

Abstract
NUT Carcinoma (NC) is a rare, distinctly aggressive subtype of squamous carcinoma defined by the presence of NUT-fusion oncogenes resulting from chromosomal translocation. In most cases, the NUT gene (NUTM1) is fused to bromodomain containing 4 (BRD4) forming the BRD4-NUT oncogene. Here, a novel fusion partner to NUT was discovered using next-generation sequencing (NGS) and fluorescence in situ hybridization (FISH) from a young patient with an undifferentiated malignant round cell tumor. Interestingly, the NUT-fusion identified involved ZNF592, a zinc finger containing protein, which was previously identified as a component of the BRD4-NUT complex. In BRD4-NUT-expressing NC cells, wild-type ZNF592 and other associated “Z4” complex proteins, including ZNF532 and ZMYND8, co-localize with BRD4-NUT in characteristic nuclear foci. Furthermore, ectopic expression of BRD4-NUT in a non-NC cell line induces sequestration of Z4 factors to BRD4-NUT foci. Finally, the data demonstrate the specific dependency of NC cells on Z4 modules, ZNF532 and ZNF592.
Implications:
This study establishes the oncogenic role of Z4 factors in NC, offering potential new targeted therapeutic strategies in this incurable cancer.
Introduction
NUT Carcinoma (NC, also known as NUT Midline Carcinoma) is a rare, aggressive cancer of squamous origin affecting patients of all ages, but predominantly children and young adults. NC is one of the most aggressive solid tumors known, with a median survival of 6.7 months (1), and no known effective treatment, despite the emergence of targeted inhibitors (2). NC is characterized by the presence of chromosomal translocation involving rearrangement of the NUT gene (also known as NUTM1) on chromosome 15q14 encoding the nuclear protein in testis (NUT) (3). In most cases, NUT is fused to BRD4, and its encoded fusion oncoprotein, BRD4-NUT, drives growth through the blockade of differentiation and maintenance of tumor growth (4). BRD4-NUT function depends on its binding to acetylated chromatin through the bromodomains of BRD4, as shown through mutagenesis of key acetyl-histone binding amino acid residues within the bromodomains of BRD4 (5). Indeed, the anti-oncogenic activity of the acetyl-histone mimetic prototype bromodomain inhibitor, JQ1, was first demonstrated in NC (6), and has subsequently been shown to have activity in human patients (2). The findings in NC have led to interest in this class of drug in a broad range of cancers, however its clinical efficacy in NC and other malignancies has been severely limited by toxicity (2). Thus, there remains an urgent need for effective treatment of NC, necessitating a better understanding of the molecular pathway that drives this cancer.
The function of wild type NUT, whose expression is restricted to the testis (3), is almost completely unknown. Immunoprecipitation of NUT has revealed that NUT interacts with and activates the histone acetyltransferase activity (HAT) of p300 (7). A study using LacO transgene system with Lacl-fused NUT demonstrated that tethering of NUT on the chromatin is sufficient to induce transcriptional activation through recruitment of p300, BRD4 and P-TEFb subunits (8). These findings have led to the hypothesis that BRD4 bromodomains tether BRD4-NUT to chromatin containing acetylated histones, whereby recruitment of p300 leads to further acetylation, and iterative recruitment of BRD4-NUT and p300 in a feed-forward manner that leads to expansion of BRD4-NUT occupancy across broad regions of chromatin (7,9). Indeed, comprehensive analysis of genomic regions enriched by BRD4-NUT revealed that BRD4-NUT co-occupies with p300 unprecedented 100kb-2 Mbp ‘megadomains’ of acetylated chromatin (10). Megadomains drive transcription of underlying coding and non-coding DNA. The targets of BRD4-NUT megadomains critical to the growth and blockade of differentiation of all NCs analyzed includes MYC and TP63 (5,10,11). Despite these advances, very little is known of what factors determine how and where BRD4-NUT megadomains form, or what their oncogenic functions are.
While the BET family protein, BRD4, is considered the canonical fusion partner of NUT, several variant NUT-fusions have been reported in NC, including BRD3- (4), NSD3- (12), and ZNF532-NUT (11). All of the wild type counterparts of the variant fusion partners interact with BRD4, and through this interaction are components of the BRD4-NUT oncogenic complex (11). Thus, every known variant fusion causes aberrant association of BET proteins with NUT. Moreover, like BRD4-NUT, the known variant NUT-fusion oncoproteins form megadomains that drive expression of MYC (10,11), and enforce growth and the blockade of differentiation (4,12), implicating their key oncogenic role in NC. Hence, identification of variant NUT-fusion partners has proven a powerful means to identify indispensable members of the BRD4-NUT oncogenic complex, a model analogous to the many MLL-fusion genes in infantile leukemia that each play important roles in the super elongation complex (SEC) that drives that disease (13).
To illustrate this point, ZNF532 can be fused to NUT to drive the ZNF532-NUT variant of NC, but wild type ZNF532 is also present as a BRD4-NUT complex interacting protein in classic NC through its association with BRD4 (11). Normally, ZNF532 is a component of a transcriptional coregulator complex, here termed “Z4”, with other zinc finger containing proteins ZNF592, ZNF687 and ZMYND8 (14,15). Importantly, all four Z4 factors are present within the BRD4-NUT oncogenic complex (11), and as such may pathologically affect the function of both BRD4-NUT and Z4 complexes. Here we show direct evidence of the role of Z4 proteins in NC pathogenesis.
Materials and Methods
Fluorescence in situ hybridization (FISH)
Dual color bring-together and split-apart FISH on 5 micron, formalin-fixed, paraffin embedded (FFPE) sections of surgically removed primary pelvic bone tumor (UNC) was performed as described (16), using the probes described in Supplemental Materials and Methods.
Next Generation Sequencing (Archer® FusionPlex®)
An Anchored Multiplex PCR (AMP) assay was performed with Archer FusionPlex solid Tumor Kit (ArcherDX, Boulder, CO) for detection of targeted fusion transcripts using next generation sequencing (NGS) (17). Detailed protocol is described in Supplemental Materials and Methods.
Reverse Transcriptase PCR (RT-PCR) and Sequencing
Nested RT-PCR was performed as described in Supplemental Materials and Methods. PCR products were identified by 2% agarose gel electrophoresis, and gel extracted DNA fragments (QIAquick Gel Extraction kit, Qiagen) were cloned by TOPO™ TA Cloning™ Kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions, and sent for Sanger sequencing (Genewiz, South Plainfield, NJ).
Immunoblotting and Immunofluorescence
Western blots were performed as described (4). Antibodies used are listed in Supplemental Materials and Methods. Immunofluorescence (IF) were performed as described previously (7) using the antibodies described in Supplemental Materials and Methods.
Histology and immunohistochemistry (IHC)
Hematoxylin and eosin (H&E) staining was performed using standard procedures. Immunohistochemistry was performed on FFPE as described (11) using monoclonal antibody to NUT (clone C52B1, Cell Signaling Technologies, Danvers, MA).
Cell Culture
293T, TC-797 (18), PER-403 (19), 797TRex-FLAG-BRD4-NUT-HA (10), U2OSTRex-FLAG-BRD4-NUT, 797-ZNF532-NBioTAP cell lines were cultured under conditions described in the supplemental materials and methods.
siRNA transfections
Small interfering RNA (siRNA) (50nM) was transfected using Lipofectamine® RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer’s instructions. The sequence of siRNA is listed in Supplemental Materials and Methods.
Viability assay
Cells after transfection were plated at 8000 cells per well on 96-well plates and cultured for 48 hours or 96 hours (specified in figure legends). The cellular viability was determined by CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI) following the manufacturer’s protocol. Data are shown as mean ± standard deviation from three biological replicates. Statistics used are student’s t test (two-tailed), and a p value less than 0.01 was considered as significant.
Results
Identification of ZNF592-NUT fusion
A pelvic tumor in an 18-year-old female was biopsied and revealed an undifferentiated malignant round cell tumor (Fig. 1A) that lacked immunohistochemical (IHC) staining for known epithelial, mesenchymal, or hematolymphoid markers. Because of the undifferentiated characteristics of the tumor and the patient age, we performed IHC with NUT antibody, and this revealed diffuse nuclear speckled staining diagnostic of NC (Fig. 1A). We performed FISH to identify the NUT fusion partner using probes to the known NUT fusion partners including BRD4, BRD3, ZNF532, and NSD3, and all failed to demonstrate a fusion (Fig. S1A). The patient subsequently developed pulmonary metastases despite intensive chemotherapy and radiation and died 13 months after diagnosis.
Figure 1.
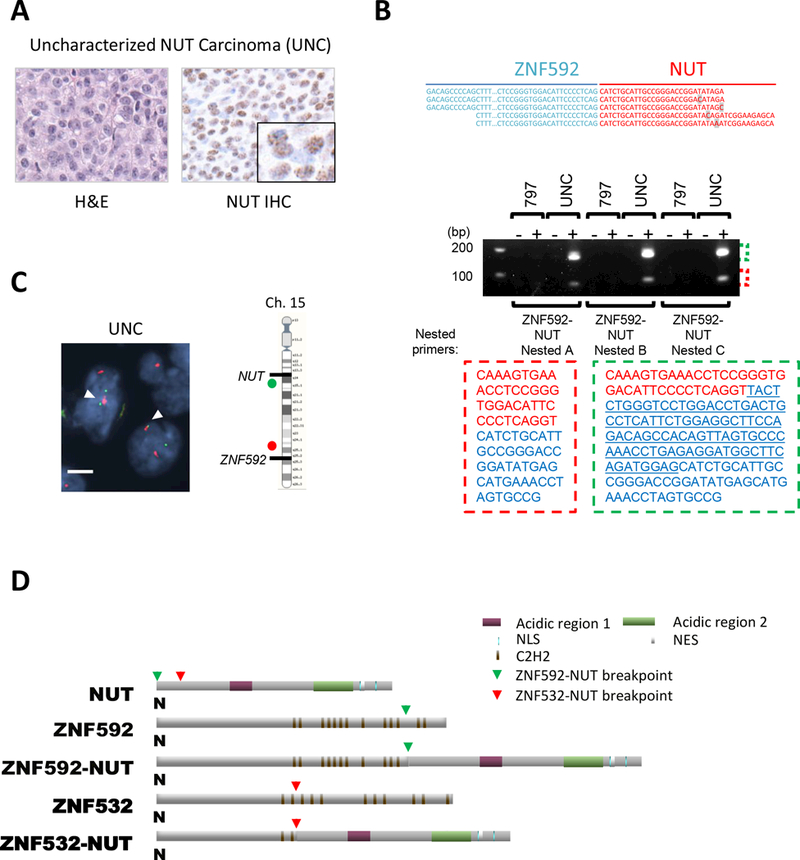
ZNF592-NUT fusion oncogene was discovered in FFPE tumor from a NC patient. A, H&E stained (left) and diagnostic anti-NUT immunohistochemistry (right) of resected NC of the pelvic bone of the 18 year old patient with uncharacterized NUT carcinoma (UNC) (magnification: 400×; inset, 1707×). B, Top: The result of Archer® FusionPlex® analysis indicating the sequence spanning the breakpoint of ZNF592 and NUT. Unique nucleotides in each read were highlighted in grey. Bottom: Gel electrophoresis of nested RT-PCR of TC-797 and UNC with (+) and without (–) reverse transcriptase. Nested PCR using three different nested primer sets (ZNF592-NUT Nested A/ B/ C) on TC-797 and UNC. The sequences corresponding to each band are indicated with red or green dashed lines. ZNF592 and NUT sequences are indicated by red and blue letters, respectively. The underlined sequence was detected only in the larger 200 bp band. C, Dual-color FISH bring-together assay using centromeric 5’ ZNF592 probes (red) and telomeric 3’ NUT (green). White arrows indicate the ZNF592-NUT fusion. magnification: 1000×, scale bar: 5 μm. D, Schematic of the ZNF592-NUT predicted encoded protein in comparison with ZNF532-NUT. Green arrows and red arrows denote ZNF592-NUT breakpoints and ZNF532-NUT breakpoints, respectively.
We then used a next-generation sequencing approach in an effort to identify the fusion partner to NUT using RNA extracted from archival FFPE sections of the patient’s tumor. Archer® FusionPlex®, a next-generation sequencing derivation of rapid amplification of cDNA ends (RACE), was employed and included targeting primers to the 3’ end of known NUT fusion breakpoints (exon 3). Analysis revealed sequences consistent with a fusion between NUT and a zinc finger protein encoding gene, ZNF592 (Fig. 1B).
Reverse transcriptase-polymerase chain reaction (RT-PCR) on the FFPE-extracted total RNA revealed two products, the larger of which is an in-frame ZNF592-NUT fusion, and the smaller is an out-of-frame sequence corresponding with the one detected by Archer® FusionPlex® (Fig. 1B). Of note, Archer® FusionPlex® performed on FFPE is often biased towards shorter fragments generated by RNA fragmentation from formalin fixation and does not always identify the in-frame functional product, as in this case. FISH confirmed a ZNF592-NUT fusion (Fig. 1C, S1B).
The identified ZNF592-NUT transcript is 7327 bp and contains the 5’ coding sequence of ZNF592 up to exon 10 fused with exons 2–10 of NUT (variant 1, RefSeq NM_001284292), and it is predicted to encode a 2249 amino acid protein containing amino acids 1–1091 of ZNF592 and 3–1160 of NUT. The ZNF592 moiety of the fusion protein retains eleven C2H2 zinc finger domains (Fig. 1D). The NUT portion that is essential for interaction between BRD4-NUT and p300 is entirely conserved in ZNF592-NUT, therefore ZNF592-NUT likely drives aberrant histone hyperacetylation through recruitment of p300, the oncogenic mechanism characteristic of other NCs (7,10,11). Indeed, immunofluorescence (IF) performed on the ZNF592-NUT positive UNC FFPE tumor revealed enrichment of histone H3 acetylated at lysine 27 (H3K27ac) at ZNF592-NUT foci (Fig. S1C), supporting our hypothesis that ZNF592-NUT could induce histone hyperacetylation through the mechanism similar to that of BRD-NUT and ZNF532-NUT.
Z4 (ZNF592, ZNF532, ZMYND8) components co-localize with and are sequestered by BRD4-NUT
We and others had previously shown that ZNF592 and other zinc finger proteins, ZNF532, ZNF687 and ZMYND8, collectively termed ‘Z4’, co-purify with BRD4/BRD4-NUT chromatin complex proteins (11,20). We hypothesized that if ZNF592 is associated with BRD4-NUT megadomains, it should form foci similar to those of BRD4-NUT foci observed within the nuclei of NC cells expressing endogenous BRD4-NUT. To determine the pattern of distribution of ZNF592 within the nuclei of NC cells that are driven by endogenous BRD4-NUT, we first performed IF on the NC patient-derived cell line, TC-797. IF revealed that ZNF592 forms discrete foci within the nuclei of TC-797 similar to those of BRD4-NUT (Fig. 2A) and ZNF592-NUT (Fig. 1A). Next, to test whether the nuclear distribution of Z4 factors overlaps with that of BRD4-NUT foci, we used a derivative of TC-797, 797TRex-FLAG-BRD4-NUT-HA (10), that can be induced to express a single dual HA-/FLAG- tagged BRD4-NUT (FLAG-BRD4-NUT-HA) transgene in the presence of tetracycline, to allow co-IF staining of tagged BRD4-NUT and ZNF592 or other Z4 factors. Of note, 797TRex also retains expression of endogenous BRD4-NUT, therefore untagged BRD4-NUT is constitutively expressed regardless of the induction of the tagged protein. We found that FLAG-BRD4-NUT-HA formed overlapping foci with ZNF592, as it did with ZNF532 and ZMYND8 (Fig. 2B). The overlap of these factors with FLAG-BRD4-NUT-HA foci was predicted based on the known association of these factors shown by proteomic and biochemical analyses, and indicates that these Z4 factors co-localize with BRD4-NUT (11,20). To test whether BRD4-NUT is sufficient to cause recruitment of Z4 factors to BRD4-NUT foci, we expressed FLAG-tagged BRD4-NUT in non-NC U2OSTRex cells containing a single isogenic copy of the transgene. We observed strong accumulation of ZNF592 and ZMYND8 at nuclear foci upon induction of the FLAG-BRD4-NUT (Fig. 2C). Of note, induction of FLAG-BRD4-NUT had minimal or no effect on levels of ZNF592 and ZMYND8 (Fig. S2).
Figure 2.
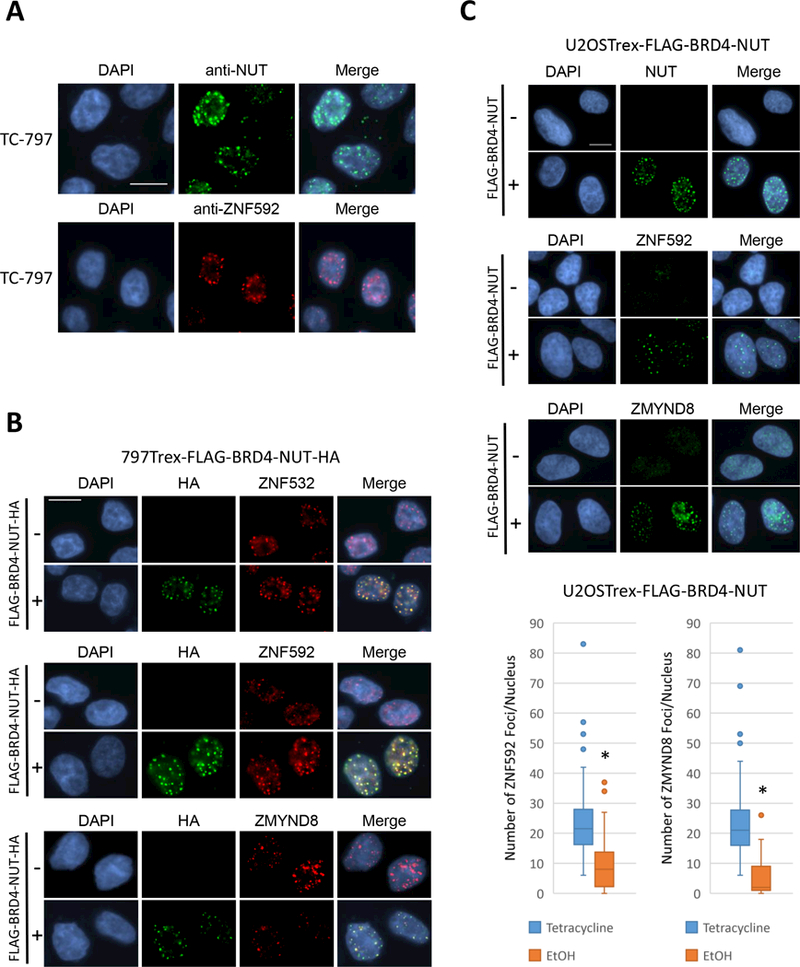
Z4 proteins co-localize with and are sequestered by BRD4-NUT. A, Immunofluorescence of TC-797 cells using anti-NUT and anti-ZNF592 antibodies. (magnification: 1000×, scale bar = 10 μm.) B, Immunofluorescence of 797TRex cells with (+) or without (–) tetracycline induction of FLAG-BRD4-NUT-HA expression using anti-HA (visualizing FLAG-BRD4-NUT-HA), anti-ZNF532, anti-ZNF592 and anti-ZMYND8 antibodies. (magnification:1000×, scale bar = 10 μm.) C, Top: Immunofluorescence of U2OSTRex cells with (+) or without (–) tetracycline induction of FLAG-BRD4-NUT expression using anti-NUT, anti-ZNF592 and anti-ZMYND8 antibodies. (magnification:1000×, scale bar = 10 μm.) Bottom: Boxplots showing the number of foci formed by ZNF592 (left) or ZMYND8 (right) in U2OSTRex-FLAG-BRD4-NUT cells treated with ethanol vs. tetracycline. FLAG-BRD4-NUT expression was induced by tetracycline treatment for 36 hours. Up to 40 nuclei were counted. *p<0.01, Student’s t test.
These findings, together with previous biochemical and proteomic studies (11,20), suggest that Z4 factors associate with BRD4-NUT within megadomains. Moreover, BRD4-NUT-driven foci formation of Z4 factors suggests a mechanism of pathological sequestration that may alter the normal function of Z4 proteins while enhancing those of BRD4-NUT, as first proposed for the sequestration of p300 by BRD4-NUT (7,8,21).
Knockdown of ZNF532 or ZNF592 reduces cell viability in NC cells
Considering the potential functional association of Z4 factors and BRD4-NUT, we tested the effect of siRNA knockdown of the two Z4 factors identified as oncogenic fusions to NUT in NC, ZNF532 and ZNF592, on viability of NC cells. We were unable to obtain sufficient knockdown of ZNF532 using seven siRNAs targeting the gene, therefore we utilized an alternative approach. Using CRISPR/Cas9, we genomically inserted a BioTAP tag into the 5’ end of both ZNF532 alleles in TC-797, and knocked the gene down using siRNA targeting the BioTAP-tag (Fig. S3A). TC-797s lacking the BioTAP tag were also transfected with siBioTAP as control. siBioTAP efficiently knocked down ZNF532-NBioTAP after 48 hours from the transfection and strongly reduced the viability of 797-ZNF532-NBioTAP cells after 96 hours from transfection, while control TC-797 cells were unaffected (Fig. 3A, S3B). Similarly, knockdown of ZNF592 using two siRNAs targeting this gene in TC-797and another NC cell line, PER-403, caused a strong decrease in cellular viability, while non-NC 293T cells were unaffected (Fig. 3B). These results indicate that ZNF532 and ZNF592 are critical factors for NC growth and/or viability.
Figure 3.
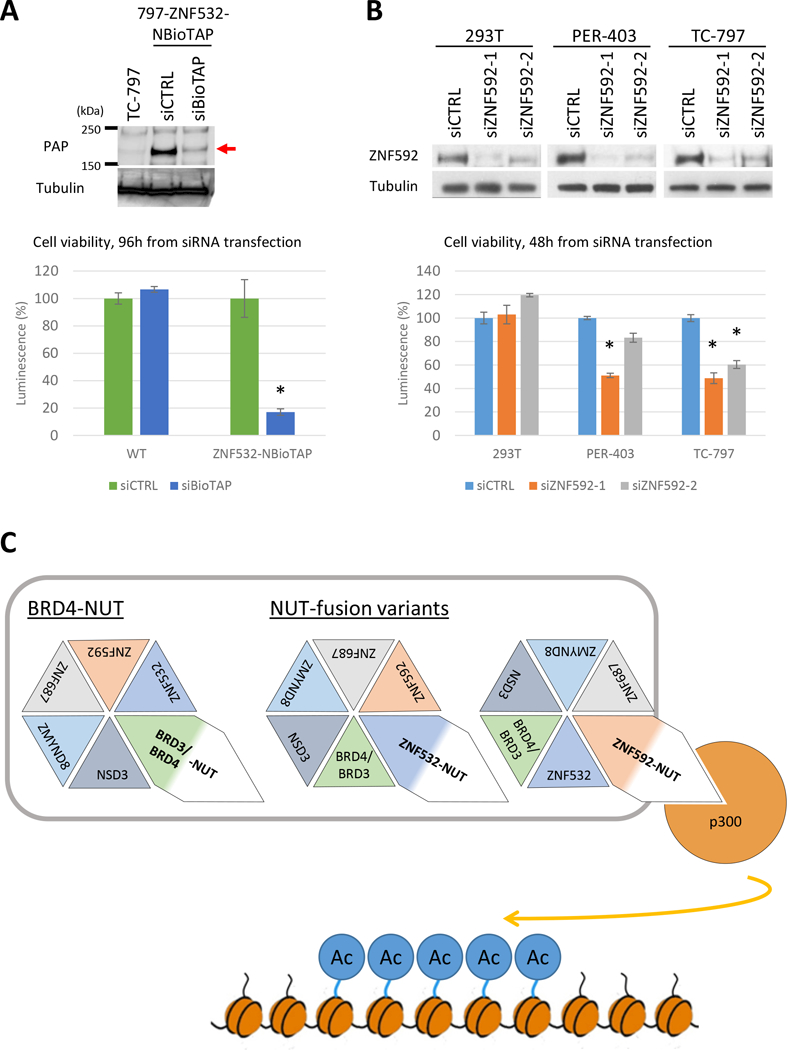
ZNF592 and ZNF532 are critical for NC cell viability. A, Top: Immunoblot of TC-797, 797-ZNF532-NBioTAP at 48 hours after siCTRL or siBioTAP using Peroxidase Anti-Peroxidase (PAP) antibody, which binds the protein A moiety of BioTAP and visualizes ZNF532-NBioTAP protein on the blot. ZNF532-NBioTAP protein band is indicated by a red arrow. Tubulin was used as a loading control. Bottom: Viability assays performed on TC-797 or 797-ZNF532-NBioTAP cells at 96 hours following transfection with siCTRL or siBioTAP. 8000 cells were plated per well in a 96-well plate. Error bars represent standard deviation (SD) from three biological replicates. *p<0.01, student’s t test. B, Top: Immunoblot of 293T, PER-403 and TC-797 at 48 hours after transfected with siCTRL, siZNF592–1 or siZNF592–2 with ZNF592 antibody. Tubulin was used as a loading control. Bottom: Viability assay performed on TC-797, PER-403 and 293T cells 48 hours following transfection with siCTRL, siZNF592–1 or siZNF592–2. 8000 cells were plated per well in a 96-well plate. Error bars represent SD from three biological replicates. *p<0.01, student’s t test. C. Mechanistic model of how NUT-fusions function. Associations of NUT, BET protein and p300 through NUT-fusions are essential to drive proliferation and blockade of differentiation by inducing ectopic histone hyperacetylation on broad region of the chromatin (“megadomain”). This association is mediated by interaction of NUT-fusion protein with other associated proteins (BRDs, ZNF532, ZNF592, ZMYND8, ZNF687, NSD3) or formation of BRD3/4-NUT, NSD3-NUT (not shown here), ZNF532-NUT or ZNF592-NUT fusion.
Discussion
In this study, we discover a novel ZNF592-NUT fusion in a NC patient and together with previous work demonstrate the importance of Z4 factors ZNF532 and ZNF592 in function of the BRD4-NUT oncogenic complex. Archer® FusionPlex® analysis is a powerful tool for diagnosis in cancers harboring multiple fusion gene partners, such as NC, where fresh tissue is often not available, and where FISH for multiple partner genes is not clinically feasible. The present analysis enabled the identification of a novel NUT-fusion variant, and the method could be used to identify NUT-fusions in the future both clinically and as a discovery tool.
Figure 3C shows the working model of the function of NUT-complex. In our previous study, we proposed that forced cooperation of NUT and BRD4 (or BRD3), through the BRD4-NUT fusion, or fusion with other subunits in the NUT complex such as ZNF532, drives p300-dependent histone hyperacetylation and megadomain formation (11). The findings in this study add another key factor in this complex, ZNF592, that facilitates linkage between NUT to BET through the ZNF592-NUT fusion. Furthermore, wild type ZNF532 and ZNF592, two of four Z4 factors found in NUT complex, are essential for growth of BRD4-NUT+ NC cells. Strikingly, siRNA targeting ZNF592 decreased cell viability specifically in NC cell lines, indicating that ZNF592 plays a key role in BRD4-NUT-dependent growth, and may be a therapeutic target in this disease.
Although understanding of Z4 factor function is limited, some Z4 proteins have been shown to have a role in cancer pathogenesis, either as oncogenes or tumor suppressor genes. RUNX1-ZNF687 and ZMYND8-RELA fusion oncoproteins have been reported in acute myeloid leukemia (AML) (22,23). Moreover, it has been shown that ZMYND8 may function to promote tumor growth through the activation of tumor angiogenesis (24). Conversely, ZMYND8 has been shown to repress the expression of metastasis-linked genes in prostate cancer through combinatorial binding to H3K14ac and H3K4me1 through its PHD finger-bromodomain cassette, and acting as a co-repressor of JARID1D, a histone demethylase often mutated in this malignancy (25). Another important study demonstrated that ZMYND8, together with histone demethylase KDM5C, suppress overactivation of enhancers/ super-enhancers of oncogenic genes, including S100, in a breast cancer cell line (26). These studies suggest that ZMYND8 can act either against cancer progression, or in other settings may promote tumor growth. One could speculate that in NC, BRD4-NUT sequesters Z4, and effectively short-circuits its tumor suppressive function.
The multiple interaction domains of Z4 proteins may also be important in BRD4-NUT function. All components of Z4 contain zinc finger domains known to modulate protein-DNA and/or protein-protein interactions. ZMYND8 contains additional interaction modules that allow for both chromatin reading, DNA-binding, and protein-protein interactions, including PHD, bromodomain, PPWP, and MYND domains. Thus, the multimodality of Z4 suggests a possible function as an interaction hub that could play an important role in the stability of the BRD4-NUT complex. Moreover, the presence of zinc finger DNA-binding modules could impart DNA-sequence specificity and influence the genomic localization of BRD4-NUT, which otherwise lacks DNA sequence specific recognition.
In a clinical context, there is no effective therapy for NC, and there is an immediate unmet need for rational approaches to combating this aggressive cancer. Uncovering the mechanism by which BRD4-NUT may alter Z4 function, and vice versa, is likely to identify targetable factors that are specific for the BRD4-NUT oncogenic pathway.
Supplementary Material
Acknowledgements
We thank Tim Martin (Harvard Medical School) for providing the pAAV-tagBFP U6-gRNA expression vector. This work was supported by the Samuel Waxman Cancer Research Foundation, NIH R01 CA124633, R01CA124633–10S1, R01CA124633–10S2 (CAF), NIH R01 GM101958 (MIK) and by a GSK post-doctoral fellowship (HS).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Bauer DE, Mitchell CM, Strait KM, Lathan CS, Stelow EB, Lüer SC, et al. Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin Cancer Res. 2012;18:5773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stathis A, Zucca E, Bekradda M, Gomez-Roca C, Delord J-P, de La Motte Rouge, Thibault Uro-Coste E, et al. Clinical Response of Carcinomas Harboring the BRD4-NUT Oncoprotein to the Targeted Bromodomain Inhibitor OTX015/MK-8628. Cancer Discov. 2016;6:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-atayde AR, Fletcher J a. BRD4-NUT Fusion Oncogene : A Novel Mechanism in Aggressive Carcinoma. Cancer Res. 2003;63:304–7. [PubMed] [Google Scholar]
- 4.French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, et al. BRD–NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27:2237–42. [DOI] [PubMed] [Google Scholar]
- 5.Grayson AR, Walsh EM, Cameron MJ, Godec J, Ashworth T, Ambrose JM, et al. MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene. 2013;1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynoird N, Schwartz BE, Delvecchio M, Sadoul K, Meyers D, Mukherjee C, et al. Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. EMBO J. 2010;29:2943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang R, You J. Mechanistic analysis of the role of bromodomain-containing protein 4 (BRD4) in BRD4-NUT oncoprotein-induced transcriptional activation. J Biol Chem. 2015;290:2744–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.French CA. Pathogenesis of NUT midline carcinoma. Annu Rev Pathol. 2012;7:247–65. [DOI] [PubMed] [Google Scholar]
- 10.Alekseyenko AA, Walsh EM, Wang X, Grayson AR, Hsi PT, Kharchenko PV, et al. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015;29:1507–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alekseyenko AA, Walsh EM, Zee BM, Pakozdi T, Hsi P, Lemieux ME, et al. Ectopic protein interactions within BRD4–chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proc Natl Acad Sci. 2017;114:E4184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.French CA, Rahman S, Walsh EM, Kühnle S, Grayson AR, Lemieux ME, et al. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: Implications for a novel oncogenic mechanism. Cancer Discov. 2014;4:929–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012;13:543–7. [DOI] [PubMed] [Google Scholar]
- 14.Spruijt CG, Luijsterburg MS, Menafra R, Lindeboom RGH, Jansen PWTC, Edupuganti RR, et al. ZMYND8 Co-localizes with NuRD on Target Genes and Regulates Poly(ADP-Ribose)-Dependent Recruitment of GATAD2A/NuRD to Sites of DNA Damage. Cell Rep. 2016;17:783–98. [DOI] [PubMed] [Google Scholar]
- 15.Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, et al. Analysis of the human endogenous coregulator complexome. Cell. 2011;145:787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.French CA, Kutok JL, Faquin WC, Toretsky JA, Antonescu CR, Griffin CA, et al. Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol. 2004;22:4135–9. [DOI] [PubMed] [Google Scholar]
- 17.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84. [DOI] [PubMed] [Google Scholar]
- 18.Toretsky JA, Jenson J, Sun C-C, Eskenazi AE, Campbell A, Hunger SP, et al. Translocation ( 11 ; 15 ; 19 ): a Highly Specific Chromosome Rearrangement Associated With Poorly Differentiated Thymic Carcinoma in Young Patients. Am J Clin Oncol. 2003;26:300–6. [DOI] [PubMed] [Google Scholar]
- 19.Kees U, Mulcahy M, Willoughby M. Intrathoracic carcinoma in an 11-year-old girl showing a translocation t(15;19). Am J Pediatr Hematol Oncol. 1991;13:459–646. [DOI] [PubMed] [Google Scholar]
- 20.Rahman S, Sowa ME, Ottinger M, Smith J a, Shi Y, Harper JW, et al. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of pTEFb by Recruiting Multiple Proteins, Including NSD3. Mol Cell Biol. 2011;31:2641–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan J, Diaz J, Jiao J, Wang R, You J. Perturbation of BRD4 protein function by BRD4-NUT protein abrogates cellular differentiation in NUT midline carcinoma. J Biol Chem. 2011;286:27663–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen TT, Ma LN, Slovak ML, Bangs CD, Cherry AM, Arber DA. Identification of Novel RUNX1 (AML1) Translocation Partner Genes SH3D19, YTHDF2, and ZNF687 in Acute Myeloid Leukemia TuDung. Genes Chromosomes Cancer. 2006;45:918–32. [DOI] [PubMed] [Google Scholar]
- 23.Panagopoulos I, Micci F, Thorsen J, Haugom L, Buechner J, Kerndrup G, et al. Fusion of ZMYND8 and RELA Genes in Acute Erythroid Leukemia. PLoS One. 2013;8:6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuroyanagi J, Shimada Y, Zhang B, Ariyoshi M, Umemoto N, Nishimura Y, et al. Zinc finger MYND-type containing 8 promotes tumour angiogenesis via induction of vascular endothelial growth factor-A expression. FEBS Lett. 2014;588:3409–16. [DOI] [PubMed] [Google Scholar]
- 25.Li N, Li Y, Lv J, Zheng X, Wen H, Shen H, et al. ZMYND8 Reads the Dual Histone Mark H3K4me1-H3K14ac to Antagonize the Expression of Metastasis-Linked Genes. Mol Cell. 2016;63:470–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, et al. Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell. 2016;165:331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.