

Abstract
Background:
HIV-positive women are at substantial risk of HPV-associated cervical neoplasia caused by high-risk (HR) HPVs. Methylation of the HPV genome is associated with cervical intraepithelial neoplasia grade 3 (CIN3) in HIV-negative women, yet it is unknown whether this holds true for HIV-positive women.
Methods:
We designed a case-control study within the WIHS cohort comparing HIV-positive CIN3 cases (N=72) to HIV-positive controls without detectable CIN2+. The unit of analysis and matching was HPV type infection. Cases with ≥2 HR-HPV types (N=23; 32%) had a separate control for each HR-HPV type. We developed and utilized next generation sequencing (NGS) methylation assays for 12 different HR-HPVs, focusing on CpG sites in the L1/L2 regions.
Results:
Significant case-control differences in individual CpG site methylation levels were observed for multiple alpha-9 (HPV16/31/35/58) and alpha-7 HPV (HPV18/39/45) types, based on dichotomization of tertile levels (T3 versus T1 and T2). Analyses combining homologous CpG sites (e.g., HPV16-L1–5608/HPV31-L1–5521/HPV35-L2L1–5570; odds ratio [OR]=7.28; 95% CI:2.75–19.3), and (e.g., HPV18-L1–7062/HPV45-L1–7066 OR=6.94; 95% CI:1.23–39.3) were significant in separate case-control comparisons. In cases with multiple HR-HPVs, we tested and confirmed the hypothesis that one HR-HPV type would have higher methylation than other types detected, consistent with there being a single HR-HPV causally-related to a lesion.
Conclusion:
CIN3 is associated with elevated L1/L2 CpG methylation levels in HIV-positive women.
Impact:
HPV DNA CpG methylation is a promising triage option in HIV-positive women testing positive for HR-HPV types and provides risk attribution in women with multiple HPV type infections.
Keywords: HPV, HIV, viral methylation, multiple infections, cervical precancer, CIN3+
INTRODUCTION
Human papillomavirus (HPV) infection is a common sexually transmitted infection (STI) and most sexually active women will acquire at least one HPV infection at some point during their lives (1). Persistent infection by a high-risk HPV (HR-HPV) type is the cause of nearly all cervical cancers (2). However, only a small number of HR-HPV infections result in cervical precancerous lesions, of which a third go on to become cancers (1, 3). Worldwide, cervical cancer is the fourth most common cancer and cause of cancer deaths in women (4), and there is especially high incidence of cervical precancer and cancer among women with HIV (5).
Cervical cancer screening has largely depended on cytology and more recently HPV DNA testing (6). However, their combined (co-test) positive predictive value (PPV) remains moderate (7). The majority of women who are co-test positive do not have clinically relevant cervical disease upon colposcopy and biopsy. Therefore, biomarkers capable of improving cervical cancer screening specificity and PPV are greatly needed, especially as several generations of women are too old to receive HPV vaccination, both in high-resource and limited-resource settings.
Epigenetic changes of the HPV viral genome have been identified as a promising biomarker for cervical precancer and cancer (8). Viral genome methylation can influence protein expression, replication, and susceptibility to immune surveillance, cell proliferation, and risk of virus-related disease. DNA methylation occurs preferentially in regions where a cytosine occurs next to a guanine nucleotide linked to one another by a phosphate bond (termed a CpG site). As we recently reviewed (8), there are no classical CpG islands within the HPV genome, but there are regions of high CpG density, and conservation of CpG sites across HPV types suggest these sites are biologically important (8–11). A correlation has been made between high levels of CpG methylation within the HPV genome and cervical precancer development (7, 12), especially methylation levels in the HPV capsid L1 and L2 genes (13).
Despite the high rates of HPV and cervical disease in HIV-positive women, recent prospective studies have shown that HPV co-testing in HIV-positive women has high sensitivity and negative predictive values (NPV), as in HIV-negative women; strongly discriminating between those at high and low-risk of current as well as subsequent cervical precancer (14). Therefore, recent cervical cancer screening guidelines in the US recommend the use of HR-HPV co-testing in women infected with HIV (15); albeit, as in the general population, the specificity and PPV of co-testing in HIV-positive women are moderate. Given the much higher rate of positive co-tests in HIV-infected women than in the general population, the need for biomarkers capable of improving the accuracy of cervical cancer screening in HIV-positive women is especially great.
However, there are little data regarding HPV CpG methylation and precancer in HIV-positive women. This issue is compounded by the diversity of HR-HPV types detected in HIV-positive precancer and cancer cases (16), as it requires methylation assays for a wide range of HR-HPV types to appropriately characterize the relation of HPV CpG methylation with precancer. Therefore, we developed high-throughput next generation sequencing (NGS) HPV CpG methylation assays able to assess HR-HPV types 16/18/31/33/35/39/45/51/52/56/58/59. The current investigation was designed as a proof-of-concept study of these new assays, and to determine whether there are differences in HPV CpG methylation levels between HIV-positive women with and without cervical precancer. We also evaluated whether HPV CpG methylation could be used in precancer cases with multiple concurrent HR-HPV infections to identify the type causally-related to the lesion. That is, we hypothesized that one HR-HPV type would have markedly higher methylation than other types detected, consistent with there being a single HR-HPV type causally-related to each lesion.
MATERIALS and METHODS
Study Population and Specimen Collection
The WIHS Study is a large ongoing cohort of HIV-positive (n=2,793) and HIV-negative (n=975) women enrolled from six clinical sites (Brooklyn, NY; Bronx, NY; Washington, DC; Chicago, IL; San Francisco, CA; Los Angeles, CA), as previously described (14, 17). These women are followed every 6 months with questionnaires, general physical and pelvic examinations, including Papanicolaou (Pap) smear, and cervicovaginal lavage (CVL) for HPV testing. Written informed consent was obtained from all participants, and the WIHS Study was approved by each local institutional review board.
The current case-control study was performed only in HIV-infected women. As CIN3 is the established precancerous lesion, cases were limited to CIN3 (N=72, CIN3) selected from women with available HPV DNA results. Controls had no CIN2 or greater at any time in the WIHS and were initially matched 1:1 to cases based on age, CD4 count, and HPV type. The HPV type present was defined as the most oncogenic type present in each precancer case defined hierarchically (HPV16 > HPV18 > other HPV types) and selected so that cases would include N=20 positive for HPV16, and N=25 other alpha-9 HPV species (i.e., HPV31/33/35/52/58); a total of N=25 alpha-7 species (i.e., HPV18/39/45/59); and as many alpha-5/alpha-6 (i.e., HPV51 and 56) as available. Those precancers and controls with the fewest HPV types present were prioritized for inclusion. Overall, 1 fewer HPV16 (N=19), and 1 greater alpha-7 (N=26) than planned were available for the current pilot study. Too few alpha-5/alpha-6 were available to analyze individually. Further, approximately 32% (23/72) of cases and 13% (9/72) controls had multiple HR-HPV infections (i.e., ≥ 2 HR-HPV types), and all data were used to maximize the efficiency of this proof-of-concept investigation (see Statistical Methods).
HPV Detection and Typing
The HPV DNA detection methods have been described in detail elsewhere (16, 18, 19). Briefly, HPV DNA was detected using a well-established MY09/11 primer system PCR assay and probed for more than 40 individual HPV types using filters hybridized with type-specific biotinylated oligonucleotides. Consistent with recommendations from the International Association for Research on Cancer (IARC), HPV types 16/18/31/33/35/39/45/51/52/ 56/58/59 were considered oncogenic (20, 21).
Bisulfite Conversion and HPV DNA Methylation Assays
Isolated cervicovaginal DNA was treated with bisulfite using the Qiagen Epitect Bisulfite Kit (Qiagen, Valencia, CA) as recommended by the manufacturer. Negative and positive controls, DNase-free water and HPV-positive DNA, respectively, were processed as described for the clinical samples. SiHa (ATCC® HTB-35™) and HeLa (ATCC® CCL-2™) cells were obtained from ATCC in January 2017, shipped frozen to Albert Einstein College of Medicine (Bronx, NY), and maintained for 20 passages in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. The cells tested negative for Mycoplasma in March 2017 using the e-Myco™ Mycoplasma PCR Detection Kit 2.0 (iNtRON Biotechnology, Kyungki-Do, North Korea) prior to DNA extraction using QIAamp DNA Blood Mini kit (Qiagen). The cell lines were authenticated in May 2017 by mapping HPV integration sites previously described (22, 23) using a deep-sequencing NGS methodology. Following bisulfite treatment and subsequent PCR, unmethylated cytosines (C’s) were converted to uridine (U’s) and then thymidine (T’s), whereas methylated C’s remain unmodified (8).
Up to three regions within the viral genome (E2E4/L1/L2) of HR-HPV types 16/18/31/33/35/39/45/51/52/56/58/59) were selected for quantitative site-specific CpG methylation using NGS assays. Methylation-specific primers (Integrated DNA Technologies IDT, Coralville, IA) for bisulfite converted DNA were designed using MethPrimer (24) (http://www.urogene.org/methprimer/index1.html). Primers were designed to amplify fragments comprising at least 3 CpG sites per selected region. Each forward primer contained a unique 12 base pair (bp) Golay barcode attached to the 5’ end of the forward primer sequence (7) (see Bioinformatics). Reverse primers were also labeled with a 12 bp barcode to distinguish each sample in downstream bioinformatics analyses while increasing the number of multiplexing combinations (7).
Barcoded PCR was performed using the Qiagen PyroMark PCR Kit, allowing enhanced specificity of primer annealing to increase amplification efficiency in bisulfite converted DNA sequences. Each PCR reaction contained 1μL of bisulfite converted DNA, 2.5 mM MgCl2, 1× PyroMark Master Mix, 5 μM of forward and reverse primers in a total volume of 25 μL. The conditions and annealing temperatures for each assay were as follows: initial denaturation step at 95°C for 15 min followed by 50 cycles: 30 sec at 94°C; 30 sec at the optimized primer-specific annealing temperature (56°C to 58°C); 30 sec at 72°C, and a final extension for 10 min at 72°C.
PCR products were evaluated by 3% gel electrophoresis stained with ethidium bromide to confirm the expected amplicon sizes (range=148–292 bp). Samples with and without visible bands in the gel were pooled together as part of each assay using 2 μL or 20 μL, respectively, (i.e., independent pools per each PCR assay) to adjust for disproportionate numbers of reads from samples with a visible DNA band. PCR amplicons from the pooled samples were purified by gel excision using Qiagen QIAquick Gel Extraction Kit according to manufacturer’s guidelines, and then used to create libraries for NGS.
Libraries were constructed using KAPA LTP Library Preparation Kit for Illumina platforms (Kapa Biosystems, Boston, MA) according to the manufacturer’s instructions. Libraries were sequenced using 300 bp paired-end reads on an Illumina MiSeq at the Albert Einstein College of Medicine Epigenomics Core Facility (Bronx, NY). Overall, percent methylation of 117 CpG sites was evaluated for the selected HR-HPV types, including those previously reported (13, 25, 26).
Bioinformatic Analysis
Illumina sequencing data files (13.4 M reads) were first filtered for low quality reads using Prinseq v0.20.4 (27) for a minimum average Phred score of 25 (11.4 M reads) (28). After pre-processing of the Illumina sequencing files, an in-house pipeline was used to extract CpG methylation percentage information by demultiplexing the reads using the novobarcode computer package (Novocraft Technologies Sdn Bhd), based on the 12 bp Golay forward and reverse barcodes uniquely assigned to each sample as described above (7.1 M reads). Bismark v0.15.3 and Bowtie v2.2.3 (29, 30) with samtools v1.2 were used for paired-end mapping to HR-HPV reference genomes bisulfite converted in silico to determine the methylation state of CpG and non-CpG cytosines. On average, the number of reads per fragment was 17,713 (range 1,409–120,461) with the exception of HPV58, with average read counts less than 5,000. Custom R v3.2.1 (31) scripts were incorporated in the pipeline to process the Bismark output by: i) producing a methylation pattern for every sequenced molecule, indicating whether cytosines were methylated (+), unmethylated (–), missing (o) or in disagreement for paired-end reads (x); ii) generating counts of each unique pattern per sample; and iii) calculating site-specific CpG methylation percentages for each assayed cytosine per sample by comparing the ratio of methylated C’s to the total number of methylated and unmethylated (C+T) at each CpG site screened. The ratio of “C/(C + T)” indicates the proportion of methylated cytosines at each CpG site for the assayed sample.
Statistical Methods
Reproducibility of intra-plate duplicate PCR-positive samples for each assay was evaluated based on Pearson and Spearman correlation coefficients and all were R2 > 0.7. Individual site-specific CpG methylation levels were analyzed by HR-HPV type using median and range within each PCR fragment assayed using the Mann-Whitney U test. Standard logistic regression (rather than conditional logistic regression) was performed to determine case-control differences in percent methylation distributions for every CpG site by defining a tertile cut-off (T3 versus T1 and T2, i.e. high methylation versus low methylation) based on data in controls. This approach made it possible to incorporate data for multiple different HPV types from cases and controls with ≥ 2 HPV types, and thereby maximize statistical power in this proof-of-concept pilot study, which would not have been possible with conditional logistic regression models. Note however that for HPV type-specific analyses each case and control only contributed a single observation (i.e., for that one HPV type). Thus, statistical methods to address multiple observations per subject were not applicable.
Conversely, for analyses involving multiple homologous CpG sites we used logistic regression models that incorporated a generalized estimating equation (GEE), as women could contribute data to more than one HPV type even if from the same phylogenetic group, resulting in multiple observations, and all models were adjusted for the matching variables, age and CD4 count. To conduct these analyses, tertiles were separately determined for each HPV type and CpG site using the methylation level of controls to define cut-off values. The data for all HPV types and CpG sites within a given grouping of homologous sites were then combined and the GEE models were fitted to estimate the odds ratios, 95% confidence intervals, and p-values. Thus, the primary a priori hypothesis formally tested using multivariate models in this study was that phylogenetically related HPV types with homologous CpG sites would, as a group, show greater methylation at selected CpG sites amongst cervical precancer cases than controls; an approach which maximized statistical power. SAS 9.3 (SAS Institute Inc) was used to conduct all analyses. All statistical tests were two-sided, with p<0.05 used to define statistical significance.
We hypothesized that in precancer cases one HR-HPV type would have markedly higher methylation than other types detected, consistent with there being a single HR-HPV type causally-related to each lesion, whereas among controls (in the absence of disease) we predicted there would be no standout HR-HPV type with high methylation levels relative to the other HPV types detected. Therefore, we assessed the heterogeneity in methylation levels between HR-HPV types within individuals and then summary differences between cases and controls. Briefly, for each case and control with multiple HR-HPV types a Z-score was calculated as ‘Z = (m - μcontrol)/σcontrol’ for each type, where μ and σ were respectively the mean and standard deviation of the methylation values. The Z-scores measured how high the methylation level was relative to the mean level across all control samples with that HPV type for a given CpG site / HR-HPV type. A heatmap of these values among cases and controls with multiple HR-HPV types was created using a custom python script incorporating matplotlib and seaborn software packages (32, 33), and then used to visualize the heterogeneity of methylation levels. We hypothesized that cases but not controls would have one HR-HPV type with markedly higher methylation than the other types detected. Therefore, we used the Kruskal-Wallis test to compare the average differences in Z-scores among cases relative to that in controls. The expectation was that cases would have significantly greater differences than controls, as cases would more often have a single HR-HPV type with much higher methylation levels than all other HR-HPV detected.
RESULTS
Subjects
Table 1 shows selected characteristics of cases and matched controls at the time of case diagnosis. These subjects were matched by age (median = 38 and 37 years, for cases and controls, respectively), race/ethnicity (e.g., 61% and 63% Black), current CD4 count (median = 237 and 278), and HR-HPV type (e.g., HPV16 was detected in 26% of cases and 26% of controls). Among the other variables assessed, the only differences that reached statistical significance were parity (median = 3 versus 2 live births).
Table 1.
Selected Baseline Characteristics of the Precancer Cases and Controls* from the WIHS Study
| Precancer Cases (n = 72) | Controls (n = 72) * | p value | |
|---|---|---|---|
| Age, median (IQR) | 38.4 (34.0–45.4) | 36.6 (32.5–42.0) | 0.08 |
| Race, n (%) | 0.98 | ||
| Black | 44 (61) | 45 (63) | |
| Hispanic | 21 (29) | 20 (28) | |
| White | 7 (10) | 7 (10) | |
| Other | 0 (0) | 0 (0) | |
| Risk factors | |||
| Lifetime number of sex partners, median (IQR) | 10 (5–50) | 20 (6–100) | 0.16 |
| Number of sex partners past 6 months, median (IQR) | 1 (0–1) | 1 (0–1) | 0.09 |
| Parity, median (IQR) | 3 (1–4) | 2 (1–3) | 0.03 |
| Current Smoking, n (%) | 48 (67) | 39 (54) | 0.13 |
| Hierarchical HPV genotype, n (%)** | 1.00 | ||
| HPV16 | 19 (26) | 19 (26) | |
| HPV18 | 11 (15) | 11 (15) | |
| Other high risk (HR) HPV (no HPV16 or 18) | 42 (58) | 42 (58) | |
| Multiple HR-HPV Types, n (%)*** | 23 (32) | 9 (13%) | 0.01 |
| CD4 count, median (IQR) | 237 (133–456) | 278 (149–454) | 0.71 |
Controls had no CIN2 or greater at any time in the WIHS Study, and were matched to cases based on age, CD4 count, and HPV.
HPV16 > HPV18 > other HPV types. Approximately 32% (23/72) of cases and 13% (9/72) had multiple HR-HPV infections (i.e., ≥ 2 HR-HPV types).
Details of cases and controls with multiple HR-HPV types are shown in Figure 3.
NGS Site-Specific CpG Methylation Levels by HR-HPV Type
To evaluate the association of HPV CpG methylation with CIN3, site-specific CpG methylation levels were determined at a total of 117 CpG sites across each of 12 HR-HPV types (Figure 1 and Supplemental Table S1). For HPV16, cases were significantly more likely to have higher median CpG methylation levels at each CpG site within the L1 fragment (all sites p≤0.01). The ORs comparing the highest to the lowest two tertiles (T3 versus T1 and T2) for the four L1 CpG sites ranged from 4.69 (95% CI: 1.20–18.4; p=0.03) to 8.12 (95% CI: 1.87–35.2; p=0.005). In contrast, the L2 CpG methylation levels for HPV16 were not significantly associated with precancer (Supplemental Table S1).
Figure 1. HPV type- and site-specific CpG methylation comparison by case-control status.
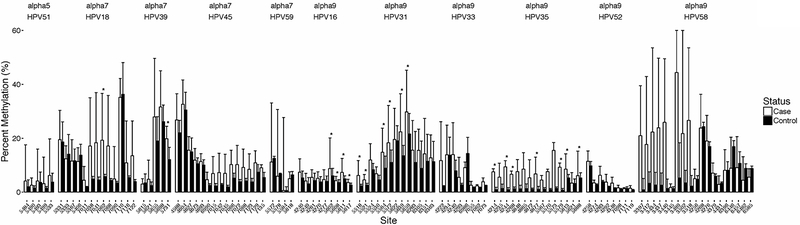
The figure displays a histogram showing the median level of the percent methylation and interquartile range of cases and controls (also see Supplemental Table S1). Median level differences were evaluated by the Mann-Whitney U test. The asterisks (*) indicate the CpG sites with significantly elevated levels of CpG methylation compared to controls (Wilcoxon test, p ≤ 0.05). There were 19 cases and 19 controls of HPV16; 11 cases and 11 controls of HPV18; 15 cases and 15 controls of HPV31; 8 cases and 2 controls of HPV33; 12 cases and 6 controls of HPV35; 8 cases and 8 controls of HPV39; 4 cases and 4 controls of HPV45; 3 cases and 5 controls of HPV51; 5 cases and 4 controls of HPV52; 8 cases and 4 controls of HPV58; and, 3 cases and 3 controls of HPV59. HPV56 was excluded from the figure because it was only detected in cases harboring multiple infections.
Other alpha-9 HR-HPV types (HPV31/35/52/58) also showed significant case-control differences at specific CpG sites (Supplemental Table S1). For example, there were five HPV31 L2 CpG sites with median methylation levels positively associated with case status (p≤0.01), and based on tertiles, the strongest association was OR=28.0 (95% CI: 2.82–278; p=0.004) (see Supplemental Table S1). HPV35 L2 had five CpG sites with significant differences in median methylation levels between cases and controls, and by tertiles the strongest association was OR=22.0, 95% CI: 1.54–314 (p=0.02). Similarly to our observations in HPV16 L1 region, HPV35 L2/L1 region showed significant associations in median levels between cases and controls (Supplemental Table S1). HPV58 had several CpG sites in the E2/E4 region (rather than L1/L2) that showed significant positive associations of methylation levels with case status by the Mann-Whitney U test (Supplemental Table S1).
Similarly, for alpha-7 HPV types (HPV18/39/45) there were significant case-control differences at specific CpG sites. HPV18 had increased methylation at two L1 CpG sites (e.g., HPV18-L1–7062: p=0.02; OR=7.87, 95% CI: 1.10–56.1, p=0.04), and HPV39 methylation was positively associated with case status at a CpG site located at the transition between the L1 and L2 genes (CpG#: HPV39-L2L1–5731). For HPV45 there were an insufficient number of cases and controls to adequately analyze, although the results were in the expected direction (Supplemental Table S1).
Homologous CpG Sites Amongst Phylogenetically-related HR-HPV Types
We scanned CpG locations within L1 and L2 of the alpha-9 species group, and in the alpha-7 species group, to identify homologous sites that could be combined for analysis. Figure 2A shows the fragment alignments for all 12 HR-HPV types surveyed and identifies multiple homologous CpG sites present in some or all of the genomes (vertical red bars). We posited that phylogenetically-related HR-HPV types should have similar case-control associations at these homologous CpG sites. Tertile methylation levels (T3 versus T1 and T2) were used to categorize these data, rather than absolute values (which may vary between HR-HPV types and CpG sites), as discussed in the Statistical Methods. The alpha-9 species analyses showed significant results for the combined data from HPV16L1, HPV31L2, HPV35L2L1, and similar results were observed for other HPV types (see Figure 2B). For example, significant case-control differences were observed across the homologous alpha-9 CpG sites HPV16-L1–5608/HPV31-L1–5521/HPV35-L2L1–5570 (OR=7.28; 95% CI: 2.75–19.3; p=0.0001), and alpha-7 CpG sites HPV18-L1–7062/HPV45-L1–7066 (OR=6.94; 95% CI: 1.23–39.3; p=0.03).
Figure 2. Multiple HPV sequence alignment to identify and test homologous CpG sites associated with cervical precancer.
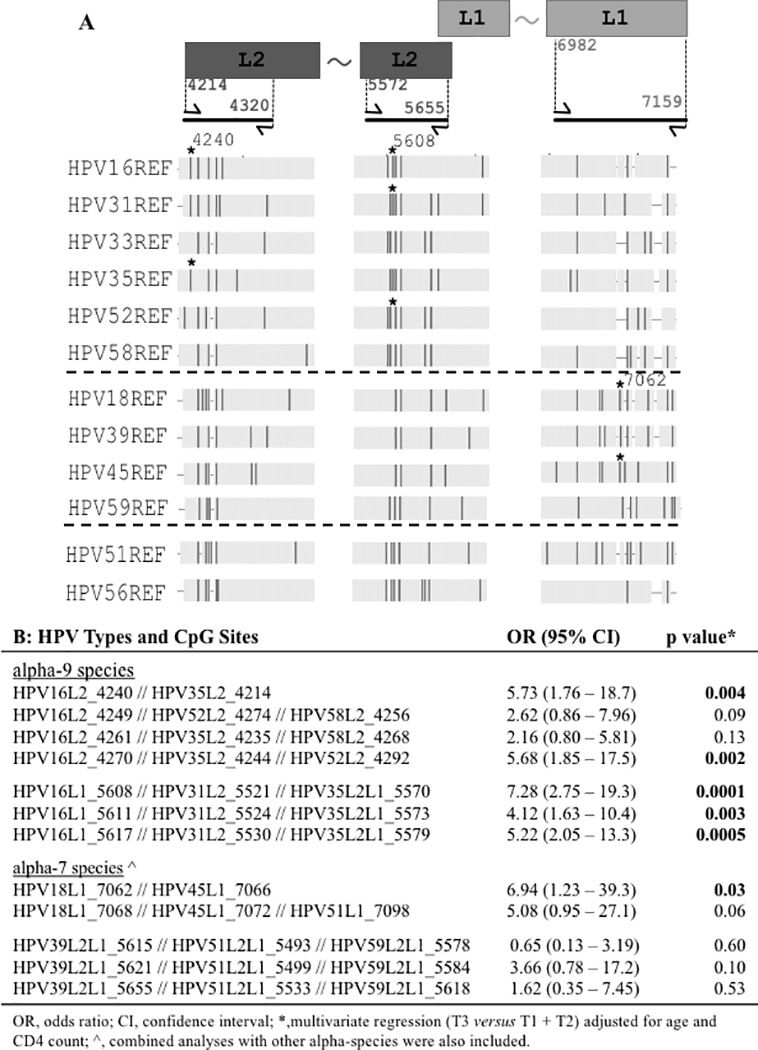
Panel A shows a schematic of the L1 and L2 genes (not to scale) at the top right and top left, respectively. The PCR amplicons of bisulfite treated DNA are shown below the rectangles as thick black lines. The flanking arrows represent forward and reverse primers with the 5’ positions of the primers indicated between the rectangular boxes and the corresponding arrows. The two amplicons with the L2 region use HPV16 nucleotide positions; the amplicon region within the L1 gene corresponds to HPV18 nucleotide positions. The tilde between the rectangles indicates continuity of the gene. The vertical dark bars indicate the location of individual CpG sites for the HR-HPV types shown to the left of the figure. The asterisk (*) marks CpG sites (nucleotide positions 4240 and 5608 for HPV16, and 7062 for HPV18) listed in panel B with the highest significant OR. Alpha species groups are separated by dashed lines alpha-9, alpha-7, and alpha-5/alpha-6 from top to bottom. Panel B shows combinations of homologous CpG sites analyzed for association with cervix precancer. The ORs were calculated by dichotomizing the CpG methylation levels into high or low based on the top tertile (T3) compared to the lower tertiles (T1 and T2). The tertiles were determined for each HPV type and CpG site separately: the methylation of the controls for the HPV type at the CpG site was used to select the cut-off values, which were then used to define the tertiles for the HPV type at the corresponding CpG site. The data for the HPV types and CpG sites within the homologous CpG sites was combined and the GEE (generalized estimating equation) models were adjusted for age and CD4 count to calculate the ORs, 95% confidence interval and p-values.
Methylation and The “Causal” HR-HPV Type in Precancer Cases with Multiple HR-HPV Types
We addressed the hypothesis that, for cases but not controls, one HR-HPV type would have markedly higher methylation than the other types detected, consistent with there being a single HR-HPV type causally-related to each lesion. First, a Z-score was determined for each CpG site in each HR-HPV in all women with multiple HR-HPV types. A heatmap of Z-score levels (Figure 3) was then generated, and confirmed that most precancer cases had a single HR-HPV with much higher methylation levels than all other HR-HPV detected, whereas this was not true for controls. To formally quantify this case-control difference, for each individual woman, we calculated the absolute (subtracted) difference between the highest Z-score and the Z-score for each of the other HR-HPVs (using the mean of these subtracted differences to represent that woman). To summarize these results, we then compared the average of the individual differences for cases and controls using the Kruskal-Wallis test. As predicted, the average difference among cases was significantly greater than for controls (see Figure 3 insert; p=3.041e−5).
Figure 3. Attribution of the possible “causal” HR-HPV in cervical precancer cases with multiple HR-HPV types.
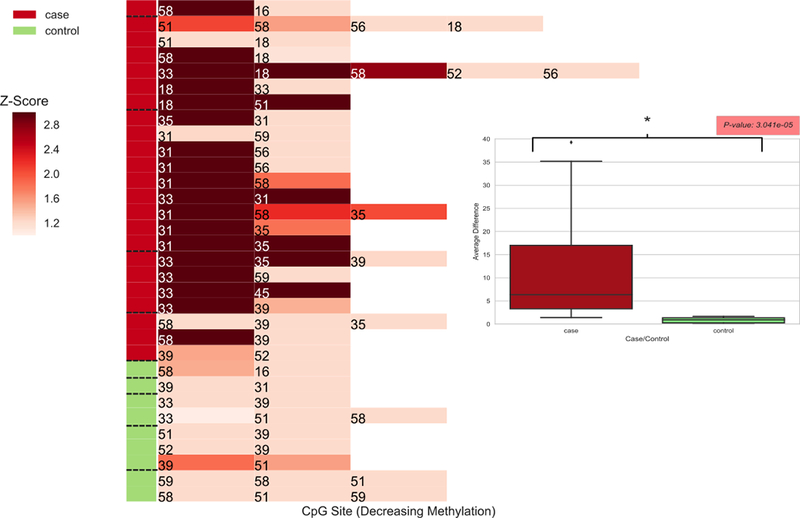
The heatmap shows the highest Z-scores for each HR-HPV type detected in precancer cases and controls with multiple HPV types, calculated as ‘Z = (m – μcontrol) / σcontrol’, where μ and σ are the mean and standard deviation, respectively, of the methylation values for the selected CpG site representing each HPV type across all control samples. This heatmap addresses the hypothesis “Most precancer cases will have a single (standout) HR-HPV with much higher methylation levels at a relevant CpG site than all other HR-HPV detected”. As shown in the Figure, this proved true for most cases, as would be expected for the HPV type causally related to the precancer, whereas no controls with multiple HR-HPV had a HR-HPV type with high methylation levels compared to the other HR-HPV present. To formally quantify these relationships we determined the differences between Z-scores from the highest compared to all other HR-HPV for each individual, and contrasted the average size of these differences between cases and controls, using the Kruskal-Wallis test. A statistically significant difference would indicate that cases were more likely than controls to have a single (standout) HR-HPV type with greater methylation levels than all other HR-HPV detected. The insert in the Figure shows that the differences between cases and controls are statistically significant (p = 3.041e−5).
DISCUSSION
This study found significant differences in HR-HPV CpG methylation levels between HIV-positive women with and without cervical precancer. Prior studies in HIV-negative populations also found that HR-HPV methylation levels are strongly associated with detection of cervical precancer and cancer, and might be a promising biomarker with potential to improve the specificity and PPV of primary HR-HPV testing or of co-testing (HR-HPV testing in combination with Pap tests) (7, 8, 12, 25, 34–40). Improving the accuracy of cervical cancer screening is important since the majority of HIV-positive and HIV-negative women who are HR-HPV or co-test positive do not have clinically relevant cervical disease upon colposcopy and biopsy. Because a diversity of HR-HPV causes cervical precancer in HIV-positive women, we developed NGS methylation assays for 12 different HR-HPVs. The development and use of these NGS assays contributes to the clinical relevance of this proof-of-concept study, since these high-throughput assays allow specimens from a large number of women to be concurrently assessed, which will be important for use in real-world cervical cancer screening programs (7). In addition, the current data provide interesting preliminary evidence that HR-HPV CpG methylation levels might be useful in determining the causal HPV-type in precancer and cancer cases who test positive for multiple HR-HPV types. From a biological perspective, furthermore, the study also provided evidence that homologous CpG sites across phylogenetically-related HR-HPV types share similar associations with cervical disease, and probably epigenetic mechanisms of carcinogenesis.
More specifically, we observed several strong cervical precancer case-control associations in HIV-positive women for alpha-9 HR-HPV types, especially HPV16, 31 and 35, as well as moderate associations for HPV58. The data were also suggestive of case-control differences related to methylation of HPV18 and other alpha-7 HR-HPV types, HPV39 and 45. We then combined tertile data across homologous L1/L2 CpG sites of related HR-HPVs, and found case-control differences across homologous alpha-9 CpG sites, and alpha-7 CpG sites, indicating a similar role in cervical disease for related HR-HPV, though the mechanisms underlying these relationships remain an important area of investigation.
Multiple concurrent HR-HPV infections are common in HIV-positive women, but have not been extensively studied in the context of type-specific CpG methylation levels. Since several sources of evidence have shown that each CIN3 and cancer lesions are caused by a single HR-HPV type (25, 40), we used a novel approach to determine whether HPV CpG methylation patterns could be used to identify the HR-HPV causally associated with CIN3 in HIV-positive cases with multiple concurrent HPV. First we evaluated each HR-HPV genome for the CpG methylation site most strongly associated with precancer based on Z-scores. Then we tested and confirmed the hypothesis that one HR-HPV type would have markedly higher methylation than all other types detected, consistent with there being a single HR-HPV type causally-related to each lesion. While this was not true in every precancer case with multiple HR-HPV types, it was observed in most. In fact, for a few cases it was not possible to ascertain the causal HR-HPV type using the Z-score even though it was more accurate than comparing the heatmap color coding. This could possibly be due to the development of a recent cervical precancerous lesion that might still accumulate increasing viral methylation over time (i.e., for cases where the Z-score was not elevated above control levels). These findings now warrant validation in studies that involve HPV PCR testing of microdissected histologic specimens to confirm the “causal” HPV type inferred from HPV CpG methylation levels of exfoliated cervical cells. This was not possible in the current study since we did not have histologic specimens from WIHS women. If confirmed, though, the findings may be particularly relevant for use in limited resource settings, where inexpensive high throughput molecular HPV assays are increasingly being used, as recommended by the World Health Organization (41). Persistence of the same HR-HPV following precancer treatment is an important risk factor for disease recurrence (42–44).
Although this study had several strengths, including a well-characterized cohort of HIV-positive women with detailed cervical HPV, cytology, and histologic data, there were also limitations that warrant discussion. In particular, as a proof-of-concept study, sample size and power were inadequate to conduct subgroup analyses, some effect estimates were based on few cases, and we did not correct for multiple comparisons. The main a priori hypothesis formally tested using multivariate models was that phylogenetically related HPV types with homologous CpG sites would, as a group, show greater methylation at selected CpG sites amongst cervical precancer cases than controls. By prioritizing selection of cases and controls with few HPV types present for inclusion in this pilot study, our design under-represented the true proportion with multiple HPV types in the WIHS cohort, which will need to be addressed in future larger and more comprehensive studies. On the other hand, a number of a priori predicted associations were highly significant, and most additional results were in keeping with our initial hypotheses. The cross-sectional design was another major limitation and, therefore, the case-control associations we observed may represent reverse causality. That said, even if the HPV methylation – precancer relationships we characterized are not causal associations, our findings and assays could still be clinically important as biomarkers for secondary cancer prevention in cervical cancer screening (15, 16), whereas causality will need to be studied in appropriate prospective studies.
Overall, the current study presents novel initial data showing that cervical precancer in HIV-positive women is associated with high levels of methylation at CpG sites in the L1/L2 region. In combination with prior data in the general population, these findings raise the possibility that with continued improvements the high-throughput NGS methylation assays we utilized could help improve the accuracy of HR-HPV testing and co-testing in cervical cancer screening. These assays might even be useful in determining the HPV-type causally associated with precancer in those cases with multiple HR-HPV. However, future laboratory studies are warranted to understand the mechanisms underlying the relation of L1/L2 methylation and cervical disease development. Larger, more comprehensive prospective molecular epidemiologic studies will be needed to understand temporality between methylation and cervical disease, to determine whether early risk stratification is possible, and understand the possible role of L1/L2 methylation in the natural history of HR-HPV and cervical disease progression.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge the contribution of the Epigenomics Core Facility at the Albert Einstein College of Medicine for providing assistance with the Illumina sequencing and helpful discussions on this work.
Financial Support: HPV testing and the current analyses were supported by the National Cancer Institute (NCI) (grant numbers R01-CA-085178 and R01-CA-174634 to H.D. Strickler and P30-CA-013330). Data in this manuscript were collected by the Women’s Interagency HIV Study (WIHS). The contents of this publication are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health (NIH). WIHS (Principal Investigators): UAB-MS WIHS (M. Saag, M.C. Kempf and D. Konkle-Parker), U01-AI-103401; Atlanta WIHS (I. Ofotokun and G. Wingood), U01-AI-103408; Bronx WIHS (K. Anastos), U01-AI-035004; Brooklyn WIHS (H. Minkoff and D. Gustafson), U01-AI-031834; Chicago WIHS (M. Cohen and A. French), U01-AI-034993; Metropolitan Washington WIHS (S. Kassaye), U01-AI-034994; Miami WIHS (M.A. Fischl and L. Metsch), U01-AI-103397; UNC WIHS (A. Adimora), U01-AI-103390; Connie Wofsy Women’s HIV Study, Northern California (R. Greenblatt, B. Aouizerat and P. Tien), U01-AI-034989; WIHS Data Management and Analysis Center (S. Gange and E. Golub), U01-AI-042590; Southern California WIHS (J. Milam), U01-HD-032632 (WIHS I – WIHS IV). The WIHS is funded primarily by the National Institute of Allergy and Infectious Diseases (NIAID) (grant number P30-AI-050410 to R. Swanstrom), with additional co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the National Cancer Institute (NCI), the National Institute on Drug Abuse (NIDA), and the National Institute on Mental Health (NIMH). Targeted supplemental funding for specific projects is also provided by the National Institute of Dental and Craniofacial Research (NIDCR), the National Institute on Alcohol Abuse and Alcoholism (NIAAA), the National Institute on Deafness and other Communication Disorders (NIDCD), and the NIH Office of Research on Women’s Health. WIHS data collection is also supported by UL1-TR000004 (UCSF CTSA) and UL1-TR000454 (Atlanta CTSA).
Footnotes
Conflict of Interest: A study of H.D.S. involves free-blinded testing using HPV E6/E7 protein assays by Arbor Vita, p16/Ki67 cytology by MTM Laboratories/Ventura-Roche, and MCM-2/TOP2A cytology by BD Diagnostics; no financial payments to H.D.S. or his home institution were received.
REFERENCES
- 1.Schiffman M, Wentzensen N, Wacholder S, Kinney W, Gage JC, Castle PE. Human papillomavirus testing in the prevention of cervical cancer. J Natl Cancer Inst. 2011;103:368–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. [DOI] [PubMed] [Google Scholar]
- 3.McCredie MR, Sharples KJ, Paul C, Baranyai J, Medley G, Jones RW, et al. Natural history of cervical neoplasia and risk of invasive cancer in women with cervical intraepithelial neoplasia 3: a retrospective cohort study. Lancet Oncol. 2008;9:425–34. [DOI] [PubMed] [Google Scholar]
- 4.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 5.Chen CH, Chung CY, Wang LH, Lin C, Lin HL, Lin HC. Risk of cancer among HIV-infected patients from a population-based nested case-control study: implications for cancer prevention. BMC Cancer. 2015;15:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schiffman M, Doorbar J, Wentzensen N, de Sanjose S, Fakhry C, Monk BJ, et al. Carcinogenic human papillomavirus infection. Nat Rev Dis Primers. 2016;2:16086. [DOI] [PubMed] [Google Scholar]
- 7.Gradissimo A, Burk RD. Molecular tests potentially improving HPV screening and genotyping for cervical cancer prevention. Expert Rev Mol Diagn. 2017;17:379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke MA, Wentzensen N, Mirabello L, Ghosh A, Wacholder S, Harari A, et al. Human papillomavirus DNA methylation as a potential biomarker for cervical cancer. Cancer Epidemiol Biomarkers Prev. 2012;21:2125–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez AF, Esteller M. Viral epigenomes in human tumorigenesis. Oncogene. 2010;29:1405–20. [DOI] [PubMed] [Google Scholar]
- 10.Lorincz AT. The Promise and the Problems of Epigenetics Biomarkers in Cancer. Expert Opin Med Diagn. 2011;5:375–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu P, Iden M, Fye S, Huang YW, Hopp E, Chu C, et al. Targeted, Deep Sequencing Reveals Full Methylation Profiles of Multiple HPV Types and Potential Biomarkers for Cervical Cancer Progression. Cancer Epidemiol Biomarkers Prev. 2017;26:642–50. [DOI] [PubMed] [Google Scholar]
- 12.Brandsma JL, Sun Y, Lizardi PM, Tuck DP, Zelterman D, Haines GK 3rd, et al. Distinct human papillomavirus type 16 methylomes in cervical cells at different stages of premalignancy. Virology. 2009;389:100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirabello L, Schiffman M, Ghosh A, Rodriguez AC, Vasiljevic N, Wentzensen N, et al. Elevated methylation of HPV16 DNA is associated with the development of high grade cervical intraepithelial neoplasia. Int J Cancer. 2013;132:1412–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller MJ, Burk RD, Xie X, Anastos K, Massad LS, Minkoff H, et al. Risk of cervical precancer and cancer among HIV-infected women with normal cervical cytology and no evidence of oncogenic HPV infection. Jama. 2012;308:362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeronimo JC PE; Temin S; Denny L; Gupta V; Kim JJ; Luciani S; Murokora D; Ngoma T; Qiao Y; Quinn M; Sankaranarayanan R; Sasieni P; Schmeler KM; Shastri SS Secondary Prevention of Cervical Cancer: ASCO Resource-Stratified Clinical Practice Guideline. Journal of Global Oncology. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strickler HD, Burk RD, Fazzari M, Anastos K, Minkoff H, Massad LS, et al. Natural history and possible reactivation of human papillomavirus in human immunodeficiency virus-positive women. J Natl Cancer Inst. 2005;97:577–86. [DOI] [PubMed] [Google Scholar]
- 17.Barkan SE, Melnick SL, Preston-Martin S, Weber K, Kalish LA, Miotti P, et al. The Women’s Interagency HIV Study. WIHS Collaborative Study Group. Epidemiology. 1998;9:117–25. [PubMed] [Google Scholar]
- 18.Burk RD, Ho GY, Beardsley L, Lempa M, Peters M, Bierman R. Sexual behavior and partner characteristics are the predominant risk factors for genital human papillomavirus infection in young women. J Infect Dis. 1996;174:679–89. [DOI] [PubMed] [Google Scholar]
- 19.Castle PE, Schiffman M, Gravitt PE, Kendall H, Fishman S, Dong H, et al. Comparisons of HPV DNA detection by MY09/11 PCR methods. Journal of medical virology. 2002;68:417–23. [DOI] [PubMed] [Google Scholar]
- 20.Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–27. [DOI] [PubMed] [Google Scholar]
- 21.Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, et al. A review of human carcinogens--Part B: biological agents. Lancet Oncol. 2009;10:321–2. [DOI] [PubMed] [Google Scholar]
- 22.el Awady MK, Kaplan JB, O’Brien SJ, Burk RD. Molecular analysis of integrated human papillomavirus 16 sequences in the cervical cancer cell line SiHa. Virology. 1987;159:389–98. [DOI] [PubMed] [Google Scholar]
- 23.Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–31. [DOI] [PubMed] [Google Scholar]
- 25.Wentzensen N, Sun C, Ghosh A, Kinney W, Mirabello L, Wacholder S, et al. Methylation of HPV18, HPV31, and HPV45 genomes and cervical intraepithelial neoplasia grade 3. J Natl Cancer Inst. 2012;104:1738–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirabello L, Frimer M, Harari A, McAndrew T, Smith B, Chen Z, et al. HPV16 methyl-haplotypes determined by a novel next-generation sequencing method are associated with cervical precancer. Int J Cancer. 2015;136:E146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R Development Core Team. R: A language and environment for statistical computing. v3.2.1 ed. Vienna, Austria: : R Foundation for Statistical Computing; 2015. [Google Scholar]
- 32.Python Software Foundation. Python Language Reference. v2.7 ed.
- 33.Hunter JD. Matplotlib: A 2D graphics environment. Computing in Science & Engineering. 2007;9:90–5. [Google Scholar]
- 34.Badal S, Badal V, Calleja-Macias IE, Kalantari M, Chuang LS, Li BF, et al. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology. 2004;324:483–92. [DOI] [PubMed] [Google Scholar]
- 35.Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Lie K, Barrera-Saldana HA, et al. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J Virol. 2004;78:12762–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turan T, Kalantari M, Cuschieri K, Cubie HA, Skomedal H, Bernard HU. High-throughput detection of human papillomavirus-18 L1 gene methylation, a candidate biomarker for the progression of cervical neoplasia. Virology. 2007;361:185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun C, Reimers LL, Burk RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecol Oncol. 2011;121:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mirabello L, Sun C, Ghosh A, Rodriguez AC, Schiffman M, Wentzensen N, et al. Methylation of human papillomavirus type 16 genome and risk of cervical precancer in a Costa Rican population. J Natl Cancer Inst. 2012;104:556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorincz AT, Brentnall AR, Vasiljevic N, Scibior-Bentkowska D, Castanon A, Fiander A, et al. HPV16 L1 and L2 DNA methylation predicts high-grade cervical intraepithelial neoplasia in women with mildly abnormal cervical cytology. Int J Cancer. 2013;133:637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vasiljevic N, Scibior-Bentkowska D, Brentnall A, Cuzick J, Lorincz A. A comparison of methylation levels in HPV18, HPV31 and HPV33 genomes reveals similar associations with cervical precancers. J Clin Virol. 2014;59:161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santesso N, Mustafa RA, Schunemann HJ, Arbyn M, Blumenthal PD, Cain J, et al. World Health Organization Guidelines for treatment of cervical intraepithelial neoplasia 2–3 and screen-and-treat strategies to prevent cervical cancer. Int J Gynaecol Obstet. 2016;132:252–8. [DOI] [PubMed] [Google Scholar]
- 42.Gallwas J, Ditsch N, Hillemanns P, Friese K, Thaler C, Dannecker C. The significance of HPV in the follow-up period after treatment for CIN. Eur J Gynaecol Oncol. 2010;31:27–30. [PubMed] [Google Scholar]
- 43.Kreimer AR, Schiffman M, Herrero R, Hildesheim A, Gonzalez P, Burk RD, et al. Long-term risk of recurrent cervical human papillomavirus infection and precancer and cancer following excisional treatment. Int J Cancer. 2012;131:211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massad LS, Gierut DR, Meyer PMs. Predictors of compliance with colposcopy referral in an indigent urban population. J Low Genit Tract Dis. 1997;1:9–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.