

Abstract
The treatment of Ewing sarcoma has changed very little in the past two decades and novel treatment approaches are needed. We recently identified that Ewing sarcoma cells are uniquely vulnerable to inhibitors of ribonucleotide reductase (RNR), the rate limiting enzyme in the synthesis of deoxyribonucleotides. We subsequently found that the inhibition of checkpoint kinase 1 (CHK1) increases the sensitivity of Ewing sarcoma cells to inhibitors of RNR, such as gemcitabine. However, Ewing sarcoma cells exhibit high levels of the CHK1 protein, which may represent an adaptive response to elevated levels of endogenous DNA replication stress. Consequently, we began this work with the aim of determining the impact of CHK1 levels on drug sensitivity, as well as identifying the mechanisms and pathways that regulate CHK1 levels in Ewing sarcoma cells. In this report, we show that the high levels of the CHK1 protein in Ewing sarcoma cells limit the efficacy of CHK1 inhibitors. However, inhibition of mTORC1/2 activates the translational repressor 4E-BP1, reduces protein synthesis, and decreases levels of the CHK1 protein in Ewing sarcoma cells. Similarly, we identified that the CHK1 inhibitor prexasertib also activates 4E-BP1, inhibits protein synthesis, and reduces CHK1 protein levels in Ewing sarcoma cells. Moreover, the combination of prexasertib and gemcitabine was synergistic in vitro, caused tumor regression in vivo, and significantly prolonged mouse survival in a Ewing sarcoma xenograft experiment. Overall, our results provide insight into Ewing sarcoma biology and support further investigation of the CHK1 pathway as a therapeutic target in Ewing sarcoma tumors.
INTRODUCTION
Ewing sarcoma is a highly aggressive bone and soft tissue cancer that is caused by the EWS-FLI1 fusion protein (1). EWS-FLI1 is required for tumor growth and survival (2). However, directly targeting EWS-FLI1 with drugs has been challenging and an alternative therapeutic approach is to identify unique vulnerabilities incurred by the oncoprotein (3). In previous work, we used a human embryonic stem cell model to identify that Ewing sarcoma cells are vulnerable to gemcitabine, hydroxyurea, ciclopirox, and other drugs that inhibit ribonucleotide reductase (RNR), which is the rate limiting enzyme in the synthesis of deoxyribonucleotides (4, 5).
More recently, we found that the inhibition of checkpoint kinase 1 (CHK1), a downstream effector of Ataxia Telangiectasia and Rad3-Related Protein (ATR) and a critical mediator of the response to DNA replication stress, increases the sensitivity of Ewing sarcoma cells to RNR inhibitors in vitro and in vivo (5). Ewing sarcoma cells also exhibit sensitivity to CHK1 and ATR inhibitors as single-agents (6–8). However, Nieto-Soler et al. has shown that Ewing sarcoma cells exhibit high levels of the CHK1 protein, which may reflect a response to elevated levels of endogenous DNA replication stress (6). In addition, Wayne et al. has shown that the complete and sustained inhibition of CHK1 is necessary to inhibit the growth of sensitive cell lines (9). Consequently, we began this work with the aim of determining the effect of CHK1 protein levels on drug sensitivity and identifying the mechanisms that regulate CHK1 levels in Ewing sarcoma cells.
In this report, we show that the high levels of the CHK1 protein in Ewing sarcoma cells limit the efficacy of CHK1 inhibitors. Notably, multiple mechanisms are known to regulate CHK1 protein levels and activity in cancer cells, but recent work has identified that the mTOR pathway can regulate levels of CHK1 via both transcriptional and translational mechanisms (10–15). The mTOR pathway is also known to have a critical role in Ewing sarcoma tumors and mTOR inhibitors reduce the growth of Ewing sarcoma cells in vitro and in xenograft experiments (16–18). Moreover, in addition to CHK1, the mTOR pathway has also been reported to regulate the abundance of the RRM1 and RRM2 subunits of RNR (19, 20). Consequently, based on our previous work demonstrating synergy between RNR and CHK1 inhibitors in Ewing sarcoma, we initially focused our investigation on the role of mTOR signaling in the regulation of CHK1 levels in Ewing sarcoma cells (5).
We identified that the inhibition of mTORC1/2, but not mTORC1, activates the protein translation repressor 4E-BP1, reduces protein synthesis, and decreases levels of the CHK1 protein in Ewing sarcoma cells. In addition, prexasertib, a catalytic CHK1 inhibitor that also inhibits the mTOR pathway, activates 4E-BP1, inhibits protein synthesis, and reduces CHK1 protein levels in Ewing sarcoma cells. Moreover, the combination of prexasertib and gemcitabine was synergistic in vitro and significantly prolonged mouse survival in a xenograft experiment. Overall, our results provide insight into Ewing sarcoma biology and identify a candidate pathway that can be targeted in Ewing sarcoma tumors.
MATERIAL AND METHODS
Cell lines and culture:
Cell lines were maintained at 37° C in a 5% CO2 atmosphere. The A673, TC32, TC71, SK-NEP, TTC466 and EW8 cell lines were kindly provided by Dr. Kimberly Stegmaier (Dana-Farber Cancer Institute, Boston, MA). The BJ-tert, HEK-293T, HT1080, RPE-tert, HeLa, and U2OS cell lines were obtained from ATCC. The RH30, RD, and SAOS cell lines were provided by Dr. Munir Tanas (University of Iowa, Iowa City, IA). The Panc-1 and PaCa-2 cell lines were provided by Dr. Garry Buettner (University of Iowa, Iowa City, IA). Cells were cultured as previously described (4, 5). Cell lines were authenticated by DNA fingerprinting using the short tandem repeat (STR) method and used within 8–10 passages of thawing.
Chemical compounds:
Chemical compounds were purchased from Sigma (gemcitabine and temsirolimus), Selleckchem (prexasertib, LY2603618, U0126, and TAK-228), APExBio (VE-822), Thermo Fisher Scientific (puromycin), and MedChemExpress (Torin2, MK-8776, olaparib, and 4EGI-1).
Puromycin labeling:
Protein synthesis was assessed using puromycin labeling (SUnSET technique), as described (21). For in vitro labeling, puromycin (2 μg/mL, Thermo Fisher Scientific) was added to cells at a 1:400 dilution. The cells were then incubated with the puromycin for one hour before cell lysates were collected, as described in the Immunoblotting section. Protein loading for the immunoblots was normalized using cell number.
Cell viability:
Cell proliferation was measured using the resazurin (AlamarBlue) fluorescence assay as previously described (5). The combination index (CI) as a measure of drug synergy was determined using the method of Chou and Talalay with five drug concentrations at a fixed dose ratio (22). The data were analyzed using the CompuSyn software (http://www.combosyn.com/).
siRNA transfection:
Cells (1.5–3 × 105) were plated one day prior to transfection in six-well plates. Cells were transfected with siRNA using Lipofectamine RNAiMax (Thermo Fisher Scientific) as previously described (5). siCHK1.1, siCHK1.2, and siCHK1.3 were obtained from IDT (Coralville, IA). siCHK1.pool was a SMARTpool ON-TARGETplus reagent (GE Dharmacon) and siRRM2 was described previously (4). siControl was purchased from Cell Signaling Technology (#6568). siEWS-FLI1 (5’-GCAGAACCCUUCUUAUGACUU-3’) was synthesized by IDT (23).
Doxycycline-inducible shCHK1:
A shERWOOD UltramiR Lentiviral Inducible shRNA plasmid targeting CHK1 (ULTRA342152) was obtained from Transomic Technologies (Huntsville, AL). Lentivirus was produced by transfecting HEK-293T cells with the shRNA plasmid and packaging plasmids (psPAX2 and pMD2.G) according to the FuGENE 6 (Roche) protocol. For the lentiviral transduction, Ewing sarcoma cells were incubated with 2 mL of virus and 6 μg/mL of polybrene (Sigma-Aldrich) for 12–16 hours. Cells were selected in 1 μg/mL puromycin 48 hours after transduction.
Xenograft:
The Institutional Animal Care and Usage Committee at the University of Iowa approved the animal studies. The studies were conducted in adherence with the NIH Guide for the Care and Use of Laboratory Animals. Approximately 1.0 × 106 TC71 cells were mixed with 30% matrigel and injected subcutaneously into the flanks of 6-week old, female NCr mice. When tumors were palpable (~100–200 mm3), the mice were divided into cohorts (8–9 mice per cohort) and treated with drug or vehicle. Mice cohorts were treated with vehicle, gemcitabine (150 mg/kg, intraperitoneal, day 1), prexasertib (10 mg/kg, subcutaneous, BID, days 1, 2, 3 and 14, 15, 16), and the combination of gemcitabine and prexasertib (dosing as for the single agents). Tumor volumes were measured periodically using calipers (volume = 0.5×length×width2). Animals were sacrificed when a tumor reached 20 mm in any dimension. GraphPad Prism was used to generate survival curves. For the in vivo puromycin labeling experiment, NCr mice were subcutaneously injected with two Ewing sarcoma cell lines (TC71 and EW8) and allowed to develop measurable tumors. The mice were then divided into four cohorts, with two mice per cohort, and treated with drugs, as described above. On Day 2, the mice were injected with puromycin (0.5 mg) and then sacrificed 90 minutes later (24).
Immunoblotting:
Immunoblots were performed as previously described (4, 5). Protein loading for the immunoblots was normalized using cell number. Antibodies to the following proteins were used in the immunoblots: phospho-Histone-139 H2A.X (Cell Signaling, #9718, 1:1000), phospho-Chk1–345 ( Cell Signaling, #2348, 1:1000), phospho-Chk1–296 (Cell Signaling, #12302, 1:1000), Chk1 (Cell Signaling, #2360, 1:1000), c-MYC (Cell Signaling, #5605, 1:1000), puromycin (Millipore, #AF488, 1:2000), 4E-BP1 (Cell Signaling, #9644, 1:1000), PARP (Cell Signaling, #9532, 1:1000), cleaved caspase-3 (Cell Signaling, #9664, 1:1000), p-4E-BP1–37/46 (Cell Signaling, #2855, 1:1000), RRM1 (Cell Signaling, #8637, 1:1000), RRM2 (Santa Cruz, #398294, 1:500), CHK2 (Cell Signaling, #6334, 1:1000), S6 (Abcam, #40820, 1:1000), P-S6–235/236 (Cell Signaling, #4858, 1:1000) and tubulin (Proteintech, 66031–1, 1:2000).
Immunohistochemistry:
Tumor xenografts were fixed in formalin for immunohistochemical staining. Immunostaining for Ki67 was conducted using a rabbit anti-human Ki67 antibody (Cell Signaling, #9027, 1:400) on tumor sections. Staining was performed using the Vectastain ABC Kit (Vector Laboratories), according to the manufacturer’s instructions.
Cancer Cell Line Encyclopedia (CCLE):
Data for 4E-BP1 expression were obtained from the CCLE (Broad Institute; https://portals.broadinstitute.org/ccle) (25). A Mann-Whitney test was performed to compare the Ewing sarcoma cell lines to the other cell lines.
Clonogenic assay:
Cells (500 cells/well) were plated in triplicate in a 6-well plate and allowed to adhere overnight. The cells were then treated with prexasertib, or vehicle, for six hours. Prexasertib was then removed and the cells were washed three times with PBS. The cells were allowed to grow for ~10–14 days and then fixed and counted as described (5).
γH2AX flow cytometry:
Cells (3 × 105 cells/well) were plated in a 6-well plate and allowed to adhere overnight. The cells were then treated with drugs, or vehicle, for six hours. Flow cytometry for γH2AX was then performed as previously described (5).
Annexin V assay:
Annexin V was measured by flow cytometry using a FITC Annexin V/Dead Cell Apoptosis Kit (ThermoFisher) (5).
Quantitative reverse transcriptase PCR:
Total RNA was extracted from cells using a RNeasy kit (Qiagen) following the manufacturer’s instructions. 1 μg of total RNA was reverse-transcribed into first-strand cDNA using random hexamer primers and the SuperScript III Reverse Transcriptase (Invitrogen). RT-qPCR was performed on the ViiA 7 Real-Time PCR System (Life Technologies) using SYBR Select Master Mix (Thermo Fisher Scientific). Reactions were performed in triplicate and gene expression was normalized to GAPDH. The PCR primer sequences for CHEK1 and GAPDH were 5’-ATATGAAGCGTGCCGTAGACT-3’ and 5’-CTGGGCTACACTGAGCACC-3’, respectively.
Statistical Analysis:
Student’s t-test was used to calculate P-values for the comparison of two groups and analyses for more than two groups were conducted with a one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test. P-values for the tumor volume measurements in the xenograft experiment were determined by 2-way ANOVA and the Log-rank (Mantel-Cox) test was used to calculate P-values comparing the mouse survival curves. Statistical analyses were conducted using GraphPad Prism 5.0.
RESULTS
Reducing CHK1 protein levels sensitize Ewing sarcoma cells to inhibitors of CHK1.
Ewing sarcoma cell exhibit high levels of CHK1 protein (6). We used multiple siRNAs to knockdown CHK1 in Ewing sarcoma cells (Figures 1A and Supplementary Figure 1). However, the ~70% reduction in CHK1 protein caused by the siRNAs did not reduce the growth of the Ewing sarcoma cells, which is consistent with our previous data and work from Wayne et al. showing that the complete and sustained inhibition of CHK1 is necessary to inhibit the growth of sensitive cell lines (Figure 1B) (5, 9). Similar results were obtained using a doxycycline-inducible shRNA that targets CHK1 (Figures 1C and 1D). Next, we used immunoblotting for the CHK1 auto-phosphorylation site Ser-296 to test the ability of three CHK1 inhibitors, LY2603618, MK-8776, and prexasertib, to inhibit CHK1 function (26–29). Figure 1E shows that the three CHK1 inhibitors caused a dose-dependent inhibition of CHK1–296 auto-phosphorylation when the cells were also treated with gemcitabine, which inhibits ribonucleotide reductase and activates CHK1 (5). Notably, prexasertib was significantly more potent at inhibiting CHK1 than either LY2603618 or MK-8776 and inhibited CHK1 activity at drug concentrations <1 nM.
Figure 1.

Reducing CHK1 protein levels sensitizes Ewing sarcoma cells to inhibitors of CHK1. (A) Immunoblot showing the effect of a control siRNA and four unique siRNAs targeting CHK1 on CHK1 protein levels. (B) Viability of EW8 Ewing sarcoma cells treated with the control siRNA and the four siRNAs targeting CHK1. Cell viability was assessed 48 hours after transfection of the siRNAs using the AlamarBlue Fluorescence Assay. The results are representative of three independent experiments. Error bars represent the mean ± SD of three technical replicates. (C) Immunoblot showing the effect of a doxycycline-inducible shRNA that targets CHK1 on CHK1 protein levels. (D) Viability of doxycycline-inducible shCHK1 cells treated with doxycycline or vehicle. Cell viability was assessed 72 hours after addition of doxycycline. (E) EW8 cells were treated with 50 nM gemcitabine in combination with increasing doses of prexasertib, LY2603618, or MK-8776 for 6 hours. Cell lysates were then collected and blotted for p-CHK1–296. (F) EW8 and TC71 cells were treated with increasing doses of prexasertib for 6 hours. Cell lysates were then collected and blotted for p-CHK1–345. (G) Immunoblot analysis of p-CHK1–345 in EW8 cells treated with prexasertib in the presence or absence of an ATR inhibitor (VE-822). (H) Knockdown of CHK1 in EW8 cells using four unique siCHK1 sensitizes cells to the CHK1 inhibitors prexasertib (1 nM), LY2603618 (250 nM), and MK-8776 (250 nM). Cells were transfected with siRNA on day 1, treated with drug for 24 hours starting on day 2, and then analyzed for growth on day 4 using the AlamarBlue Fluorescence Assay. The results are representative of two independent experiments. Error bars represent mean ± SD of three technical replicates. (I) Knockdown of CHK1 using doxycycline-inducible shCHK1 sensitizes cells to the CHK1 inhibitors prexasertib, LY2603618, and MK-8776. Error bars represent mean ± SD of three technical replicates. (J) Immunoblot showing the effect of siCHK1.pool and prexasertib (1 nM) on the markers of apoptosis, cleaved PARP and cleaved caspase-3, in four Ewing sarcoma cell lines. (K) Immunoblot showing the effect of the combination of siCHK1.pool and doxycycline-inducible shCHK1on cleaved PARP and cleaved caspase-3. ****, P-value < 0.0001; ***, P-value < 0.001; NS, not significant.
Inhibition of CHK1, in addition to impairing the response to DNA replication stress, also causes DNA replication stress by altering origin licensing, which results in the phosphorylation of CHK1 itself at Ser-345 via ATR (30). Figures 1F and 1G show that treating Ewing sarcoma cells with prexasertib resulted in the phosphorylation and activation of CHK1, which was blocked by co-treatment with the ATR inhibitor VE-822 (31). We then tested whether reducing CHK1 levels, using siRNA knockdown, would increase the sensitivity of Ewing sarcoma cells to the CHK1 inhibitors. Figures 1H and 1I show that reducing CHK1 levels in Ewing sarcoma cells using either siRNA or shRNA significantly sensitized the cells to concentrations of CHK1 inhibitors that blocked CHK1 activity (Figure 1E) but did not impair cell growth as single agents. This reduction in cell growth was associated with cleavage of PARP and caspase-3, markers of apoptosis, in multiple Ewing sarcoma cell lines (Figures 1J and 1K).
Inhibition of mTORC1/2 reduces protein synthesis and CHK1 levels in Ewing sarcoma cells.
The mTOR pathway, which has been reported to regulate CHK1 levels, is known to be active in Ewing sarcoma cells (16, 17). To test whether the mTOR pathway regulates levels of CHK1 in Ewing sarcoma cells we treated cell lines with a mTORC1/2 inhibitor (TAK-228) that was previously shown by Slotkin et al. to inhibit the growth of Ewing sarcoma cells in vitro and in xenograft experiments (17). Figure 2A shows that the treatment of three Ewing sarcoma cell lines, A673, EW8, and TC71, with TAK-228 caused a dose-dependent reduction in cell growth. Treatment of Ewing Sarcoma cells with TAK-228 reduced protein levels of CHK1, but not the related protein CHK2 (Figure 2B). Based on reports in the literature, as well as the sensitivity of Ewing sarcoma cells to inhibitors of RNR, we also tested whether inhibition of the mTOR pathway reduces levels of RRM1 and RRM2 (4, 19, 20). Figure 2B shows that inhibition of mTOR reduced levels of both RRM1 and RRM2, although the effect was larger for RRM2 than RRM1. TAK-228 also reduced CHK1 levels in three non-Ewing sarcoma cell lines, HT1080, RD, and U2OS, but did not alter CHK1 levels in HEK-293T cells (Figure 2C).
Figure 2.

Inhibition of mTORC1/2 reduces protein synthesis and CHK1 levels in Ewing sarcoma cells. (A) Dose-response curves for cell lines treated with different concentrations of TAK-228 for 72 hours. Cell viability was then assessed using the AlamarBlue Fluorescence Assay. The results are representative of two independent experiments. Error bars represent mean ± SD of three technical replicates. (B, C) Immunoblot showing the effect of vehicle (DMSO) or TAK-228 on CHK1, CHK2, RRM1, and RRM2 levels in four Ewing sarcoma cell lines (B) and four other cell lines (C). (D, E) Immunoblot showing the effect of TAK-228 on protein synthesis, as assessed using puromycin labeling, in Ewing sarcoma (D) and other (E) cell lines. (F) Immunoblot assessing protein synthesis, using puromycin labeling, in Ewing sarcoma cells treated with mTORC1 (temsirolimus) and mTORC1/2 (TAK-228 and Torin2) inhibitors. (G) Immunoblot of the mTOR targets 4E-BP1 and ribosomal protein S6 (S6) in Ewing sarcoma cell treated with TAK-228, temsirolimus, and Torin2. (H) Immunoblot assessing protein synthesis, using puromycin labeling, and CHK1 levels of cell lines treated with the protein translation inhibitor 4EGI-1. (I) Immunoblot analysis of c-MYC and p-4E-BP1–37/46 in EW8 cells treated with different doses of TAK-228. (J) RT-qPCR for CHK1 mRNA in two Ewing sarcoma cell lines treated with 100 nM TAK-228 or DMSO. In all immunoblots, protein loading was normalized using cell number.
The mTOR pathway regulates protein synthesis via multiple mechanisms (32). Consequently, we used puromycin labeling (SUnSET technique) to directly detect the synthesis of nascent proteins in Ewing sarcoma cells in the presence and absence of mTOR inhibition (21). Figure 2D shows that TAK-228 reduced protein synthesis in four Ewing sarcoma cell lines, similar to results obtained with the protein translation inhibitor cycloheximide (Supplementary Figure 2). TAK-228 also reduced protein synthesis in several non-Ewing sarcoma cell lines with the exception of HEK-293T cells, similar to the results for CHK1 protein levels (Figure 2E).
In order to delineate the impact of inhibition of mTORC1 versus mTORC1/2 we also tested an mTORC1 inhibitor (temsirolimus) and an additional ATP-competitive, mTORC1/2 inhibitor (Torin2) (33, 34). Figure 2F shows that the mTORC1/2 inhibitors (TAK-228 and Torin2), but not the mTORC1 inhibitor temsirolimus, suppressed protein synthesis in the EW8 and TC71 Ewing sarcoma cell lines. We then evaluated the effect of the TAK-228 on the phosphorylation of two mTOR targets, 4E-BP1 and ribosomal protein S6 (S6), that are critical regulators of protein translation (32). Figure 2G shows that temsirolimus reduced phosphorylation of ribosomal protein S6, but not 4E-BP1. In contrast, the mTORC1/2 inhibitors TAK-228 and Torin2 inhibited the phosphorylation of both 4E-BP1 and S6. Similarly, the mTORC1/2 inhibitors TAK-228 and Torin2, but not temsirolimus, also reduced levels of CHK1. These findings are consistent with work that has shown that the ATP-competitive mTORC1/2 inhibitors suppress cap-dependent protein translation through 4E-BP1 protein dephosphorylation, while the effects of temsirolimus on this pathway are more variable (35–38).
Analysis of data generated by the Cancer Cell Line Encyclopedia project (CCLE, Broad Institute) demonstrate that Ewing sarcoma cell lines express high levels of 4E-BP1 mRNA, total 4E-BP1 protein, and phospho-4E-BP1 protein relative to other cancer types (Supplementary Figure 3) (25). Active (dephosphorylated) 4E-BP1 protein negatively regulates protein synthesis by binding the cap-binding protein eIF4E, inhibiting eIF4G binding, and preventing assembly of the eIF4F complex (39). To further evaluate the role of cap-dependent protein translation in the regulation of levels of CHK1 we treated Ewing sarcoma cells with 4EGI-1, a drug that disrupts the eIF4F complex and inhibits cap-dependent translation (40). Figure 2H shows that 4EGI-1 reduced total protein synthesis, as assessed using puromycin labeling, and specifically reduced levels of CHK1 in Ewing sarcoma and osteosarcoma cells. 4E-BP1 is known to regulate the translation of c-Myc, which has a well-described role in Ewing sarcoma tumorigenesis, and Figure 2I shows that TAK-228 caused a dose-dependent reduction in the c-Myc protein (39). Finally, to test whether a gene transcription mechanism may also contribute to the regulation of CHK1 protein levels, we isolated mRNA from EW8 and TC71 cells treated with TAK-228 and performed RT-qPCR to measure CHEK1 mRNA. Figure 2J demonstrates that TAK-228 caused a small, ~20% reduction in CHEK1 mRNA in both EW8 and TC71 cells, suggesting that inhibition of protein synthesis is the primary mechanism regulating CHK1 levels.
Prexasertib inhibits the growth of Ewing sarcoma cells.
TAK-228 was previously shown by Slotkin et al. to inhibit the growth of CHP100 Ewing sarcoma cells in vivo in xenograft experiments (17). Consequently, in order to identify additional drugs that regulate CHK1 levels in Ewing sarcoma, we turned our focus to the CHK1 inhibitor prexasertib that was reported by Sen et al. to also target the mTOR and protein synthesis pathways in small-cell lung cancer cells (27, 41). Figure 3A shows that treatment of Ewing sarcoma cells with prexasertib caused a decrease in phosphorylation (activation) of 4E-BP1 and a depletion of CHK1 protein in two Ewing sarcoma cell lines. Figures 3B and 3C show that prexasertib also caused a dose-dependent inhibition of protein synthesis in the Ewing sarcoma and osteosarcoma cells, respectively. We then evaluated the effect of prexasertib on the growth of Ewing sarcoma cells, as well as additional transformed (RH30, SAOS, HEK-293T, and HT1080) and non-transformed (BJ-tert and RPE-tert) cell lines. In a dose-response assay, Ewing sarcoma cells were sensitive to treatment with prexasertib (Figure 3D) with IC50 values in the low nanomolar range. In comparison, the other cancer cell lines showed variable sensitivity, while the non-transformed cells lines were relatively resistant to prexasertib (Figure 3E). Prexasertib also significantly inhibited the growth of Ewing sarcoma cells, but not non-transformed cell lines, in long-term clonogenic assays (Figure 3F and Supplementary Figure 4). Similarly, treatment of Ewing sarcoma cells with prexasertib resulted in cleavage of PARP and an increase in cells positive for annexin-V and propidium iodide, both markers of apoptosis (Supplementary Figure 5). These results are consistent with work by Lowery et al., and others, that has shown that a wide range of adult and pediatric cancer cell lines, including Ewing sarcoma, are sensitive to prexasertib (8, 27, 42).
Figure 3.

Prexasertib inhibits the growth of Ewing sarcoma cells. (A) Immunoblot analysis of CHK1 and p-4E-BP1–36/47 in EW8 and TC71 cells treated with prexasertib. (B, C) Immunoblot assessing protein synthesis, using puromycin labeling, in two Ewing sarcoma cell lines (B) and U2OS osteosarcoma cells (C) treated with prexasertib. (D) Dose-response curves for 6 Ewing sarcoma cell lines treated with different concentrations of prexasertib for 24 hours. Cell viability was then assessed 48 hours later using the AlamarBlue Fluorescence Assay. The results are representative of two independent experiments. Error bars represent mean ± SD of three technical replicates. (E) Dose-response curves for 7 non-Ewing sarcoma cell lines treated with different concentrations of prexasertib for 24 hours. Experiment was performed as described in (E). (F) Cell lines were treated with prexasertib (5 nM) for 24-hours and cell growth was then quantified in colony formation assays. Error bars represent mean ± SD of three technical replicates ( ***, P-value < 0.0001; **, P-value < 0.001; *, P-value < 0.01). (G) Dose-response curve for EW8 cells treated for 6 hours with different concentrations of gemcitabine in the presence or absence of 1 nM prexasertib. Cell viability was then assessed 42 hours after drug removal using the AlamarBlue Fluorescence Assay. The results are representative of two independent experiments. Error bars represent mean ± SD of three technical replicates. (H) EW8 and TC71 cells were treated with different concentrations of gemcitabine and prexasertib, using a constant drug ratio, for 6 hours after which the drugs were removed and cell viability was measured 42 hours later. Data were analyzed using the CompuSyn software. Combination Index (CI) versus Fraction Affected (Fa) plot shows the effect of the combination of gemcitabine and prexasertib. CI < 0.9 indicates synergism. (I) The phosphorylation of H2AX was assessed using flow cytometry in EW8 cells treated with prexasertib (1 nM), gemcitabine (10 nM), or the combination of drugs.
Prexasertib and gemcitabine cause synergistic toxicity.
Next, we tested whether prexasertib would increase the sensitivity of Ewing sarcoma cells to gemcitabine, which impairs DNA replication and is synergistic with the CHK1 inhibitor LY2603618 in Ewing sarcoma cells (5). Figure 3G shows that prexasertib (1 nM) increased the sensitivity of EW8 Ewing sarcoma cells to gemcitabine, with a ~4-fold reduction in the IC50 value. We also used the method of Chou and Talalay to calculate a combination index (CI) to test if the combination of gemcitabine and prexasertib was synergistic (22). The combination of gemcitabine and prexasertib demonstrated synergism (CI<0.9) in two Ewing sarcoma cell lines (EW8 and TC71) with CI values ranging from 0.25 to 0.80 (Figure 3H). Consistent with this cell viability data, the combination of prexasertib (1 nM) with gemcitabine (10 nM) caused robust phosphorylation of H2AX, a marker of DNA damage and double strand breaks (Figure 3I) (43).
The combination of gemcitabine and prexasertib inhibits protein synthesis and causes tumor regression in Ewing sarcoma xenografts.
We then evaluated whether gemcitabine and prexasertib could inhibit the growth of tumor cells in a mouse xenograft experiment. NCr mice were subcutaneously injected with Ewing sarcoma (TC71) cells and allowed to develop measurable tumors. The mice were then divided into four cohorts and treated with either vehicle, gemcitabine, prexasertib, and the combination of gemcitabine and prexasertib. Figures 4A and 4B shows that there was a statistically significant difference in tumor volumes between the control and drug treated groups. Notably, both prexasertib and the combination of gemcitabine and prexasertib resulted in tumor regression over the first ten days of the experiment without apparent systemic toxicity (Figure 4B and Supplementary Figure 6). However, the effect of gemcitabine in combination with prexasertib resulted in a more sustained inhibition of tumor growth and prolongation of mouse survival compared to prexasertib alone (Figure 4C). The median survival of the mice treated with vehicle, gemcitabine, prexasertib, and the combination of gemcitabine and prexasertib was 21, 27, 34, and 99 days, respectively.
Figure 4.

The combination of gemcitabine and prexasertib inhibits protein synthesis and causes tumor regression in Ewing sarcoma xenografts. TC71 cells were engrafted in nude mice. After developing tumors, the mice were divided into four cohorts and treated with either vehicle, gemcitabine (150 mg/kg, intraperitoneal, daily, days 1 and 14), Prexasertib (10 mg/kg, subcutaneous, BID, days 1, 2, 3 and 14, 15, 16), and the combination of gemcitabine and prexasertib (dosing as for the single agents). (A) Tumor size was quantified using caliper measurements. Growth curves for each drug treatment cohort are shown until mice were removed from that cohort due to tumor size. (B) Comparison of tumor volumes at Day 1 and Day 10 for each mouse cohort. Prexasertib and the combination of gemcitabine and prexasertib caused tumor regression ( ***, P-value < 0.0001; **, P-value < 0.001). (C) Kaplan-Meier survival curves for the different mouse cohorts. Log-rank (Mantel-Cox) test was used to calculate P-values comparing the survival curves. Vehicle versus the combination of gemcitabine and prexasertib, P-value < 0.0001. (D, E) NCr mice were subcutaneously injected with two Ewing sarcoma cell lines (TC71 and EW8) and allowed to develop measurable tumors. The mice were then divided into four cohorts, with two mice per cohort, and treated with either vehicle, gemcitabine (150 mg/kg, intraperitoneal, day 1), prexasertib (10 mg/kg, subcutaneous, BID, day 1), and the combination of gemcitabine and prexasertib (dosing as for the single agents). On Day 2, the mice were injected with puromycin and then sacrificed 90 minutes later. Tumors were collected, protein lysates isolated, and protein synthesis assessed using immunoblotting for puromycin. (E) Tumor lysates, collected as described in (D), were also analyzed for activation of CHK1 (P-CHK1), DNA damage (γH2AX), and apoptosis (cleaved PARP and cleaved caspase-3). (F) Immunohistochemical staining for the proliferation marker Ki-67.
Next, we used in vivo puromycin labeling to assess the effect of gemcitabine, prexasertib and the combination of the drugs on protein synthesis in vivo in a xenograft experiment (24). NCr mice were subcutaneously injected with two Ewing sarcoma cell lines (TC71 and EW8) and allowed to develop measurable tumors. The mice were then divided into four cohorts and treated with drugs, as described above. On Day 2, the mice were injected with puromycin and then sacrificed 90 minutes later. Figure 4D shows that the combination of prexasertib and gemcitabine reduced protein synthesis in vivo in the tumor xenografts. In addition, gemcitabine, and to a lesser extent prexasertib, also reduced protein synthesis in vivo. Similarly, Figure 4E shows that the combination of prexasertib and gemcitabine caused phosphorylation of CHK1 and gH2AX, as well as cleavage of PARP and caspase-3, in vivo in the xenografts. The combination of gemcitabine and prexasertib also reduced expression Ki-67, a marker of cell proliferation (Figure 4F).
Gemcitabine inhibits protein synthesis in Ewing sarcoma cells.
Based on the unexpected finding that gemcitabine reduced protein synthesis in Ewing sarcoma cells in vivo, we then treated Ewing sarcoma (TC71) and osteosarcoma (U2OS) cells with gemcitabine in vitro and assessed protein synthesis using puromycin-labeling. Figure 5A shows that treatment of Ewing sarcoma cells, but not osteosarcoma cells, with gemcitabine (100 nM) reduced protein synthesis. Additional Ewing sarcoma cell lines (A673, TC32, and EW8) also showed a reduction in protein synthesis after treatment with gemcitabine (Figure 5B). The effect of gemcitabine on protein synthesis in Ewing sarcoma cells was dose-dependent (Figure 5C) and evident by the end of the 6 hour drug incubation (Figure 5D). We then tested additional non-Ewing sarcoma cell lines, including BJ-tert, RPE-tert, and HEK-293T, and did not observe a reduction in protein synthesis after treatment with gemcitabine (Figure 5E and Supplementary Figure 7). To evaluate for a dose-dependency effect in the non-Ewing sarcoma cells lines, we treated U2OS and RPE-tert cells with 10-fold and 100-fold higher concentrations of gemcitabine, but did not observe any reduction in protein synthesis (Figure 5F). Finally, we used siRNA to knockdown the EWS-FLI oncogene in Ewing sarcoma A673 cells, which tolerate a reduction in EWS-FLI1 with a decrease in tumorigenicity but not viability, and observed that knockdown of EWS-FLI rescued protein synthesis in cells treated with gemcitabine (Figure 5G) (44). In addition, based on the xenograft results, we also tested the effect of the combination of gemcitabine and prexasertib on protein synthesis. Figure 5H shows that gemcitabine (10 nM) and prexasertib (1 nM or 2 nM) also suppressed protein synthesis, in contrast to either drug as a single agent (at the specified doses) (Figure 3B and Figure 5C).
Figure 5.

Gemcitabine inhibits protein synthesis in Ewing sarcoma cells. (A) Immunoblot assessing protein synthesis, using puromycin labeling, in a Ewing sarcoma (TC71) and osteosarcoma (U2OS) cell line treated with gemcitabine or the protein translation inhibitor 4EGI-1. Cells were treated with 100 nM gemcitabine for six hours, allowed to recover for 18 hours, labeled with puromycin, and then subjected to immunoblotting. (B) Four Ewing sarcoma cell lines were treated with gemcitabine, as described in (A), and then labeled with puromycin to assess protein synthesis. (C) EW8 and TC71 cells were treated with different doses of gemcitabine, as described for (A), and then labeled with puromycin to assess protein synthesis. (D) EW8 and TC71 cells were treated with 100 nM gemcitabine for 6 hours and then protein synthesis was assessed at 0, 6, and 24 hours after the drug treatment. (E) Four non-Ewing sarcoma cell lines were treated with 100 nM gemcitabine, as described in (A), and then labeled with puromycin to assess protein synthesis. (F) U2OS and RPE-tert cells were treated with increased doses of gemcitabine (1 μM and 10 μM ) and then protein synthesis was assessed using puromycin labeling. (G) EWS-FLI1 was knocked down in A673 cells using siRNA and then the effect of gemcitabine on protein synthesis was assessed using puromycin labeling. (H) EW8 cells were treated with gemcitabine, prexasertib, or the combination of the drugs and then protein synthesis was evaluated using puromycin labeling. In all immunoblots, protein loading was normalized using cell number.
Gemcitabine activates 4E-BP1 in Ewing sarcoma cells.
Based on the high levels of expression of 4E-BP1 in Ewing sarcoma cells, as well as the activation of 4E-BP1 by mTORC1/2 inhibitors and prexasertib, we then tested whether gemcitabine results in activation of 4E-BP1 in Ewing sarcoma cells. Figure 6A shows that treatment of Ewing sarcoma cell lines (A673, EW8, TC32, and TC71) with gemcitabine resulted in an increase in the active (unphosphorylated) form of 4E-BP1, which suppresses protein translation. However, in contrast to the Ewing sarcoma cells, treatment of other cell types with gemcitabine did not alter 4E-BP1 phosphorylation, with the exception of a modest increase in the unphosphorylated form of 4E-BP1 in BJ-tert cells (Figure 6B and Supplementary Figure 8). Next, we tested whether inhibition of protein synthesis by gemcitabine would also reduce CHK1 levels. Figure 6C shows that treatment of Ewing sarcoma cells with gemcitabine caused phosphorylation of CHK1 at 6-hours, but a loss of total CHK1 protein at the 24-hr time point. In contrast, treatment of U2OS osteosarcoma cells with gemcitabine resulted in phosphorylation of CHK1 at 6-hours, but no change in total CHK1 protein levels. Figure 6D shows that gemcitabine caused a dose-dependent activation of 4E-BP1 and reduction in both CHK1 and c-Myc protein levels. In addition, the knockdown of EWS-FLI1 in A673 cells reversed the effect of gemcitabine on 4E-BP1 phosphorylation (Figure 6E). Knockdown of EWS-FLI1 also caused a reduction in total 4E-BP1 levels, which may provide an explanation for the high levels of 4E-BP1 in Ewing sarcoma cells (Supplementary Figure 3). In an approach complementary to gemcitabine treatment, we also used siRNA (siRRM2) to knockdown RRM2 in two Ewing sarcoma cell lines and identified a reduction in protein synthesis (Figure 6F), as well as a loss of CHK1 (Figure 6G) and c-Myc (Figure 6H) proteins (4). Finally, to evaluate whether the inhibition of protein synthesis is a general stress response in Ewing sarcoma cells we treated two cell lines with olaparib (PARP inhibitor) and U0126 (MEK inhibitor), drugs that are known to cause toxicity in Ewing sarcoma (45). Neither of these drugs, though, had an effect on protein synthesis or 4E-BP1 phosphorylation (Figure 6I).
Figure 6.

Gemcitabine reduces the phosphorylation of 4E-BP1 in Ewing sarcoma cells. (A, B) Immunoblot for p-4E-BP1–37/46 and total 4E-BP1 in Ewing sarcoma (A) and control (B) cell lines treated with gemcitabine. Cells were treated with 100 nM gemcitabine for six hours, allowed to recover for 18 hours, and then subjected to immunoblotting. (C) Immunoblot for p-CHK1–345 and total CHK1 in Ewing sarcoma and osteosarcoma cells treated with gemcitabine, as described in (A). Lysates were collected at 0, 6, and 24 hours after drug was added. (D) Immunoblot showing the dose-dependent effects of gemcitabine on c-Myc, CHK1, 4E-BP1, and p-4E-BP1–37/46. (E) EWS-FLI1 was knocked down in A673 cells using siRNA and then the effect of gemcitabine on phosphorylation of 4E-BP1 was assessed using immunoblotting. (F) Protein synthesis was assessed, using puromycin labeling, in Ewing sarcoma and osteosarcoma cells treated with siRNA targeting RRM2 (siRRM2) or a control siRNA (siControl). (G, H) Immunoblot for total CHK1 (G) and c-Myc (H) in cells treated with siRRM2. In all immunoblots, protein loading was normalized using cell number. (I) EW8 and TC71 cells were treated with olaparib (5 μM) or U0126 (5 μM) for 24 hours and then protein synthesis was assessed using puromycin labeling.
DISCUSSION
The treatment of Ewing sarcoma, in particular metastatic and recurrent disease, is challenging and requires highly intensive, cytotoxic chemotherapy in combination with surgery and/or radiation (1). We previously used a human embryonic stem cell model of Ewing sarcoma to identify that the combination of gemcitabine, an irreversible inhibitor of RNR, and a checkpoint kinase 1 (CHK1) inhibitor is synergistic in vitro and significantly prolongs mouse survival in xenograft experiments (4, 5, 46). In this work, we identified that high levels of the CHK1 protein in Ewing sarcoma cells limit the efficacy of CHK1 inhibitors. However, from a mechanistic standpoint, we discovered that inhibition of mTORC1/2 decreases levels of CHK1 protein via a reduction in protein synthesis. Inhibition of mTORC1/2 also reduced levels of the subunits of RNR, RRM1 and RRM2, in Ewing sarcoma cells. This reduction of RRM1, RRM2, and CHK1 levels by inhibition of mTORC1/2 may provide an explanation, in part, for the toxicity of mTOR inhibitors toward Ewing sarcoma cells, although other targets and mechanisms likely contribute as well (16, 17).
We identified a novel pharmacological approach to modulate CHK1 levels in Ewing sarcoma cells using prexasertib. Prexasertib is an ATP-competitive, catalytic inhibitor of CHK1 that also reduces CHK1 protein levels by inhibiting protein synthesis in Ewing sarcoma cells (27). Although the decrease in CHK1 levels caused by prexasertib contributes to the synergy of this drug with gemcitabine, we also expect that the effect of prexasertib on overall protein synthesis is a critical factor in modulating the drug sensitivity and synergy. Notably, the combination of prexasertib and gemcitabine was synergistic in vitro, decreased protein synthesis in vivo in Ewing sarcoma xenografts, and significantly prolonged mouse survival in the xenograft experiment. We do note that gemcitabine is a nucleoside analogue that causes DNA chain termination, in addition to inhibiting the RRM1 subunit of RNR, and that this effect of the drug on cellular proliferation may also contribute to its mechanism of action.
Prexasertib is currently being tested in clinical trials in children and adults (47). Although the combination of prexasertib and gemcitabine has not been tested in a clinical trial, the combination of gemcitabine and other CHK1 inhibitors has been tested in several clinical trials (clinicaltrials.gov). The combination of prexasertib with other targeted agents, including WEE1, ATR and PARP inhibitors, may also offer a novel therapeutic approach for treating Ewing sarcoma tumors. For example, Sen et al. recently demonstrated that prexasertib improves the response to the PARP inhibitor olaparib in small cell lung cancer xenograft models (41). Several groups have identified that Ewing sarcoma cells are sensitive to PARP inhibitors, which suggests that prexasertib in combination with a PARP inhibitor may warrant investigation (48).
We also identified that multiple drugs, including TAK-228, prexasertib, and gemcitabine, activate 4E-BP1 and inhibit protein synthesis in Ewing sarcoma cells. TAK-228 is an ATP-competitive inhibitor of mTORC1/2 and known activator of 4E-BP1 (17). Similarly, prexasertib was reported by Sen et al. to block mTOR signaling via direct inhibition of p90RSK (27, 41). However, the finding that gemcitabine and/or knockdown of RRM2 activated 4E-BP1 and inhibited protein synthesis in Ewing sarcoma cells was unexpected. But, other groups have reported similar findings. For example, Williams et al. demonstrated that gemcitabine activates 4E-BP1 in non-small cell lung cancer cells and Jacobson et al. showed that the toxicity of gemcitabine is enhanced by concurrent blockade of cap-dependent translation (49, 50). Similarly, Tee et al. identified that the treatment of cells with multiple DNA-damaging agents, including etoposide, mitomycin-C, and cisplatin, can activate 4E-BP1 and reduce protein synthesis (51). Overall, based on this published work and our data demonstrating that activation of 4E-BP1 is not a general response to gemcitabine in all cell types, we believe that the regulation of 4E-BP1 is likely a complex interplay between cell backgrounds, drug mechanisms, and oncogenic signaling pathways. Work is currently underway in our laboratory investigating how these factors regulate 4E-BP1 in Ewing sarcoma cells and, in particular, whether the high levels of 4E-BP1 mRNA and protein in Ewing sarcoma contribute to this phenotype.
We have not yet identified the upstream pathway that mediates the effects of gemcitabine on protein synthesis and 4E-BP1 activation in Ewing sarcoma cells, but inhibition of mTOR signaling can occur via multiple mechanisms (32). Interestingly, Zhou et al. recently showed that ciclopirox, which inhibits RNR and the growth of Ewing sarcoma cells in vitro and in vivo, inhibits mTOR signaling by activation of AMP-activated protein kinase (AMPK) (52). In addition, Kruiswijk et al. identified that genotoxic stress can activate AMPK and inhibit protein synthesis via regulation of eukaryotic elongation factor 2 kinase (eEFK2) (53). Consequently, we are currently investigating whether RNR inhibitors, including ciclopirox and gemcitabine, activate the AMPK/tuberous sclerosis/raptor pathways in Ewing sarcoma cells. In addition, although mTOR is the best known upstream regulator of 4E-BP1 phosphorylation, other kinases have also been reported to phosphorylate 4E-BP1, including CDK1, MAP Kinase p38, GSK3beta, PIM2, and ATM (54).
Similarly, alterations in multiple proteins and pathways that regulate protein synthesis have also been described in Ewing sarcoma tumors. Consequently, there may be underlying cellular abnormalities driven by EWS-FLI1 that predispose Ewing sarcoma cells to inhibition of protein synthesis. For example, Erkizan et al. first reported a critical role for RNA helicase A (RHA), which functions in mRNA translation, in Ewing sarcoma tumors (55). Similarly, Wilky et al. demonstrated that Ewing sarcoma cells express high levels of the RNA helicase DDX3, which is critical for ribosome assembly and the translation of mRNAs with complex 5’ untranslated regions (56). In addition, Y-box binding protein 1 (YB-1), which regulates the translation of specific mRNAs, is also overexpressed in Ewing sarcoma cells (57). Overall, the pleiotropic effects of the EWS-FLI1 oncogene may generate a cell state that is particularly prone to inhibition of protein synthesis in the setting of DNA replication stress, DNA damage, or other stressors.
In summary, we have identified that protein translation is a critical regulator of CHK1 levels and the response to DNA replication stress in Ewing sarcoma cells. Overall, we believe that our preclinical data provides a rationale testing the combination of gemcitabine and ATR-CHK1 pathway inhibitors in patients with relapsed and refractory Ewing sarcoma.
Supplementary Material
{kind=link}
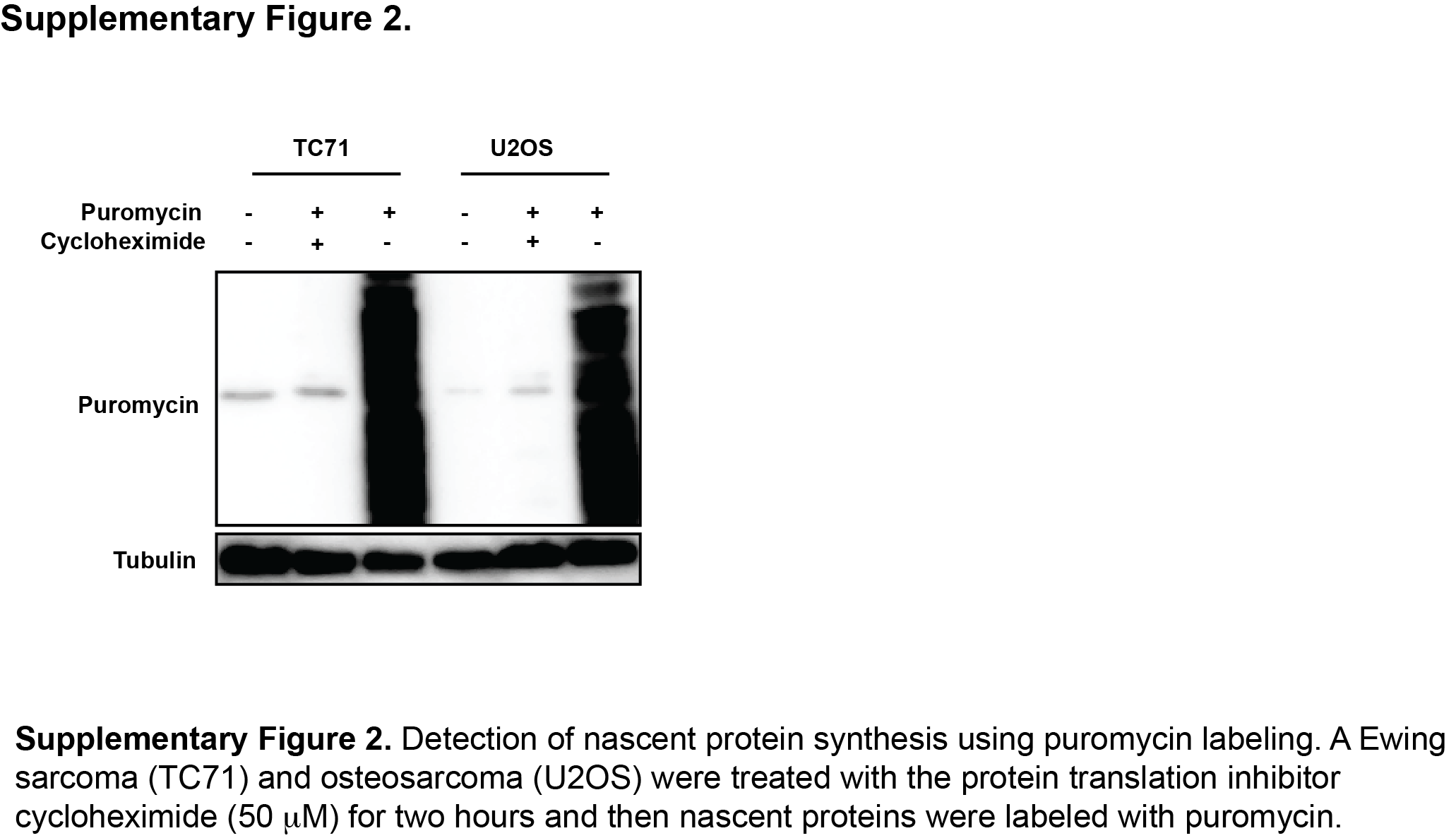{kind=link}
{kind=link}
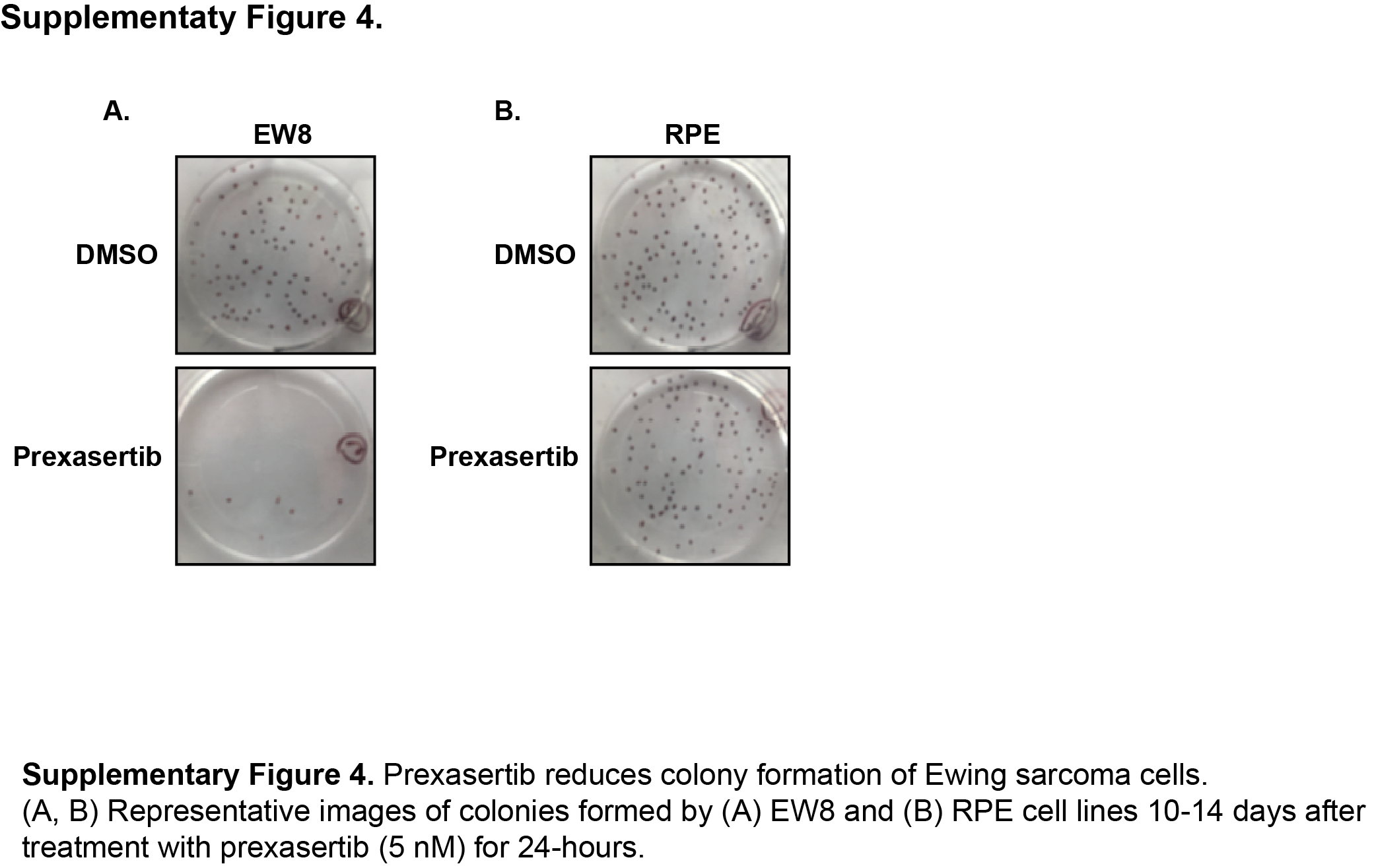{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank Mohammed Milhem, Varun Monga, Munir Tanas, Rebecca Dodd, and Yasmeen Rose for helpful discussions. DJG is supported by a University of Iowa Dance Marathon Award, a Holden Comprehensive Cancer Center Sarcoma Multidisciplinary Oncology Group Seed Grant, a University of Iowa Oberley Seed Grant, a University of Iowa Stead Family Research Award, The Matt Morrell and Natalie Sanchez Pediatric Cancer Research Foundation, and NIH Grant R37-CA217910.
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Balamuth NJ, Womer RB. Ewing’s sarcoma. The Lancet Oncology. 2010;11:184–92. [DOI] [PubMed] [Google Scholar]
- 2.Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annual review of pathology. 2012;7:145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovar H Blocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapy. Expert opinion on therapeutic targets. 2014;18:1315–28. [DOI] [PubMed] [Google Scholar]
- 4.Goss KL, Gordon DJ. Gene expression signature based screening identifies ribonucleotide reductase as a candidate therapeutic target in Ewing sarcoma. Oncotarget. 2016;7:63003–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goss KL, Koppenhafer SL, Harmoney KM, Terry WW, Gordon DJ. Inhibition of CHK1 sensitizes Ewing sarcoma cells to the ribonucleotide reductase inhibitor gemcitabine. Oncotarget. 2017;8:87016–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieto-Soler M, Morgado-Palacin I, Lafarga V, Lecona E, Murga M, Callen E, et al. Efficacy of ATR inhibitors as single agents in Ewing sarcoma. Oncotarget. 2016;7:58759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, et al. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Science translational medicine. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, Stewart J, et al. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23:4354–4363. [DOI] [PubMed] [Google Scholar]
- 9.Wayne J, Brooks T, Massey AJ. Inhibition of Chk1 with the small molecule inhibitor V158411 induces DNA damage and cell death in an unperturbed S-phase. Oncotarget. 2016;7:85033–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou X, Liu W, Hu X, Dorrance A, Garzon R, Houghton PJ, et al. Regulation of CHK1 by mTOR contributes to the evasion of DNA damage barrier of cancer cells. Scientific reports. 2017;7:1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvarajah J, Elia A, Carroll VA, Moumen A. DNA damage-induced S and G2/M cell cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget. 2015;6:427–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selvarajah J, Moumen A, Carroll VA. Role of mTOR-Chk1 in enhancing DNA-damaging therapy. Cell cycle (Georgetown, Tex). 2015;14:1989–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Musa F, Alard A, David-West G, Curtin JP, Blank SV, Schneider RJ. Dual mTORC1/2 Inhibition as a Novel Strategy for the Resensitization and Treatment of Platinum-Resistant Ovarian Cancer. Molecular cancer therapeutics. 2016;15:1557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, et al. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:2625–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tapia-Alveal C, Calonge TM, O’Connell MJ. Regulation of chk1. Cell division. 2009;4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamhamedi-Cherradi SE, Menegaz BA, Ramamoorthy V, Vishwamitra D, Wang Y, Maywald RL, et al. IGF-1R and mTOR Blockade: Novel Resistance Mechanisms and Synergistic Drug Combinations for Ewing Sarcoma. Journal of the National Cancer Institute. 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slotkin EK, Patwardhan PP, Vasudeva SD, de Stanchina E, Tap WD, Schwartz GK. MLN0128, an ATP-competitive mTOR kinase inhibitor with potent in vitro and in vivo antitumor activity, as potential therapy for bone and soft-tissue sarcoma. Molecular cancer therapeutics. 2015;14:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subbiah V, Brown RE, Jiang Y, Buryanek J, Hayes-Jordan A, Kurzrock R, et al. Morphoproteomic profiling of the mammalian target of rapamycin (mTOR) signaling pathway in desmoplastic small round cell tumor (EWS/WT1), Ewing’s sarcoma (EWS/FLI1) and Wilms’ tumor(WT1). PloS one. 2013;8:e68985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Z, Hu X, Liu W, Dorrance A, Garzon R, Houghton PJ, et al. P53 suppresses ribonucleotide reductase via inhibiting mTORC1. Oncotarget. 2017;8:41422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen C, Lancaster CS, Shi B, Guo H, Thimmaiah P, Bjornsti MA. TOR signaling is a determinant of cell survival in response to DNA damage. Molecular and cellular biology. 2007;27:7007–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nature methods. 2009;6:275–7. [DOI] [PubMed] [Google Scholar]
- 22.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research. 2010;70:440–6. [DOI] [PubMed] [Google Scholar]
- 23.Grunewald TG, Diebold I, Esposito I, Plehm S, Hauer K, Thiel U, et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Molecular cancer research : MCR. 2012;10:52–65. [DOI] [PubMed] [Google Scholar]
- 24.Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, et al. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okita N, Minato S, Ohmi E, Tanuma S, Higami Y. DNA damage-induced CHK1 autophosphorylation at Ser296 is regulated by an intramolecular mechanism. FEBS letters. 2012;586:3974–9. [DOI] [PubMed] [Google Scholar]
- 27.King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, et al. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Molecular cancer therapeutics. 2015;14:2004–13. [DOI] [PubMed] [Google Scholar]
- 28.Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Molecular cancer therapeutics. 2011;10:591–602. [DOI] [PubMed] [Google Scholar]
- 29.King C, Diaz H, Barnard D, Barda D, Clawson D, Blosser W, et al. Characterization and preclinical development of LY2603618: a selective and potent Chk1 inhibitor. Investigational new drugs. 2014;32:213–26. [DOI] [PubMed] [Google Scholar]
- 30.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Molecular and cellular biology. 2001;21:4129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charrier JD, Durrant SJ, Golec JM, Kay DP, Knegtel RM, MacCormick S, et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. Journal of medicinal chemistry. 2011;54:2320–30. [DOI] [PubMed] [Google Scholar]
- 32.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Ahmed T, et al. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2( 1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. Journal of medicinal chemistry. 2011;54:1473–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Xu C, Kirubakaran S, Zhang X, Hur W, Liu Y, et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer research. 2013;73:2574–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. The Journal of biological chemistry. 2009;284:8023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.So L, Lee J, Palafox M, Mallya S, Woxland CG, Arguello M, et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Science signaling. 2016;9:ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS biology. 2009;7:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nature medicine. 2010;16:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Musa J, Orth MF, Dallmayer M, Baldauf M, Pardo C, Rotblat B, et al. Eukaryotic initiation factor 4E-binding protein 1 (4E-BP1): a master regulator of mRNA translation involved in tumorigenesis. Oncogene. 2016;35:4675–88. [DOI] [PubMed] [Google Scholar]
- 40.Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–67. [DOI] [PubMed] [Google Scholar]
- 41.Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, et al. CHK1 inhibition in small cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer research. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Molecular cancer therapeutics. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Firsanov DV, Solovjeva LV, Svetlova MP. H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clinical epigenetics. 2011;2:283–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer cell. 2007;11:421–9. [DOI] [PubMed] [Google Scholar]
- 45.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. The Journal of biological chemistry. 1998;273:18623–32. [DOI] [PubMed] [Google Scholar]
- 46.Gordon DJ, Motwani M, Pellman D. Modeling the initiation of Ewing sarcoma tumorigenesis in differentiating human embryonic stem cells. Oncogene. 2016;35:3092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016;34:1764–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vormoor B, Curtin NJ. Poly(ADP-ribose) polymerase inhibitors in Ewing sarcoma. Current opinion in oncology. 2014;26:428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams BW, Chang JJ, Chi RM, Marker PH, Frethem CD, Le CT, et al. Cap-dependent translation blockade and fixed dose-rate gemcitabine: interaction in an in vitro bioreactor system. Cancer letters. 2009;284:37–46. [DOI] [PubMed] [Google Scholar]
- 50.Jacobson BA, Alter MD, Kratzke MG, Frizelle SP, Zhang Y, Peterson MS, et al. Repression of cap-dependent translation attenuates the transformed phenotype in non-small cell lung cancer both in vitro and in vivo. Cancer research. 2006;66:4256–62. [DOI] [PubMed] [Google Scholar]
- 51.Tee AR, Proud CG. DNA-damaging agents cause inactivation of translational regulators linked to mTOR signalling. Oncogene. 2000;19:3021–31. [DOI] [PubMed] [Google Scholar]
- 52.Zhou H, Shang C, Wang M, Shen T, Kong L, Yu C, et al. Ciclopirox olamine inhibits mTORC1 signaling by activation of AMPK. Biochemical pharmacology. 2016;116:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kruiswijk F, Yuniati L, Magliozzi R, Low TY, Lim R, Bolder R, et al. Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Science signaling. 2012;5:ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell cycle (Georgetown, Tex). 2016;15:781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nature medicine. 2009;15:750–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilky BA, Kim C, McCarty G, Montgomery EA, Kammers K, DeVine LR, et al. RNA helicase DDX3: a novel therapeutic target in Ewing sarcoma. Oncogene. 2016;35:2574–83. [DOI] [PubMed] [Google Scholar]
- 57.El-Naggar AM, Veinotte CJ, Cheng H, Grunewald TG, Negri GL, Somasekharan SP, et al. Translational Activation of HIF1alpha by YB-1 Promotes Sarcoma Metastasis. Cancer cell. 2015;27:682–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.