate the in vivo effects of everolimus and sapanisertib on mTOR pathway targets, we harvested PDX-PNET tissues and performed western blot analysis. Everolimus completely inhibited the phosphorylation of mTORC1 target RPS6, but not 4EBP1 (Fig. 4B, lanes 4–6). AKT phosphorylation was increased, consistent with rapalogs having no activity towards mTORC2 targets and the feedback activation of AKT that has been observed in human trials with other rapamycin derivatives (11). In contrast, sapanisertib inhibited the phosphorylation of both mTORC1 targets 4EBP1 and RPS6 (Fig. 4B, lanes 7–9). Also in contrast to everolimus, sapanisertib inhibited the phosphorylation of the mTORC2 target AKT. These findings demonstrate that sapanisertib more completely inhibits the mTOR pathway than everolimus in the PDX-PNETs.
The PDX-PNET Model for Studying Acquired Drug Resistance in PNETs
The development of drug resistance is a major limitation of PNET clinical response to everolimus treatment (53). We therefore sought to generate everolimus-resistant PDX-PNETs by treating a large cohort of tumor bearing animals once daily with everolimus for several months (n=34). Because this was a single-arm study, everolimus resistance was defined as a doubling in the tumor volume from the day of treatment. The baseline volume was defined as the tumor size at the start of treatment, and the smallest tumor volume recorded during treatment was defined as the best response (34). If the tumor failed to respond, then the largest change in tumor size up to 100% was used for the best response. In Fig. 5A, animals are listed in order of increasing percentage response to everolimus. Most PDX-PNET tumors (29/34) experienced a reduction in tumor volume during everolimus treatment (Fig. 5A, numbers 6–34), five failed to respond (Fig. 5A, numbers 1–5), and seven experienced complete regression (Fig. 5A, numbers 28–34). The variability in response may be due to the heterogeneous nature of the PDX-PNET model (23–25) or experimental variation. From this group, ten everolimus-resistant PDX-PNETs were identified (Fig. 5B). While all ten everolimus-resistant tumors doubled in size during everolimus treatment, their time to resistance was variable, ranging between 7–236 days, and with a median time to resistance of 110 days. The best response of the ten everolimus-resistant PDX-PNET tumors to everolimus was also variable, with some not responding at all (Fig. 5A, numbers 1 and 3), and others responding with a greater than 50% reduction in tumor volume (Fig. 5A, numbers 22 and 23).
Figure 5.
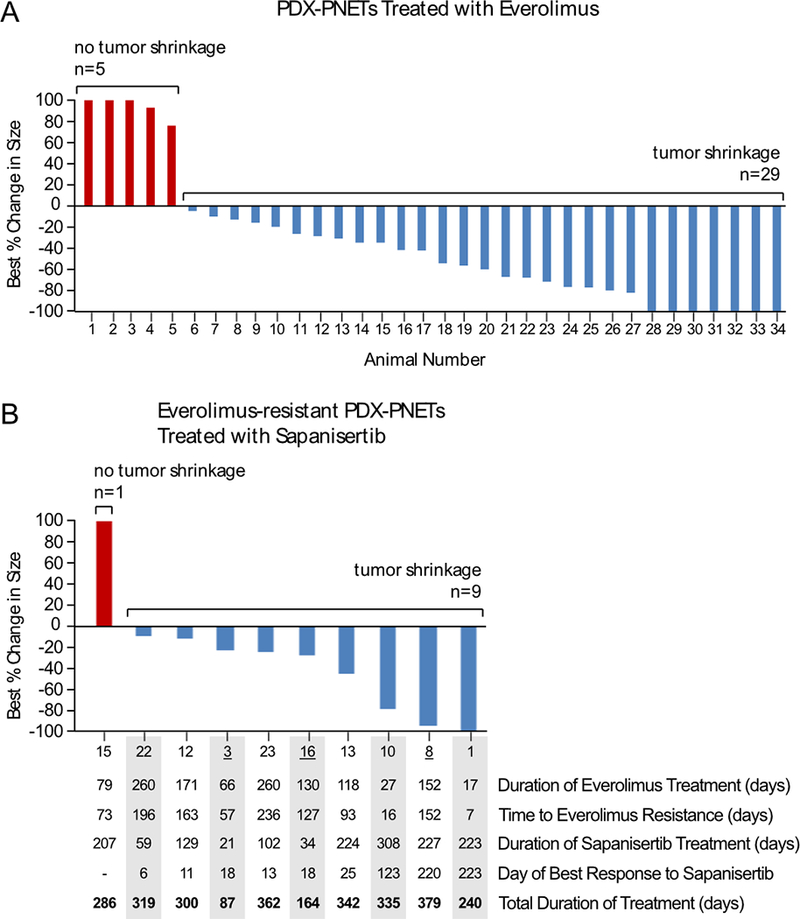
Response of everolimus-resistant PDX-PNET tumors to sapanisertib. A, the best response of tumors treated with everolimus, as compared with pretreatment baseline (n=34). Numbers along the x-axis indicate arbitrarily assigned animal numbers in order of increasing percentage response to everolimus. The bars indicate the percent change in tumor burden from baseline. B, the best response to sapanisertib in 10 animals with everolimus-resistant PDX-PNET tumors. Selected tumor characteristics are listed in the table below the graph. Animals are listed in order of increasing percentage response to sapanisertib, with listed numbers corresponding to those in Figure 5A. Sapanisertib treatment ended when tumors either regressed (n=1), developed sapanisertib resistance and exceeded 5-times the original volume (n=6), or animals died (n=3). Underlined numbers indicate animals that died during treatment.
Everolimus partially inhibits mTORC1 and fails to inhibit mTORC2, suggesting that resistance to everolimus may be overcome by the more potent mTOR pathway inhibitor drug sapanisertib, which inhibits both mTORC1 and mTORC2. We therefore evaluated the effect of sapanisertib treatment on everolimus-resistant PDX-PNETs (Fig. 5B). In Fig. 5B, animals are listed in order of increasing percentage response to sapanisertib, with listed numbers corresponding to those in Figure 5A. We found that most everolimus-resistant PDX-PNET tumors (9/10) experienced a reduction in tumor volume during sapanisertib treatment, one failed to respond, and one experienced complete regression. The everolimus-resistant PDX-PNET tumor that completely regressed during sapanisertib treatment had not responded to everolimus treatment (Fig. 5, number 1), while the everolimus-resistant PDX-PNET tumor that did not respond to sapanisertib had a moderate response to everolimus (Fig. 5, number 15).
Discussion
Patient-derived xenograft (PDX) models have proven to be extremely useful in drug discovery research (18–21). Unlike cell lines and many genetically engineered mouse models, PDX models retain the cellular and genetic heterogeneity of the primary human tumor (23–25) and may therefore better model many important aspects of the disease, such as drug resistance (30, 31). We report the establishment of the first PDX model for pancreatic neuroendocrine tumors (PDX-PNETs). We also refer to this as the HNV PDX-PNET model as the implanted tumor tissue had been resected from segment V of the patient’s liver. There have been many previous attempts by us and other groups to establish a PDX model of PNET. We do not know why this one was successful, but the PNET was unusually aggressive, had metastasized and ultimately killed the patient. We speculate that the unique aggressiveness of the tumor enabled it to successfully survive and grow in a foreign mouse host. Identifying a molecular cause for this aggressiveness is not trivial, especially as this is currently an n of 1. Additional sequencing analyses and the establishment of additional PDX-PNET models may provide some insight.
Histological analysis, gene expression profiling and whole-exome sequencing confirmed that the PDX-PNET tumors retained the hallmark features of PNETs. Interestingly, PDX-PNET tumors harbored PTEN mutations and responded to mTOR inhibitor drugs everolimus and sapanisertib, suggesting that the PNET mutational profile may determine drug response. In addition to PTEN, PDX-PNETs harbored mutations in MEN1 and BRCA2 (Fig. 3). MEN1 encodes the tumor-suppressor menin and germline mutations in MEN1 cause multiple endocrine neoplasia type 1 (MEN1) (54). A recent study identified a synthetic lethal interaction between MEN1 mutation and MEK1/2 inhibition in neuroendocrine cells (55), suggesting that MEK1/2 inhibitor drugs FDA-approved for other cancers such as trametinib or cobimetinib may have clinical activity against PNETs. Loss of BRCA2 drives one of the mutational signatures found in PNETs (4, 56) and poly(ADP-ribose) polymerases (PARP) inhibitors have shown promise in clinical studies against BRCA2-mutated tumors (57, 58). Future studies evaluating these treatments on PDX-PNET tumors may be informative.
PDX-PNET tumors responded equally to everolimus and sapanisertib despite everolimus treatment leading to AKT activation and not suppressing p4EBP (Fig. 4), suggesting that these are not the mechanisms of resistance, at least in the short-term. Comparing time to progression may reveal a potential benefit for sapanisertib over everolimus, and would also more closely resemble endpoints used in clinical trials. One particularly promising finding from our study was that the majority of everolimus-resistant PDX-PNETs responded to sapanisertib (Fig. 5), which directly targets mTOR and inhibits the activity of both mTORC1 and mTORC2 (8). Sapanisertib is currently being evaluated in multiple cancers across 11 clinical trials, including a phase II study of rapalog-resistant advanced PNET (ClinicalTrials.gov Identifier: NCT02893930). The results from this trial may help clarify the value of the HNV PDX-PNET model in selecting PNET therapies and identifying PNET biomarkers of drug response and resistance.
Supplementary Material
Acknowledgments
The authors would like to thank Bina and Roche for use of their genomic bioinformatic platform and expert advice, the UCSF Preclinical Therapeutics Core for their help with HNV PDX-PNET model development and preclinical study execution, Pamela Derish for expert help with writing and preparation of the manuscript, the Placzek Family Foundation for their generous support, and Larry and Margaret Hauben for their vision and unwavering support.
Grant Support and Financial Information
This work was supported by grants from the Neuroendocrine Tumor Research Foundation (NETRF) to E.K. Nakakura, C.E. Chamberlain and M.S. German, the NIH (P30 DK63720 to M.S. German) and the NETRF-American Association for Cancer Research Grant to E.K. Nakakura (AACR Grant ID 12–60-33-NAKA).
Footnotes
Conflict of interest: Kevan Shokat is an inventor on UCSF patents related to sapanisertib/INK128, currently licensed to Takeda Pharmaceuticals and a consultant to Takeda Pharmaceuticals. Other authors have declared no conflict of interest.
References
- 1.Yao JC, Eisner MP, Leary C, Dagohoy C, Phan A, Rashid A, Hassan M, and Evans DB 2007. Population-based study of islet cell carcinoma. Ann Surg Oncol 14:3492–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, et al. 2008. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 26:3063–3072. [DOI] [PubMed] [Google Scholar]
- 3.Bosman FT, Carneiro F, Hruban RH & Theise ND 2010. WHO Classification of Tumours of the Digestive System: International Agency for Research on Cancer. [Google Scholar]
- 4.Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A, et al. 2017. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 543:65–71. [DOI] [PubMed] [Google Scholar]
- 5.Saxton RA, and Sabatini DM 2017. mTOR Signaling in Growth, Metabolism, and Disease. Cell 168:960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, et al. 2011. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331:1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M, Piemonti L, Capurso G, Di Florio A, delle Fave G, et al. 2010. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol 28:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benjamin D, Colombi M, Moroni C, and Hall MN 2011. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov 10:868–880. [DOI] [PubMed] [Google Scholar]
- 9.Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, et al. 2011. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 364:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, Tomasek J, Raderer M, Lahner H, Voi M, et al. 2016. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 387:968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. 2006. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66:1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. 2012. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, and Shokat KM 2009. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 7:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, and Gray NS 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284:8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, and Ruggero D 2010. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 17:249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, et al. 2010. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med 16:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh AC, and Ruggero D 2010. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin Cancer Res 16:4914–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM, et al. 2014. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 4:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siolas D, and Hannon GJ 2013. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res 73:5315–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, and Eckhardt SG 2012. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 9:338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Day CP, Merlino G, and Van Dyke T 2015. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, Yung R, Parmigiani G, Dorsch M, Peacock CD, et al. 2009. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res 69:3364–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Cora D, Di Nicolantonio F, Buscarino M, Petti C, et al. 2011. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 1:508–523. [DOI] [PubMed] [Google Scholar]
- 24.Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, et al. 2015. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 526:263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, et al. 2011. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 17:1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, et al. 2015. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 21:1318–1325. [DOI] [PubMed] [Google Scholar]
- 27.Arrowsmith J 2011. Trial watch: Phase II failures: 2008–2010. Nat Rev Drug Discov 10:328–329. [DOI] [PubMed] [Google Scholar]
- 28.Arrowsmith J, and Miller P 2013. Trial watch: phase II and phase III attrition rates 2011–2012. Nat Rev Drug Discov 12:569. [DOI] [PubMed] [Google Scholar]
- 29.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, and Schacht AL 2010. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 9:203–214. [DOI] [PubMed] [Google Scholar]
- 30.Cottu P, Bieche I, Assayag F, El Botty R, Chateau-Joubert S, Thuleau A, Bagarre T, Albaud B, Rapinat A, Gentien D, et al. 2014. Acquired resistance to endocrine treatments is associated with tumor-specific molecular changes in patient-derived luminal breast cancer xenografts. Clin Cancer Res 20:4314–4325. [DOI] [PubMed] [Google Scholar]
- 31.Ter Brugge P, Kristel P, van der Burg E, Boon U, de Maaker M, Lips E, Mulder L, de Ruiter J, Moutinho C, Gevensleben H, et al. 2016. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J Natl Cancer Inst 108. [DOI] [PubMed] [Google Scholar]
- 32.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, Shi C, Danenberg K, Danenberg PV, Kuramochi H, et al. 2006. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 12:4652–4661. [DOI] [PubMed] [Google Scholar]
- 33.Damhofer H, Ebbing EA, Steins A, Welling L, Tol JA, Krishnadath KK, van Leusden T, van de Vijver MJ, Besselink MG, Busch OR, et al. 2015. Establishment of patient-derived xenograft models and cell lines for malignancies of the upper gastrointestinal tract. J Transl Med 13:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, et al. 2010. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363:1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth A, Ding J, Morin R, Crisan A, Ha G, Giuliany R, Bashashati A, Hirst M, Turashvili G, Oloumi A, et al. 2012. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 28:907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, and Ding L 2012. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, and Sirotkin K 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR 2015. A global reference for human genetic variation. Nature 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. 2017. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 45:D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng PC, and Henikoff S 2003. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adzhubei I, Jordan DM, and Sunyaev SR 2013. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang YC, Zuraek MB, Kosaka Y, Ota Y, German MS, Deneris ES, Bergsland EK, Donner DB, Warren RS, and Nakakura EK 2010. The ETS oncogene family transcription factor FEV identifies serotonin-producing cells in normal and neoplastic small intestine. Endocr Relat Cancer 17:283–291. [DOI] [PubMed] [Google Scholar]
- 46.Wang YC, Gallego-Arteche E, Iezza G, Yuan X, Matli MR, Choo SP, Zuraek MB, Gogia R, Lynn FC, German MS, et al. 2009. Homeodomain transcription factor NKX2.2 functions in immature cells to control enteroendocrine differentiation and is expressed in gastrointestinal neuroendocrine tumors. Endocr Relat Cancer 16:267–279. [DOI] [PubMed] [Google Scholar]
- 47.Reubi JC, and Waser B 2003. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging 30:781–793. [DOI] [PubMed] [Google Scholar]
- 48.Mojtahedi A, Thamake S, Tworowska I, Ranganathan D, and Delpassand ES 2014. The value of (68)Ga-DOTATATE PET/CT in diagnosis and management of neuroendocrine tumors compared to current FDA approved imaging modalities: a review of literature. Am J Nucl Med Mol Imaging 4:426–434. [PMC free article] [PubMed] [Google Scholar]
- 49.Hyman DM, Taylor BS, and Baselga J 2017. Implementing Genome-Driven Oncology. Cell 168:584–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. 2015. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song MS, Salmena L, and Pandolfi PP 2012. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13:283–296. [DOI] [PubMed] [Google Scholar]
- 52.Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F, Gonzalez-Angulo AM, Rashid A, Crosby K, Dong M, et al. 2012. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res 18:1777–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yao JC, Phan AT, Jehl V, Shah G, and Meric-Bernstam F 2013. Everolimus in advanced pancreatic neuroendocrine tumors: the clinical experience. Cancer Res 73:1449–1453. [DOI] [PubMed] [Google Scholar]
- 54.Thakker RV 2010. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab 24:355–370. [DOI] [PubMed] [Google Scholar]
- 55.Chamberlain CE, Scheel DW, McGlynn K, Kim H, Miyatsuka T, Wang J, Nguyen V, Zhao S, Mavropoulos A, Abraham AG, et al. 2014. Menin determines K-RAS proliferative outputs in endocrine cells. J Clin Invest 124:4093–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadanandam A, Wullschleger S, Lyssiotis CA, Grotzinger C, Barbi S, Bersani S, Korner J, Wafy I, Mafficini A, Lawlor RT, et al. 2015. A Cross-Species Analysis in Pancreatic Neuroendocrine Tumors Reveals Molecular Subtypes with Distinctive Clinical, Metastatic, Developmental, and Metabolic Characteristics. Cancer Discov 5:1296–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434:913–917. [DOI] [PubMed] [Google Scholar]
- 58.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. 2015. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med 373:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.