

Abstract
Background & Aims:
Transmembrane protein 173 (TMEM173 or STING) signaling by macrophage activates the type I interferon-mediated innate immune response. The innate immune response contributes to hepatic steatosis and non-alcoholic fatty liver disease (NAFLD). We investigated wither STING regulates diet-induced in hepatic steatosis, inflammation, and liver fibrosis in mice.
Methods:
Mice with disruption of Tmem173 (STINGgt) on a C57BL/6J background, mice without disruption of this gene (controls), and mice with disruption of Tmem173 only in myeloid cells were fed a standard chow diet, a high-fat diet (HFD, 60% fat calories), or a methionine- and choline-deficient diet (MCD). Liver tissues were collected and analyzed by histology and immunohistochemistry. Bone marrow cells were isolated from mice, differentiated into macrophages, and incubated with DMXAA (an activator of STING) or cGAMP. Macrophages or their media were applied to mouse hepatocytes or human hepatic stellate cells (LX2) cells, which were analyzed for cytokine expression, protein phosphorylation, and fat deposition (oil red O staining following incubation with palmitate). We obtained liver tissues from patients with and without NAFLD and analyzed these by immunohistochemistry.
Results:
Non-parenchymal cells of liver tissues from patients with NAFLD had higher levels of STING than liver tissues from patients without NAFLD. STINGgt mice and mice with disruption only in myeloid cells developed less severe hepatic steatosis, inflammation, and/or fibrosis following a HFD or MCD than control mice. Levels of phosphorylated JNK and p65 and the mRNAs encoding TNF, IL1B, and IL6 (markers of inflammation) were significantly lower in liver tissues from STINGgt mice vs control mice after a HFD or MCD. Transplantation of bone marrow cells from control mice to STINGgt mice restored the severity of steatosis and inflammation following a HFD. Macrophages from control, but not STINGgt mice, increased markers of inflammation in response to lipopolysaccharide and cGAMP. Hepatocytes and stellate cells co-cultured with STINGgt macrophages in the presence of DMXAA, or incubated with the medium collected from these macrophages, had decreased fat deposition and markers of inflammation compared to hepatocytes incubated with control macrophages.
Conclusions:
We found levels of STING to be increased in liver tissues from patients with NAFLD mice with HFD-induced steatosis. In mice, loss of STING from liver macrophages reduces the severity of fibrosis and the inflammatory response. STING might be a therapeutic target for NAFLD.
Keywords: IFN, HSC, NASH, LPS
Graphical Abstract
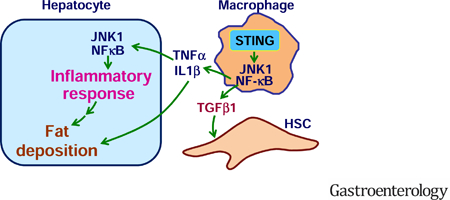
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is characterized by hepatic steatosis 1, 2. Simple steatosis may be benign, but progresses to non-alcoholic steatohepatitis (NASH) when the liver displays overt inflammatory damage 1, 2. Epidemiological data indicate that NASH affects 1.5 to 6.45 percent of the general populations 3–5. Alarmingly, the incidence of NASH in both adults and children is rising continuously due to ongoing epidemics of obesity 4, 5. NASH is one of the most common causes of liver cirrhosis and hepatocellular carcinoma, ultimately leading to liver failure 6–9. To date, there is no effective treatment for NASH 2, 10, 11.
Over the past decade, accumulating evidence validates an essential role for innate immunity in the development of hepatic steatosis and NASH 12–15. As one of the most studied cell types in innate immunity, macrophages have drawn particular attention because macrophage proinflammatory activation is highly associated with hepatic steatosis and inflammation. When proinflammatory activation status increases, macrophages are capable of generating mediators that trigger or exacerbate hepatocyte inflammatory responses and fat metabolic dysregulation 14, 15. To date, a number of regulators such as Jun-N terminal kinase 1 (JNK1), Period (Per)1/Per2, and adenosine 2A receptor are shown to alter the inflammatory status of macrophages, which in turn interact with hepatocytes to protect against or contribute to hepatic steatosis and inflammation 14–16. These findings demonstrate the importance of the innate immune system, in particular macrophages, in pathophysiology of NAFLD. However, it remains largely unknown how regulators of innate immunity regulate hepatic metabolic, inflammatory, and fibrotic programs.
Transmembrane protein 173 (TMEM173) or stimulator of interferon genes (STING) is a signaling molecule whose activation elicits powerful type I interferon (IFN) immunity. Upon viral infection or endoplasmic reticulum (ER) stress, aberrant double-stranded DNA presents in cytosol and activates cyclic GMP-AMP (cGAMP) synthase (CGAS) to generate cGAMP. The latter activates STING and recruits STING to TANK-binding kinase 1 (TBK1), leading to activation of downstream signaling cascades involving interferon regulatory factor 3 (IRF3) to enhance transcriptions of type I IFN genes 20, 21. This led to several recent studies to address how STING regulates liver injury induced by hepatitis B virus, carbon tetrachloride (CCl4), and/or nutrition stress 22–24. Interestingly, the study by Thomsen et al. suggested that human and murine hepatocytes lack STING 22. In contrast, several other studies suggest that mouse hepatocytes express STING, whose activation exerts a pro-apoptotic effect independent of inflammation 19, 25. The study by Cho et al. also pointed to the importance of TBK1 activation, by cGAS-STING pathway and other unknown mediators, in the development of NASH. However, the relevance of STING to human NAFLD/NASH has not been reported. Also, STING activation by exogenous cGAMP reveals differential effects on the proinflammatory responses of macrophages and hepatocytes 26. Given this, there is a critical need to elucidate the exact role for STING in the development and progression of NAFLD and liver fibrosis. The present study provides the primary evidence to support a deleterious role for STING in NAFLD and liver fibrosis, and this role of STING is likely mediated through enhancing macrophage proinflammatory activation.
MATERIALS AND METHODS
Animal experiments
Wild-type (WT) C57BL/6J and STING-disrupted (STINGgt) mice (C57BL/6J background) were obtained from Jackson Laboratory (Bar Harbor, ME). Chimeric mice in which STING was disrupted or intact only in myeloid cells were generated using bone marrow transplantation as described 14. Mice were maintained on 12:12-h light-dark cycles (lights on at 06:00) and fed standard chow-diet (CD), high-fat diet (HFD, 60% fat calories), low-fat diet (LFD, 10% fat calories), and/or methionine- and choline-deficient diet (MCD) as detailed in Supplementary Information (SI). All diets are products of Research Diets, Inc (New Brunswick, NJ). All study protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Texas A&M University.
Human liver samples
Liver sections of human subjects were generated from donated tissues by Sekisui-XenoTech, LLC (Kansas City, KS, USA). Subjects with NAFLD revealed severe hepatic steatosis compared with subjects without NAFLD (44.7 ± 10.6% fat content vs. 1.7 ± 1.9 %, assessed by Sekisui-XenoTech and by a board-certified pathologist (Dr. Xiangbai Chen, Baylor Scott & White Health, College Station, TX 77845)). Because of using fixed human tissues that are commercially available, the current study was exempted from the Institutional Review Board (IRB) approval.
Cell culture and treatment
Bone marrow cells were isolated from STINGgt and WT mice and differentiated into macrophages (BMDM) 14. After differentiation, STINGgt and WT BMDM were treated with 5,6-dimethylxanthenone-4-acetic acid (DMXAA, an STING activator) (75 µg/mL) 27 or cGAMP (20 µg/mL) 26 and subjected to collection of conditioned media and examination of IFNβ production and the proinflammatory activation 26. Some BMDM were trypsinized and added to primary mouse hepatocytes or LX2 cells (a cell line of human hepatic stellate cells, HSCs) at a 1:10 ratio for co-culture studies 15. Hepatocytes or LX2 cells were incubated with BMDM-conditioned media and assayed for hepatocyte fat deposition and inflammatory responses or HSC activation status. To examine the direct effect of STING on HSC activation, some LX2 were treated with DMXAA (75 µg/mL) in the absence or presence of transforming growth factor β1 (TGFβ1) (8 ng/mL for 24hr or 2.5 ng/mL for 48hr). Details were provided in SI.
Histological, biochemical and molecular assays
Liver sections were used for histological and immunohistochemical assays. Plasma parameters were measured using metabolic assay kits and ELISA kits. Also, tissue and/or cell samples were prepared for selected assays including Western blots analysis, real-time PCR, and other assays detailed in SI.
Statistical Methods
Numeric data are presented as means ± SD (standard deviation). Statistical significance was determined using unpaired, two-tailed ANOVA or Student’s t tests. Differences were considered significant at the two-tailed P < 0.05.
RESULTS
NAFLD is coupled with increased liver signaling events downstream of STING
Activating STING recruits STING to TBK1 and activates TBK1 and IRF3 19, 28, 29. We analyzed the phosphorylation states of TBK1 and IRF3 in livers of WT C57BL/6J mice fed an HFD for 12 weeks. Upon HFD feeding, WT mice displayed obesity-associated NAFLD (Figure S1A-D). Additionally, the phosphorylation states of TBK1 and IRF3 in livers of HFD-fed WT mice were significantly higher than their respective levels in livers of LFD-fed mice (Figure S1E). These results validate that NAFLD is coupled with increased liver signaling events downstream of STING, and confirm that nutrition stress activates liver STING signaling pathway 26.
STING expression is increased in livers of human subjects with NAFLD
To address the relevance of STING to human NAFLD, we examined STING expression in liver sections of human subjects. Compared with control, liver sections of NAFLD patients revealed increased fat and collagen deposition indicated by H&E and Trichrome staining, respectively (Figure 1A, B), confirming hepatic steatosis and fibrosis. When STING expression was examined, the intensity of staining in liver sections of NAFLD patients was much stronger than that in subjects without NAFLD (Figure 1A, C). Also, most STING-positive cells in liver sections of NAFLD patients were aggregated relative to those in control. Further analysis by Dr. X. Chen indicated that STING-positive cells were mainly immune cells including macrophages/Kupffer cells and endothelial cells (Figure 1A). However, human liver sections did not reveal STING-positive hepatocytes. These results suggest that, in humans, STING is expressed in non-parenchymal liver cells, mainly macrophage/Kupffer cells. Moreover, STING expression is increased in livers of human patients with NAFLD.
Figure 1. STING expression is increased in livers of human patients with NAFLD.
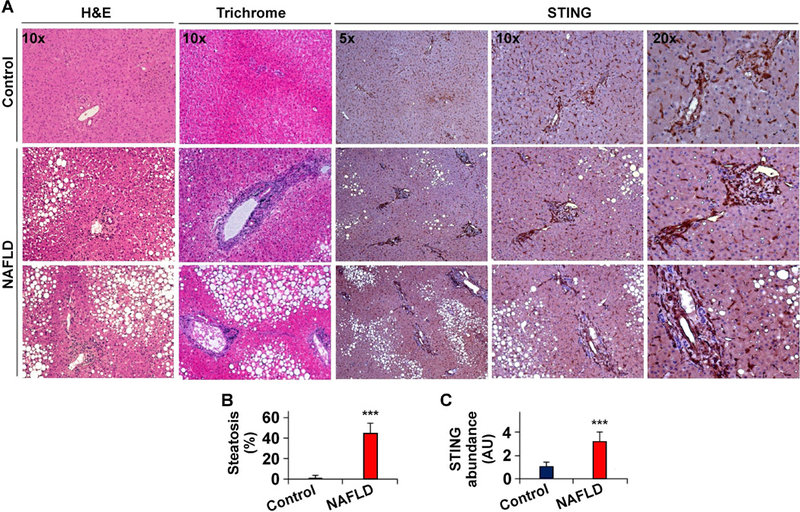
(A) Liver sections of NAFLD patients (bottom two rows) and human subjects without NAFLD (Control, top row) were stained with H&E (the very left column) and Trichrome (the second left column), and examined for STING expression (the right three columns). (B) Degrees of hepatic steatosis. (C) STING abundance in liver sections. AU, arbitrary unit. For bar graphs, data are means ± SD. n = 6 – 8. ***, P < 0.001 NAFLD vs. Control.
STING disruption protects against HFD-induced NAFLD
We explored a role for STING in NAFLD using STINGgt and WT C57BL/6J mice. Under LFD-fed conditions, STINGgt mice did not differ significantly from WT mice in most parameters related to NAFLD and system metabolic homeostasis (Figure 2A-D; Figure S2). Upon HFD feeding for 12 weeks, WT mice, but not STINGgt mice, revealed overt NAFLD aspects. Specifically, STINGgt mice displayed a smaller gain in body weight compared with WT mice (Figure S2A). Body composition analysis indicated lower fat mass in HFD-STINGgt mice than in HFD-WT mice (Figure S2A); although the values of respiratory quotient (RQ) in HFD-STINGgt mice did not differ significantly from those in HFD-WT mice (Figure S2B). Also, HFD-STINGgt mice did not display systemic insulin resistance and glucose intolerance as did HFD-WT mice (Figure S2C, D). When NAFLD aspects were examined, plasma levels of alanine aminotransferase (ALT) in HFD-STINGgt mice were lower than those in HFD-WT mice (Figure 2A). Also, HFD-STINGgt mice displayed significant decreases in liver weight and hepatic levels of triglycerides compared with HFD-WT mice (Figure 2B, C). Consistently, HFD-STINGgt mice did not develop hepatic steatosis as did HFD-WT mice, indicated by H&E and/or oil red O staining of liver sections (Figure 2D; Figure S2E). When liver inflammatory status was examined, liver sections of HFD-STINGgt mice contained fewer F4/80-postivie cells (macrophages/Kupffer cells) compared with those of HFD-WT mice (Figure 2D). Moreover, the phosphorylation states of JNK p46 and NF0κB p65 and the mRNA levels of tumor necrosis factor alpha (TNFα), interleukin (IL)-1β, and IL-6 in livers of HFD-STINGgt mice were significantly lower than their respective levels in livers of HFD-WT mice (Figure 2E, F). With regard to fat metabolic genes/enzymes, liver mRNA levels of lipogenic enzymes such as acetyl-CoA carboxylase 1 (ACC1) and fatty acid synthase (FAS) in HFD-STINGgt mice also were significantly lower than their respective levels in HFD-WT mice (Figure 2F). These results, together with those presented in Figure 1, suggest that STING plays a detrimental role in NAFLD.
Figure 2. STING disruption protects against HFD-induced NAFLD.
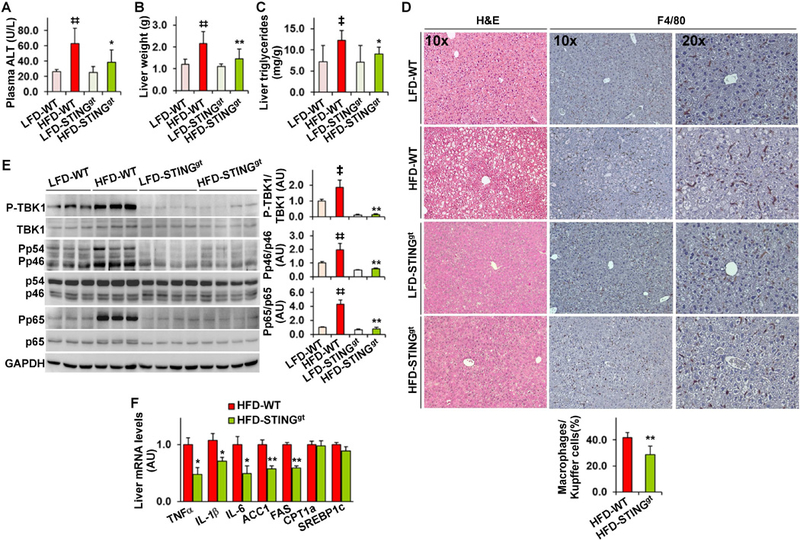
Male STING-disrupted (STINGgt) mice and WT mice, at 5 – 6 weeks of age, were fed an LFD or HFD for 12 weeks. (A) Plasma levels of alanine aminotransferase (ALT). (B) Liver weight. (C) Liver levels of triglycerides. (D) Liver sections were stained with H&E (left column) or for F4/80 expression (right two columns). Bar graph, percentages of macrophages. (E) Liver lysates were examined for inflammatory signaling using Western blot analysis. Bar graphs, quantification of blots. (F) Liver mRNA levels were examined using real-time RT-PCR. For all bar graphs, data are means ± SD. n = 10 – 12. ‡, P < 0.05 and ‡‡, P < 0.01 HFD-WT vs. LFD-WT in A - C, and E; *, P < 0.05 and **, P < 0.01 HFD-STINGgt vs. HFD-WT (in A - E) for the same gene (in F).
Myeloid cell-specific STING disruption decreases the severity of HFD-induced NAFLD
We sought to explore a role for the STING in macrophages in NAFLD using bone marrow transplantation (BMT), an approach that has been commonly used for altering macrophage gene expression 14, 30. We transplanted bone marrow cells of STINGgt mice and/or WT mice to lethally irradiated WT mice. Upon HFD feeding for 12 weeks, WT/BMT-STINGgt mice, in which STING was disrupted only in myeloid cells, and WT/BMT-WT, in which STING was intact in all cells, gained similar body weight and revealed similar body composition (Figure S3A, B). However, HFD-fed WT/BMT-STINGgt mice displayed decreased severity of insulin resistance and glucose intolerance compared with HFD-fed WT/BMT-WT mice (Figure S3C, D). When NAFLD aspects were analyzed, HFD-fed WT/BMT-STINGgt mice showed decreased levels of plasma ALT and hepatic triglycerides compared with HFD-fed WT/BMT-WT mice; although the mice displayed comparable liver weight (Figure 3A-C). Additionally, HFD-fed WT/BMT-STINGgt mice revealed decreased severity of hepatic steatosis compared with HFD-fed WT/BMT-WT mice, indicated by H&E staining of liver sections (Figure 3D). When liver inflammatory status was examined, liver sections of HFD-fed WT/BMT-STINGgt mice contained fewer F4/80-positive cells than those of HFD-fed WT/BMT-WT mice (Figure 3D). FD-fed WT/BMT-STINGgt mice also revealed significant decreases in the phosphorylation states of JNK p46 and NFκB p65 and the mRNA levels of TNFα, IL-1β, and IL-6 compared with HFD-fed WT/BMT-WT mice (Figure 3E, F). Relative to those in HFD-fed WT/BMT-WT mice, liver mRNA levels of FAS in HFD-fed WT/BMT-STINGgt mice were significantly decreased (Figure 3F); although liver mRNA levels of ACC1, CPT1a, and sterol regulatory element-binding protein 1c (SREBP1c) were not significantly altered. These results suggest that STING disruption specifically in myeloid cells (macrophages) decreases the severity of HFD-induced NAFLD.
Figure 3. Myeloid cell-specific STING disruption decreases the severity of HFD-induced NAFLD.
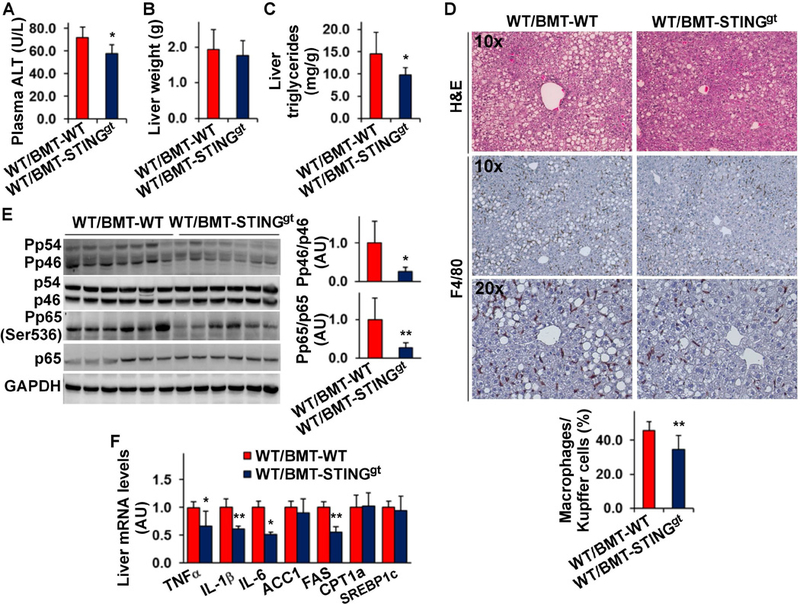
Male WT C57BL/6J mice, at 5 – 6 weeks of age, were lethally irradiated and transplanted with bone marrow cells from STINGgt and/or WT mice. After recovery for 4 weeks, the chimeric mice were fed an HFD for 12 weeks. (A) Plasma levels of ALT. (B) Liver weight. (C) Liver levels of triglycerides. (D) Liver sections were stained with H&E (top row) or for F4/80 expression (bottom two rows). Bar graph, percentages of macrophages. (E) Liver lysates were examined for proinflammatory signaling using Western blot analysis. Bar graphs, quantification of blots. (F) Liver mRNA levels were examined using real-time RT-PCR. For A - F, WT/BMT-STINGgt, WT mice received STINGgt bone marrow cells; WT/BMT-WT, WT mice received WT bone marrow cells. For all bar graphs, data are means ± SD. n = 8 – 10. *, P < 0.05 and **, P < 0.01 WT/BMT-STINGgt vs. WT/BMT-WT (in A, and C- E) for the same gene (in F).
STING presence specifically in myeloid cells exacerbates HFD-induced NAFLD
To further validate a role for the STING in macrophages in regulating the pathogenesis of NAFLD, we transplanted bone marrow cells of WT and/or STINGgt mice to lethally irradiated STINGgt mice. Upon HFD feeding, STINGgt/BMT-WT, in which STING was intact only in myeloid cells, and STINGgt/BMT-STINGgt mice, in which STING was disrupted in all cells, gained similar body weight and revealed similar body composition (Figure S4A, B). However, HFD-fed STINGgt/BMT-WT mice displayed increased severity of insulin resistance and glucose intolerance compared with HFD-fed STINGgt/BMT-STINGgt mice (Figure S4C, D). When NAFLD aspects were examined, HFD-fed STINGgt/BMT-WT mice revealed a significant increase in hepatic levels of triglycerides compared with HFD-fed STINGgt/BMT-STINGgt mice; although HFD-fed STINGgt/BMT-WT mice showed insignificant increases in plasma levels of ALT and liver weight (Figure 4A-C). Indicated by H&E staining of liver sections, HFD-fed STINGgt/BMT-WT mice revealed increased severity of hepatic steatosis compared with HFD-fed STINGgt/BMT-STINGgt mice (Figure 4D). Also, liver sections of HFD-fed STINGgt/BMT-WT mice contained significantly more F4/80-positive cells compared with those of HFD-fed STINGgt/BMT-STINGgt mice (Figure 4D). Consistently, livers of HFD-fed STINGgt/BMT-WT mice revealed significant increases in the phosphorylation states of JNK p46 and NFκB p65 and the mRNA levels of TNFα and IL-1β compared with those of HFD-fed STINGgt/BMT-STINGgt mice (Figure 4E, F). Relative to those in HFD-fed STINGgt/BMT-STINGgt mice, liver mRNA levels of FAS in HFD-fed STINGgt/BMT-WT mice were significantly increased (Figure 4F); although liver mRNA levels of ACC1, CPT1a, and SREBP1c were not significantly altered. These results suggest that STING presented only in myeloid cells (macrophages) triggers or exacerbates HFD-induced NAFLD. In combination, the results in Figures 3 and 4 strongly demonstrated a deleterious role for the STING in macrophages in the pathogenesis of NAFLD.
Figure 4. STING presence only in myeloid cells exacerbates HFD-induced NAFLD.
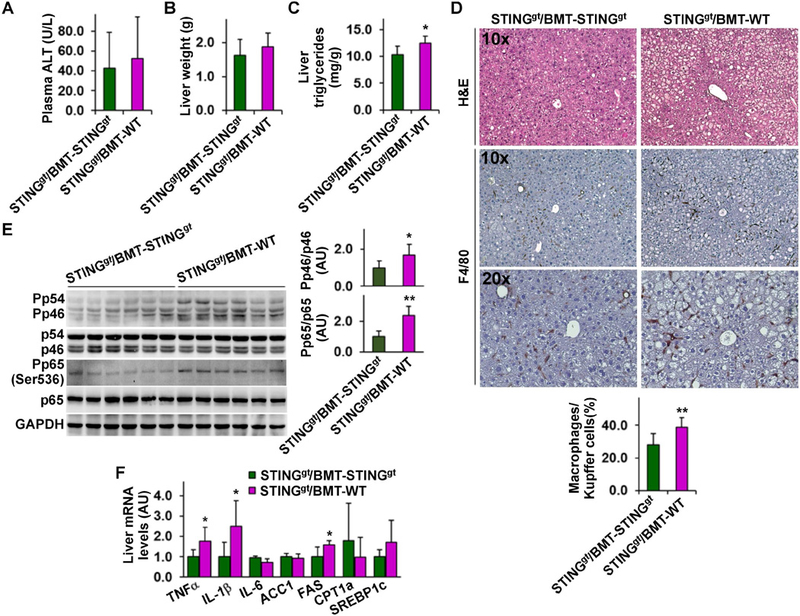
Male STINGgt mice, at 5 – 6 weeks of age, were lethally irradiated and transplanted with bone marrow cells from WT and/or STINGgt mice. After recovery for 4 weeks, the chimeric mice were fed an HFD for 12 weeks. (A) Plasma levels of ALT. (B) Liver weight. (C) Liver levels of triglycerides. (D) Liver sections were stained with H&E (top row) or for F4/80 expression (bottom two rows). Bar graph, percentages of macrophages. (E) Liver lysates were examined for proinflammatory signaling using Western blot analysis. Bar graphs, quantification of blots. (F) Liver mRNA levels were examined using real-time RT-PCR. For A - F, STINGgt/BMT-WT, STINGgt mice received WT bone marrow cells; STINGgt/BMT-STINGgt, STINGgt mice received STINGgt bone marrow cells. For all bar graphs, data are means ± SD. n = 10 – 12. *, P < 0.05 and **, P < 0.01 STINGgt/BMT-WT vs. STINGgt/BMT-STINGgt (in C - E) for the same gene (in F).
STING-driven macrophage factors enhance hepatocyte fat deposition and proinflammatory responses
To recapitulate our in vivo findings and gain mechanistic insights, we sought to validate STING regulation of macrophage activation and examine whether and how macrophage factors generated in response to STING activation or disruption regulate hepatocyte responses. In WT BMDM, cGAMP treatment caused a significant increase in LPS-induced phosphorylation states of JNK p46 (Figure S5A), confirming our previous finding 26. Strikingly, in STINGgt BMDM, cGAMP treatment caused significant decreases in LPS-induced phosphorylation states of JNK p46 and in the mRNA levels of TNFα, IL-1β, and/or IL-6 under both basal and LPS-stimulated conditions (Figure S5B, C), indicating an essential role for STING in regulating macrophage activation. Next, we treated BMDM with DMXAA to activate STING 27. As expected, WT BMDM, but not STINGgt BMDM, revealed significantly increased production of IFNβ and phosphorylation states of JNK p46 and NFκB p65 (Figure 5A, B), validating that STING activation enhances proinflammatory responses of WT macrophages. These data are consistent with previous findings 31, which suggest that the proinflammatory effects of DMXAA are STING-dependent. Upon further analyses of macrophage polarization, we demonstrated that 15 STING disruption decreased macrophage proinflammatory activation, and exhibited marginal effects on macrophage alternative (M2) activation (Figure S6).
Figure 5. STING enables macrophages to generate factors that promote hepatocyte fat deposition and proinflammatory responses.
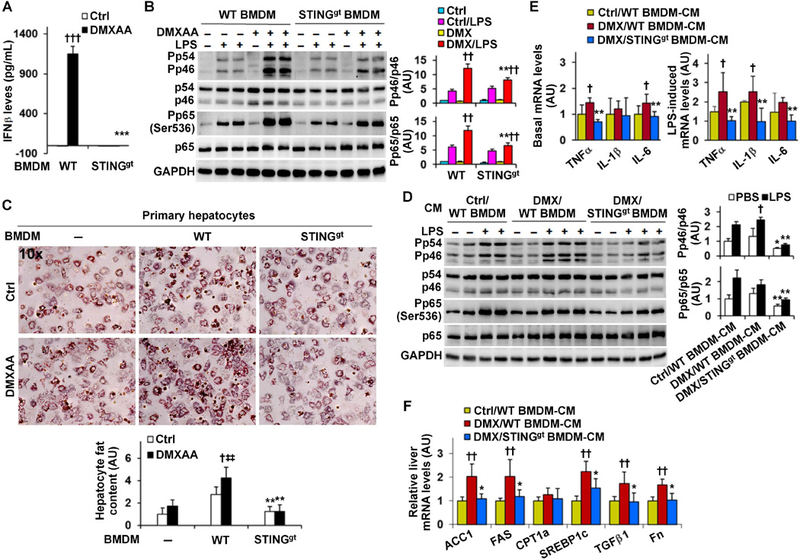
Macrophages and primary hepatocyte were prepared from male STINGgt mice and WT C57BL/6J mice as described in Methods. (A) Macrophage interferon beta (IFNβ) production. Bone marrow-derived macrophages (BMDM) were treated with DMXAA (75 µg/mL) or control (Ctrl, 7.5% NaHCO3) for 6 hr. BMDM-conditioned media were examined for IFNβ levels. (B) Macrophage proinflammatory signaling. BMDM were treated with or without DMXAA (75 µg/mL) for 24 hr in the absence or presence of LPS (100 ng/mL) for the last 30 min. (C) Hepatocyte fat deposition of co-cultures. Primary hepatocytes were incubated in the absence of macrophages or co-cultured with BMDM from WT or STINGgt mice for 48 hr, and treated with DMXAA (75 µg/mL) or control in the presence of palm itate (250 µM) for the last 24 hr. Prior to harvest, hepatocytes or co-cultures were stained with oil red O for 1 hr. Bar graph, quantification of fat content. (D, E, F) Macrophage factors regulation of hepatocyte proinflammatory responses and gene expression. Conditioned media (CM) were collected from DMXAA (DMX)-treated WT BMDM, DMX-treated STINGgt BMDM, and Ctrl-treated WT BMDM (described in B) and designated as DMX/WT BMDM-CM, DMX/STINGgt BMDM-CM, and Ctrl/WT BMDM-CM, respectively. CM were mixed with fresh media at a 1:1 ratio and supplemented to hepatocytes for 48 hr. Prior to harvest, CM-incubated hepatocytes were treated with or without LPS (100 ng/mL) for 30 min (D) or LPS (20 ng/mL) for 6 hr (E). For B and D, cell lysates were examined for proinflammatory signaling using Western blot analysis. Bar graphs, quantification of blots. For E and F, the mRNA levels were examined using real-time RT-PCR. For bar graphs in A - F, data are means ± SD. n = 6 – 8 (A, E, and F) or 4 – 6 (B, C, and D). *, P < 0.05, **, P < 0.01, and ***, P < 0.001 STINGgt vs. WT with the same treatment (Ctrl or DMXAA in A and C; Ctrl/LPS or DMX/LPS in B) or DMX/STINGgt BMDM-CM vs. DMX/WT BMDM-CM under the same condition (PBS or LPS in D) or for the same gene (in E and F); †, P < 0.05, ††, P < 0.01, and †††, P < 0.001 DMXAA vs. Ctrl (in A and C) or DMX/LPS vs. Ctrl/LPS (in B) within the same genotype, or DMX/WT BMDM-CM vs. Ctrl/WT BMDM-CM under LPS-stimulated condition (in D) or for the same gene (in E and F); ‡‡, P < 0.01 hepatocytes co-cultured with WT BMDM vs. hepatocytes alone in the presence of DMXAA (in C).
Next, we performed macrophage-hepatocyte co-cultures to examine whether STING facilitates macrophage generation of factors to promote NAFLD aspects. Palmitate-induced hepatocyte fat deposition in DMXAA-treated co-cultures of WT BMDM and hepatocytes was much greater than that in DMXAA-treated hepatocytes (without BMDM) (Figure 5C). This increase in hepatocyte fat deposition, however, was not observed in DMXAA-treated co-cultures of STINGgt BMDM and hepatocytes. We also incubated WT primary hepatocytes with macrophage-conditioned media (CM). When proinflammatory responses were analyzed, hepatocytes incubated with the CM of DMXAA-treated WT BMDM (DMX/WT BMDM-CM) displayed significant increases in LPS-stimulated phosphorylation states of JNK p46 and in the mRNA levels of TNFα, IL-1β, and/or IL-6 under both basal and LPS-stimulated conditions compared with hepatocytes incubated with the CM of control-treated WT BMDM (Ctrl/WT BMDM-CM) (Figure 5D, E). Of note, hepatocytes incubated with the CM of DMXAA-treated STINGgt BMDM (DMX/STINGgt BMDM-CM) revealed significantly decreased phosphorylation states of JNK p46 and NFκB p65 and mRNA levels of TNFα, IL-1β, and/or IL-6 under both basal and LPS-stimulated conditions compared with hepatocytes incubated with DMX/WT BMDM-CM (Figure 5D, E). With regard to the expression of fat metabolic genes/enzymes, hepatocytes incubated with DMX/WT BMDM-CM revealed significantly increased mRNA levels of ACC1, FAS, and SREBP1c compared with hepatocytes incubated with Ctrl/WT BMDM-CM (Figure 5F). These increases, however, were not observed in hepatocytes incubated with DMX/STINGgt BMDM-CM. Similar changes were observed in the mRNAs of TGFβ1 and fibronectin (Fn) (Figure 5F), which are mediators favoring liver fibrosis. Together, these results suggest that STING activation enables macrophages to generate factors that are capable of enhancing hepatocyte fat deposition and proinflammatory responses.
STING disruption decreases the severity of MCD-induced liver inflammation and fibrosis
NASH is the advanced form of NAFLD. We validated that liver TBK1 phosphorylation was increased in livers of MCD-fed mice (Figure S7). Next, we examined the effects of STING disruption on MCD-induced steatohepatitis and liver fibrosis. When fed a chow-diet, STINGgt mice did not differ significantly from WT mice in body weight, liver weight, and hepatic levels of triglycerides; although STINGgt mice revealed decreased phosphorylation states in liver JNK p46 and NFκB p65 (Figure S8). Upon MCD feeding, WT mice displayed overt hepatic steatosis and inflammation (Figures S7, S8). However, plasma levels of ALT, liver weight, and hepatic levels of triglycerides in MCD-STINGgt mice were significantly lower than their respective levels in MCD-WT mice (Figure 6A-C). Consistently, the severity of hepatic steatosis in MCD-STINGgt mice was significantly lighter than in MCD-WT mice (Figure 6D). When stained for F4/80 expression, liver sections of MCD-WT mice revealed lots of F4/80-postivie cells, many of which were aggregated. However, liver sections of MCD-STINGgt mice contained significantly fewer aggregated F4/80-postivie cells (Figure 6D). Consistently, the phosphorylation states of JNK p46 and NFκB p65 and the mRNA levels of IL-1β in MCD-STINGgt mice were much lower than their respective levels in MCD-WT mice (Figure 6E, F). When liver fibrosis was t analyzed, liver sections of MCD-STING mice contained much lesser collagen compared with those of MCD-WT mice (Figure 6D). Also, hepatic protein content of α-smooth muscle actin significantly lower than their respective levels in MCD-WT mice (Figure 6E, F). These results suggest that STING disruption decreases the severity of MCD-induced NASH.
Figure 6. STING disruption decreases the severity of MCD-induced NASH.
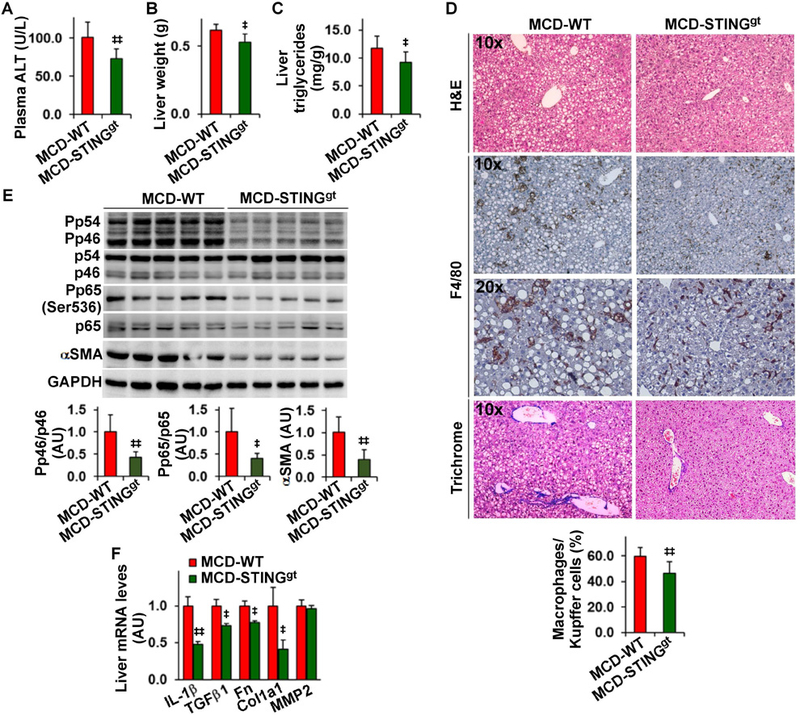
Male STINGgt mice and WT C57BL/6J mice, at 11 – 12 weeks of age, were fed an MCD for 5 weeks. (A) Plasma levels of ALT. (B) Liver weight. (C) Liver levels of triglycerides. (D) Liver sections were stained with H&E (top row), for F4/80 expression (middle two rows), or with Trichrome (bottom row). Bar graph, percentages of macrophages. (E) Liver lysates were examined for inflammatory signaling and αSMA amount using Western blot analysis. Bar graphs, quantification of blots. (F) Hepatic expression of genes related to fibrosis was examined using real-time RT-PCR. MMP2, matrix metalloproteinase 2. For bar graphs in A - F, data are means ± SD. n = 8 – 10 (A - D) or 6 – 8 (E and F)., P < 0.05 and, P < 0.01 MCD-STINGgt vs MCD-WT (in A - E) for the same gene (in F).
STING enhances the activation of hepatic stellate cells (HSCs)
To gain insights of STING regulation of NASH, we examined the direct effects of altering STING signaling on HSC activation. In LX2 cells, treatment with DMXAA in the presence of TGFβ1 significantly increased the phosphorylation states of p38 and protein amount of αSMA (Figure 7A, B). Additionally, DMXAA treatment significantly increased the mRNA levels of fibrogenic genes including Col1a1, αSMA, Fn, and TGFβ1 (Figure 7C). DMXAA treatment also caused an unexpected increase in PPARγ mRNAs, which may be a counter-regulatory response. In contrast, treatment with MRT67307, an inhibitor of TBK1, significantly decreased the effect of TGFβ1 on stimulating p38 phosphorylation (Figure S9). These results suggest a detrimental role for STING in promoting HSC activation.
Figure 7. STING increases HSC activation status and enables macrophage factors to enhance HSC activation (A, B, C).
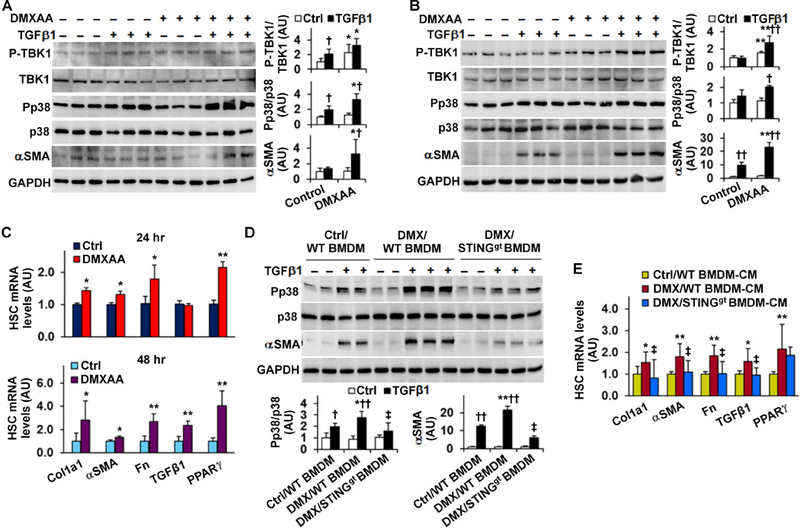
Activating STING enhances HSC activation. For A - C, LX2 cells were treated with or without DMXAA (75 µg/mL) in the absence or presence of TGFβ1 (8 ng/mL) for 24 hr (A and C (top panel)) or TGFβ1 (2.5 ng/mL) for 48 hr (B and C (bottom panel)). (D, E) STING-driven macrophage factors enhance HSC activation. For D, LX2 cells were co-cultured with STINGgt BMDM and/or WT BMDM that were pre-treated with DMXAA (75 µg/mL) or Ctrl for 24 hr. The co-cultures were incubated for 48 hr in the absence or presence of TGFβ1 (2.5 ng/mL). For E, LX2 cells were treated with conditioned media (CM) of DMXAA (DMX)- or Ctrl-treated WT BMDM and/or STINGgt BMDM for 48 hr. For A, B, and D, cell lysates were subjected to Western blot analysis. Bar graphs, quantification of blots. For C and E, the expression of fibrogenic genes was examined using real-time RT-PCR. For bar graphs in A - E, data are means SD. n = 4 – 6 (A, B, and D) or 6 – 8 (C and E). *, P < 0.05 and **, P < 0.01 DMXAA vs. Control (Ctrl) under the same condition (in A and B) or for the same gene (in C), co-cultures with DMX/WT BMDM vs. co-cultures with Ctrl/WT BMDM under the same condition (in D), or DMX/WT BMDM-CM-treated HSCs vs. Ctrl/WT BMDM-CM-treated HSCs for the same gene (in E); †, P < 0.05 and ††, P < 0.01 TGFβ1 vs. Ctrl with the same treatment (in A, B, and D); ‡, P < 0.05 DMX/STINGgt BMDM-CM-treated HSCs vs. DMX/WT BMDM-CM-treated HSCs under TGFβ1-stimulated condition (in D) or for the same gene (in E).
Since NASH was characterized by massive macrophage aggregation (Figures S7B), we sought to examine whether STING-driven macrophage factors alter HSC activation. WT BMDM and STINGgt BMDM were pre-treated with DMXAA or control for 24 hr, trypsinized, and added to LX2 cells for 48 hr. In LX2 cells co-cultured with control-treated WT BMDM (Ctrl/WT BMDM), TGFβ1 treatment caused significant increases in p38 phosphorylation states and αSMA amount, indicating increased HSC activation. These effects of TGFβ1 were significantly enhanced in LX2 cells co-cultured with DMXAA-treated WT BMDM (DMX/WT BMDM), but not in LX2 cells co-cultured with DMXAA-treated STINGgt BMDM (DMX/STINGgt BMDM) (Figure 7D). Next, we incubated LX2 cells with macrophage-CM and examined HSC gene expression. The mRNA levels of Col1a1, Fn, and TGFβ1 in LX2 cells incubated with DMX/WT BMDM-CM were significantly higher than their respective levels in LX2 cells incubated with Ctrl/WT BMDM-CM (Figure 7E). However, the mRNA levels of Col1a1, Fn, and TGFβ1 in LX2 cells incubated with DMX/STINGgt BMDM-CM were lower than their respective levels in LX2 cells incubated with DMX/WT BMDM-CM, and comparable with their respective levels in LX2 cells incubated with Ctrl/WT BMDM-CM. Together, these results suggest that macrophage factors generated in response to STING activation enhance HSC activation.
DISCUSSION
STING is a powerful regulator of innate immunity 17, 20, 21. In the present study, we validated in both animals and human subjects that liver STING is associated with hepatic steatosis and inflammation. Using various mouse models of NAFLD, we further validated that STING plays a deleterious role in NAFLD. Notably, the STING found in myeloid cells (macrophages) is needed and sufficient for nutrition stress, e.g., HFD or MCD feeding, to trigger or exacerbate NAFLD/NASH aspects in mice. Mechanistically, STING activation markedly increases macrophage proinflammatory status, which in turn increases hepatocyte fat deposition and proinflammatory responses and enhances HSC activation. Therefore, we provided compelling evidence to demonstrate that STING plays a deleterious role in NAFLD through enhancing macrophage proinflammatory activation.
We have previously shown that HFD-fed WT mice display overt hepatic steatosis and inflammation compared with control mice 15. In this model of NAFLD, we observed an increase n the phosphorylation states of liver TBK1 and IRF3, indicating increased signaling events downstream of STING. Similarly, we observed increased liver TBK1 phosphorylation in MCD-fed WT mice (a model of NASH), which was accompanied with increased liver proinflammatory status including massive macrophage aggregations. These results validated a link between STING signaling and NAFLD/NASH. Using liver sections of human subjects with NAFLD, we further validated the relevance of increased STING expression to human NAFLD. As such, we postulated a deleterious role for STING in NAFLD/NASH. In other words, we expected STING disruption to be protective and we found this to be true. In the present study, STING-disrupted mice were protected from HFD-induced NAFLD and revealed decreased severity of MCD-induced NASH compared with control mice. With regard to NAFLD/NASH pathogenesis, both hepatocytes and macrophages critically determine the development and progression of hepatic steatosis and inflammation 15, 16, 30, 32. Considering this, STING found in either hepatocytes or macrophages could contribute to NAFLD/NASH. However, the study by Thomsen et al. indicated that human and murine hepatocytes are lack of STING 22; although other studies suggest that mouse hepatocytes express STING 19, 23. We confirmed that STING is not present in human hepatocytes, but expressed at high abundance in hepatic non-parenchymal cells. Because of this, to address a role for STING in macrophages is more relevant to human disease, e.g., NAFLD.
We provided two lines of evidence from chimeric mice to support a deleterious role for the STING in myeloid cells in the pathogenesis of NAFLD. In our loss-of-function study, chimeric mice whose STING was disrupted specifically in myeloid cells revealed decreased severity of HFD-induced hepatic steatosis and inflammation compared with mice whose STING was intact in all cells. In contrast, in our gain-of-function study, chimeric mice which had intact STING specifically in myeloid cells displayed increased severity of HFD-induced NAFLD aspects compared with chimeric mice whose STING was disrupted in all cells. These complementary results suggest that macrophage-specific STING is required, and is enough to generate factors that act on other liver cells, in particular hepatocytes, to promote hepatocyte fat deposition and enhance hepatocyte proinflammatory responses. Moreover, HFD-induced NAFLD aspects in chimeric mice whose STING was disrupted only in myeloid cells were similar to those in chimeric mice whose STING was disrupted in all cells whereas HFD-induced NAFLD aspects in chimeric mice whose STING was intact only in myeloid cells were similar to those in chimeric mice whose STING was intact in all cells. This implicates a greater contribution from macrophage-specific STING during NAFLD than from the STING found in other cells. As substantial evidence, treatment with cGAMP or DMXAA significantly enhanced the effect of LPS on stimulating the proinflammatory activation of WT macrophages, but not STINGgt macrophages. More importantly, when co-cultured with primary mouse hepatocytes, DMXAA-treated WT macrophages, but not DMXAA-treated STINGgt macrophages, caused a significant increase in the degree of palmitate-induced hepatocyte fat deposition. Upon treating hepatocytes with macrophage-conditioned media, we also verified that STING-driven macrophage factors enhanced hepatocyte proinflammatory responses. In support of this, hepatocytes incubated with conditioned media of DMXAA-treated WT macrophages displayed significant increases in LPS-induced phosphorylation of JNK p46 and mRNA levels of TNFα, IL-1β, and IL-6 compared with hepatocytes incubated conditioned media of control-treated WT macrophages. These increases, however, were not observed in hepatocytes incubated with conditioned media of DMXAA-treated STINGgt macrophages, suggesting that STING disruption blunted the effect of DMXAA to drive macrophage production of factors that could enhance hepatocyte proinflammatory responses. Because of this, it is conceivable that STING-driven macrophage factors act to increase hepatocyte fat deposition and proinflammatory responses, thereby accounting for the development and progression of hepatic steatosis and inflammation.
Inflammation is key to drive NAFLD to NASH which is featured by inflammatory damage and liver fibrosis 16, 33. In the present study, we also validated a deleterious role for STING in steatohepatitis and liver fibrosis. As supporting evidence, the levels of plasma ALT and hepatic triglycerides in MCD-STINGgt mice were significantly lower than their respective levels in MCD-WT mice. Additionally, the severity of liver proinflammatory responses, as well as liver deposition of collagen and amount/expression of αSMA, Col1a1, and Fn in MCD-STINGgt were much lower their respective levels in MCD-WT mice. These results are in agreement with the report that STING disruption protected mice from CCl4-induced liver fibrosis 23. The study by Iracheta-Vellve et al., however, attributed the development of CCl4-induced liver fibrosis to STING-mediated activation of hepatocellular death pathways. This appeared to not have human relevance because STING is not present in human hepatocytes. We argue that the STING found in cells other than hepatocytes appears to trigger or exacerbate liver fibrotic program. Indeed, we validated that STING activation directly activates HSCs evidenced by the finding that treatment with DMXAA caused significant increases in TGFβ1-stimulated phosphorylation states of p38 and amount of αSMΑ in LX2 cells. This suggests the involvement of the STING in HSCs in MCD-induced liver fibrosis, unlike in CCl4-induced model where CCl4 did not alter STING signaling in HSCs. More importantly, we further validated that the STING in macrophages acted through a paracrine manner to regulate HSC activation. As supporting evidence, treatment of LX2 cells with conditioned media of DMXAA-treated WT macrophages, but not conditioned media of DMXAA-treated STINGgt macrophages, significantly increased p38 phosphorylation, αSMA amount, and the mRNA levels of αSMA, Col1a1, and Fn, which are indicative of HSC activation. At this point, it is not clear of the proportional contributions of the STING in HSC vs. macrophages in regulating HSC activation. Considering that livers of MCD-fed WT mice contained massive aggregations of macrophages/Kupffer cells, we argue a more important role for the STING in macrophages in pathogenesis of liver fibrosis through paracrine mechanisms to activate HSCs.
STING disruption also decreased HFD-induced insulin resistance, which contributes to NAFLD pathogenesis. To date, there is no available evidence indicating direct interactions between STING and the components of insulin signaling pathway. While addressing the effects of disrupting STING (in endothelial cells) and/or signaling molecules downstream of STING on regulating systemic insulin sensitivity 29, 34, 35, limited studies have suggested that STING-TBK1-IRF3 pathway(s) act through inflammatory mechanisms to impair insulin signaling. Given this, STING disruption likely acts through suppressing inflammation to improve insulin sensitivity.
In summary, we established a link between STING and NAFLD/NASH in both mouse models and human patients’ samples. Specifically, we showed increased liver signaling events downstream of STING in a mouse model of NAFLD, and revealed, for the first time, that STING expression is increased in hepatic non-parenchymal cells of human patients with NAFLD. We further demonstrated that STING plays a deleterious role in NAFLD/NASH and this role was achieved through enhancing macrophage proinflammatory activation. Therefore, targeting STING to inhibiting macrophage proinflammatory activation is a viable therapeutic or preventive approach for the management of NAFLD/NASH.
Supplementary Material
Acknowledgments
FUNDING
This work was supported in whole or in part by grants from the American Diabetes Association (1–17-IBS-145) and the National Institutes of Health (DK095862) to C.W. This work was also supported in part by the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White, a VA Research Career Scientist Award to G.A. and the NIH grants DK076898, DK110035, DK062975, AA025157, AA025997, and DK054811 to G.A, F.M. and S.G, and by a VA Merit Award (1I01BX003031) and an NIH grant (DK108959) to H.F. Also, C.W. is supported by the Hatch Program of the National Institutes of Food and Agriculture (NIFA).
Abbreviations
- ACC1
acetyl-CoA carboxylase 1
- ALD
alcoholic fatty liver disease
- αSMA
α-smooth muscle actin
- BMDM
bone marrow-derived macrophages
- BSA
bovine serum albumin
- CD
chow diet
- Col1a1
collagen Type I alpha 1
- CPT1a
carnitine palmitoyltransferase1a
- DMEM
Dulbecco’s modified Eagle’s medium
- DMXAA
5,6-dimethylxanthenone-4-acetic acid
- FAS
fatty acid synthase
- FBS
fetal bovine serum
- FFA
free fatty acids
- FN
fibronectin
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GTT
glucose tolerance test
- H&E
hematoxylin and eosin
- HFD
high-fat diet
- HSCs
hepatic stellate cells
- LFD
low-fat diet
- IFNβ
interferon beta
- IL-1β
interleukin 1β
- IL-4
interleukin 4
- IL-6
interleukin 6
- IMDM
Iscove’s Modified Dulbecco’s medium
- ITT
insulin tolerance test
- LPS
lipopolysaccharide
- IRF3
interferon regulatory factor 3
- JNK
c-Jun N-terminal kinases
- MCD
methionine- and choline-deficient diet
- MMP2
matrix metallopeptidase 2
- NAFLD
non-alcoholic fatty liver disease
- NFκB
nuclear factor kappa B
- NASH
non-alcoholic steatohepatitis
- Pp65
phosphorylated p65 subunit of NFκB
- Pp46
phosphorylated JNK1 (p46)
- PPARγ
peroxisome proliferator-activated receptor gamma
- RQ
respiratory quotient
- SREBP1c
sterol regulatory element-binding protein 1c
- STING
stimulator of interferon genes
- TBK1
TANK-binding kinase 1
- TG
triglycerides
- TNFα
tumor necrosis factor α
- TGFβ1
transforming growth factor β1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
This material, in part, is the result of work supported with resources and the use of facilities at the Central Texas Veterans Health Care System, Temple, Texas. The content is the responsibility of the author(s) alone and does not necessarily reflect the views or policies of the Department of Veterans Affairs or the United States Government.
The authors have nothing to declare.
Author names in bold designate shared co-first authorship.
REFERENCES
- 1.Sanyal AJ. Mechanisms of Disease: pathogenesis of nonalcoholic fatty liver disease. Nat Clin Pract Gastroenterol Hepatol 2005;2:46–53. [DOI] [PubMed] [Google Scholar]
- 2.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328–357. [DOI] [PubMed] [Google Scholar]
- 3.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006;43:S99–S112. [DOI] [PubMed] [Google Scholar]
- 4.Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73–84. [DOI] [PubMed] [Google Scholar]
- 5.Estes C, Razavi H, Loomba R, et al. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology (Baltimore, Md.) 2018;67:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caldwell SH, Crespo DM, Kang HS, et al. Obesity and hepatocellular carcinoma. Gastroenterology 2004;127:S97–S103. [DOI] [PubMed] [Google Scholar]
- 7.Powell EE, Jonsson JR, Clouston AD. Steatosis: Co-factor in other liver diseases. Hepatology 2005;42:5–13. [DOI] [PubMed] [Google Scholar]
- 8.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 2010;51:1820–1832. [DOI] [PubMed] [Google Scholar]
- 9.Younossi ZM, Otgonsuren M, Henry L, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015;62:1723–1730. [DOI] [PubMed] [Google Scholar]
- 10.Angulo P NAFLD, Obesity, and Bariatric Surgery. Gastroenterology 2006;130:1848–1852. [DOI] [PubMed] [Google Scholar]
- 11.Neuschwander-Tetri BA. NASH: Thiazolidinediones for NASH--one pill doesn’t fix everything. Nat Rev Gastroenterol Hepatol 2010;7:243–244. [DOI] [PubMed] [Google Scholar]
- 12.Deng Z, Liu Y, Cunren Liu C, et al. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 2009;50:1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang W, Metlakunta A, Dedousis N, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010;59:347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu H, Li H, Woo S-L, et al. Myeloid cell-specific disruption of Period1 and Period2 exacerbates diet-induced inflammation and insulin resistance. J Biol Chem 2014;289:16374–16388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai Y, Li H, Liu M, et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology 2018;68:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kodama Y, Kisseleva T, Iwaisako K, et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 2009;137:1467–1477.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishikawa H, Barber GN. STING an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature 2008;455:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petrasek J, Iracheta-Vellve A, Csak T, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proceedings of the National Academy of Sciences of the United States of America 2013;110:16544–16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng L, Liang H, Xu M, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014;41:843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paijo J, Döring M, Spanier J, et al. cGAS sense s human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog 2016;12:e1005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomsen MK, Nandakumar R, Stadler D, et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 2016;64:746–759. [DOI] [PubMed] [Google Scholar]
- 23.Iracheta-Vellve A, Petrasek J, Gyongyosi B, et al. Endoplasmic reticulum stress-induced hepatocellular death pathways mediate liver injury and fibrosis via stimulator of interferon genes. J Biol Chem 2016;291:26794–26805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho C-S, Park H-W, Ho A, et al. Lipotoxicity induces hepatic protein inclusions through TBK1-mediated p62/SQSTM1 phosphorylation. Hepatology 2017:n/a-n/a. [DOI] [PMC free article] [PubMed]
- 25.Qiao JT, Cui C, Qing L, et al. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 2018;81:13–24. [DOI] [PubMed] [Google Scholar]
- 26.Guo X, Shu C, Li H, et al. Cyclic GMP-AMP ameliorates diet-induced metabolic dysregulation and regulates proinflammatory responses distinctly from STING activation. Sci Rep 2017;7:6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conlon J, Burdette DL, Sharma S, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol 2013;190:5216–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015;347. [DOI] [PubMed] [Google Scholar]
- 29.Mao Y, Luo W, Zhang L, et al. STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler Thromb Vasc Biol 2017;37:920–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian L, Changzheng L, Guizhi Y, et al. Sphingosine kinase 1 promotes liver fibrosis by preventing miR-19b-3p-mediated inhibition of CCR2. Hepatology 2018;0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larkin B, Ilyukha V, Sorokin M, et al. Cutting Edge: Activation of STING in T cells induces Type I IFN responses and cell death. J Immunol 2017;199:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gandhi CR, Chaillet JR, Nalesnik MA, et al. Liver-specific deletion of augmenter of liver regeneration accelerates development of steatohepatitis and hepatocellular carcinoma in mice. Gastroenterology 2015;148:379–391.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuster S, Cabrera D, Arrese M, et al. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349–364. [DOI] [PubMed] [Google Scholar]
- 34.Kumari M, Wang X, Lantier L, et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest 2016;126:2839–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao P, Wong Ki, Sun X, et al. TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell 2018;172:731–743.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.