

Abstract
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is an inherited cancer syndrome associated with a highly aggressive form of type 2 papillary renal cell carcinoma (PRCC). Germline inactivating alterations in fumarate hydratase (FH) cause HLRCC and result in elevated levels of reactive oxygen species (ROS). Recent work indicates that FH−/− PRCC cells have increased activation of ABL1, which promotes tumor growth, but how ABL1 is activated remains unclear. Given that oxidation can regulate protein-tyrosine phosphatase (PTP) catalytic activity, inactivation of an ABL-directed PTP by ROS might account for ABL1 activation in this malignancy. Our group previously developed “q-oxPTPome”, a method that globally monitors the oxidation of classical PTPs. In this study, we present a refined q-oxPTPome, increasing its sensitivity by >10X. Applying q-oxPTPome to FH-deficient cell models showed that multiple PTPs were either highly oxidized (including PTPN12) or overexpressed. Highly oxidized PTP were those with relatively high sensitivity to exogenous H2O2. Most PTP oxidation in FH-deficient cells was reversible, although nearly 40% of PTPN13 was irreversibly oxidized to the sulfonic acid state. Using substrate-trapping mutants, we mapped PTPs to their putative substrates and found that only PTPN12 could target ABL1. Furthermore, knockdown experiments identified PTPN12 as the major ABL1 phosphatase, and overexpression of PTPN12 inhibited ABL1 phosphorylation and HLRCC cell growth. These results show that ROS-induced oxidation of PTPN12 accounts for ABL1 phosphorylation in HLRCC-associated PRCC, revealing a novel mechanism for inactivating a tumor suppressor gene product and establishing a direct link between pathological PTP oxidation and neoplastic disease.
Keywords: PTPN12, q-oxPTPome, Oxidation, ABL1, HLRCC
Precis:
This work demonstrates that the critical tumor-promoting ABL1 activity in HLRCC is induced by ROS-directed PTPN12 oxidation, illustrating a novel pathological mechanism of activation of an oncogenic kinase via oxidation-mediated inactivation of its cognate PTP.
Introduction
Reactive oxygen species (ROS), including superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH), are toxic by-products of aerobic respiration, but also act as second messengers in cellular signaling pathways (1). Protein-tyrosine phosphatases (PTPs) are major ROS targets under physiological and pathological conditions. The PTP superfamily has >100 members, including 38 classical PTPs, which specifically dephosphorylate phosphotyrosine (2, 3). Classical PTPs comprise non-transmembrane PTPs (NT-PTPs) and receptor-like PTPs (R-PTPs). Nearly all NT-PTPs have a single catalytic (PTP) domain, whereas most R-PTPs have two PTP domains (D1, D2), with D1 typically harboring the catalytic activity and D2 having regulatory functions. Classical PTPs are defined by a conserved signature motif (I/V)HCSAGXXR(S/T)G, containing an essential catalytic cysteinyl residue that exists as a thiolate anion (S−) at physiological pH and is quite susceptible to oxidization. Upon exposure to ROS, the PTP thiolate can be oxidized to the sulfenic acid (S-OH) state, which is labile and immediately rearranges to a disulfide (S-S) or sulfenylamide (S-N) bond. These events reversibly inhibit PTP activity and protect the catalytic cysteine from irreversible oxidation to the sulfinic (SO2H) or sulfonic (SO3H) acid. PTP oxidation reportedly occurs in physiological and pathological cellular processes, including growth factor signaling, cytokine signaling, integrin signaling, diabetes, atherosclerosis, chronic inflammation, and cancer (4).
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is an inherited syndrome, caused by germline inactivating alterations in fumarate hydratase (FH), that places patients at risk of a highly aggressive, poor prognosis form of type 2 papillary renal cell carcinoma (PRCC) (5–7). FH encodes a TCA cycle enzyme (8), whose loss-of-function results in accumulation of fumarate, which leads to inactivation of the antioxidant glutathione (GSH) and persistent oxidative stress (9, 10). Consequently, HLRCC cells have elevated ROS levels, despite hyper-activation of the antioxidant protein NRF2 (11–13). A recent study demonstrated that FH deficiency-induced ROS indirectly stimulate ABL1 phosphorylation/activation, which, in turn, is critical for FH−/− PRCC growth and survival (14). However, the precise mechanism by which ROS cause increased ABL1 activation remained undefined.
We hypothesized that the increased ROS in HLRCC-associated PRCC might induce oxidation and inactivation of the PTP that normally dephosphorylates ABL1. Several methods for detecting PTP oxidation have been developed, including the in-gel PTPase assay, alkylating agent-based assays, dimedone-based assays, and specific antibodies against oxidized forms of PTPs (15). Nevertheless, identifying endogenously oxidized cellular PTPs remains challenging. Previously, we developed a mass spectrometry (MS)-based method, q-oxPTPome, that can monitor the oxidation of all catalytically active classical PTPs (16). We substantially refined this protocol, combining it with filter-aided sample preparation (FASP) (17), and increased its sensitivity by at least 10-fold. Using this approach, we found that several PTPs were highly oxidized in FH-deficient PRCC cell lines, and identified PTPN12 oxidation as the cause of increased ABL1 tyrosyl phosphorylation, establishing a novel mechanism of tumorigenesis.
Materials and Methods
Cell Lines and Cell Culture
HEK293 T-REx cells were obtained from ThermoFisher. HeLa T-REx cells (early passage) were kindly provided by Dr. Brian Raught (Princess Margaret Cancer Centre, Canada). UOK262 FH-WT, UOK262 FH-deficient, UOK268 FH-WT and UOK268 FH-deficient cell lines (early passages) were generated in the Linehan laboratory. YUNK1 and YUNK1 FH-KD (early passages) cells were generated by the Bindra laboratory. HepG2 and THP-1 cells were purchased from ATCC. THP-1 cells were cultured in RPMI-1640, while all other cells were cultured in DMEM. All cell lines were mycoplasma-free, as assessed by the Lonza MycoAlert kit, with testing last performed on Jan 9, 2018. Swiss 3T3, HepG2, HEK293, and HeLa cells were authenticated by the CellCheck short tandem repeat profiling service (IDEXX BioResearch). Cells from ThermoFisher and ATCC were passaged for fewer than 1 month after receipt. Cell lines from other labs were used within 5–10 passages of receipt. All media contained 10% FBS (Corning) and 100 Units/mL penicillin/streptomycin (Gibco).
Immunoblotting and Immunoprecipitation
Cells were lysed in 50 mM HEPES (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1% NP40, 0.1% SDS with a protease and phosphatase inhibitor cocktail (40 μg/mL PMSF, 2 μg/mL antipain, 2 μg/mL pepstatin A, 20 μg/mL leupeptin, and 20 μg/mL aprotinin, 1 mM NaF, 1 mM β-glycerophosphate and 2 mM Na3VO4). Total protein (10–50 μg) was resolved by SDS-PAGE and analyzed by immunoblotting. Antibodies included: oxidized PTP (335636) and anti-PTP1B antibodies (AF3954) from R&D Systems; anti-ERK2 (D-2), anti-FH (H-6), anti-SHP2 (C-18 and B1) and anti-ABL1 (8E9) antibodies from Santa Cruz; anti-PSTPIP2 (HPA040944) and anti-Flag (M2) antibodies from Sigma; anti-PTPN12 antibody (A700–007) from Bethyl; anti-PSTPIP1 antibodies (ab197260) from Abcam; anti-phosphotyrosine antibodies (4G10) from Millipore; anti-pERK (9101), anti-ABL1 (2862), anti-pABL Y245 (2861), anti-PTPN18 (8311), anti-pCRKL Y207 (3181), anti-CRKL (3182), anti-pCRKII Y221 (3491) and anti-CRKII (3492) antibodies from Cell Signaling Technology. siRNAs (SMARTpools or individual siRNAs) for PTPN12, PTPN18, and ABL1 were from Dharmacon.
For immunoprecipitation, lysates (1–2 mg protein) were incubated with anti-Flag M2 affinity gel (Sigma) overnight at 4°C, washed 4× in lysis buffer, resolved by SDS-PAGE and analyzed by immunoblotting.
q-oxPTPome
A buffer containing 20 mM Tris (pH 7.35), 150 mM NaCl, 10% glycerol, 1% NP40 was degassed overnight and then transferred to a hypoxia workstation (Don Whitley Scientific). Immediately before use, 30 mM N-ethylmaleimide (NEM; Sigma), 0.5 U/μL catalase (Calbiochem), 0.125 U/μL superoxide dismutase (Sigma) and a protease inhibitor cocktail (2 μg/mL antipain, 2 μg/mL pepstatin A, 20 μg/mL leupeptin, and 20 μg/mL aprotinin) were added to the lysis buffer. Within the workstation, cells were quickly washed once with degassed PBS, immediately lysed, incubated for 1 hr at 4°C in the dark with constant rotation, and clarified at 10,000× g for 1 min at 4°C. Supernatants were collected in the hypoxia workstation and buffer-exchanged by gel filtration (GE Healthcare) into degassed solution (20 mM HEPES [pH 7.4], 100 mM NaCl, 10% glycerol, 0.05% NP40, 0.02 U/μL catalase). Desalted lysates were treated with 5 mM DTT and rotated in the dark for 1.5 hrs at 37°C. Within the workstation, lysates were again buffer-exchanged to remove DTT, freshly prepared pervanadate was added to 8.5 mM, samples were rotated overnight at 4°C in the dark and the next day, 4 mM EDTA was added.
For immunoprecipitations, proteins were denatured in 0.5% SDS at 95°C for 10 min, placed at RT for at least 30 min, and then diluted 5-fold in 50 mM HEPES (pH 7.4), 100 mM NaCl, 10% glycerol, 1% NP40 and 2 mM EDTA. Diluted samples were incubated overnight at 4°C with oxPTP antibody (5 μg antibody/mg protein), and the next day, 40 μL of protein G Sepharose (GE Healthcare) was added for another 3 hrs at 4°C.
For MS, proteins were incubated in 9 M urea with 4.5 mM DTT (Sigma) at 56°C for 30 min, cooled to RT, and then treated with 10 mM iodoacetamide (IAM) for 20 min in the dark. Samples were then loaded into a 30K Centriprep (Millipore) pre-cleaned with 50 mM ammonium bicarbonate, centrifuged at 872× g for 15 min at RT, washed 3× with 50 mM ammonium bicarbonate (pH 8–8.5), and digested with trypsin (Promega) within the Centriprep overnight at 37°C. The next day, filtrates were collected following centrifugation at 872× g, followed by 2 washes with 2.5 mL 0.5 M NaCl. Combined filtrates were acidified with trifluoroacetic acid (final pH~2) and desalted with Sep-Pak C18 Cartridges (Waters), following the manufacturer’s instructions. Desalted solutions were dried in a SpeedVac, resuspended in 1 mL 50 mM HEPES (pH 7.4)/50 mM NaCl, incubated for 30 min on a shaker at RT, and centrifuged at 10,000× g for 5 min. After pre-clearing with 40 μL of protein G Sepharose for 1 hr at 4°C, samples were incubated with oxPTP antibody overnight at 4°C, and then 50 μL of protein G Sepharose was added for 3 hrs at 4°C. Immunoprecipitates were washed 3× in 50 mM HEPES (pH 7.4)/50 mM NaCl, followed by one wash with HPLC grade H2O. Peptides were eluted in 60 μL 0.15% trifluoroacetic acid for 10 min at RT (3×) and immediately cleared using StageTips (ThermoFisher), following the manufacturer’s instructions. After vacuum-drying, eluted peptides were analyzed by MS.
qPTPome
Cells were lysed in 20 mM Tris (pH 7.35), 150 mM NaCl, 10% glycerol, 1% NP40, 2 μg/mL antipain, 2 μg/mL pepstatin A, 20 μg/mL leupeptin, and 20 μg/mL aprotinin, and then clarified at 10,000× g for 1 min at 4°C. Supernatants were treated with 5 mM DTT and rotated in the dark for 1.5 hrs at 37°C. Following a buffer exchange, freshly prepared pervanadate was added to 8.5 mM, samples were rotated overnight at 4°C in the dark, and then 4 mM EDTA was added before use for immunoblotting, immunoprecipitation or MS (as above).
PTP-SO3H Quantification
Lysis buffer containing 20 mM Tris (pH 7.35), 150 mM NaCl, 10% glycerol, 1% NP40 was degassed overnight and then transferred to a hypoxia workstation. NEM (30 mM, Sigma), 0.5 U/μL catalase, 0.125 U/mL superoxide dismutase and the protease inhibitor cocktail were added immediately before use. Within the hypoxia workstation, cells were washed quickly with degassed PBS, lysed, rotated for 1 hr at 4°C in the dark, and clarified at 10,000× g for 1 min at 4°C. Supernatants were collected and buffer-exchanged by gel filtration. The samples were then ready to proceed to MS using the q-oxPTPome protocol.
Quantitative Real-Time PCR
RNA was extracted using the miRNeasy Mini Kit (Qiagen), reverse transcribed and analyzed by Sybr green-based qPCR (Sigma), according to the manufacturer’s instructions. Forward and reverse primers for PSTPIP1 were: GCA GCA TAG ACG CCG ACA TC and CTT CCG TGC AGC AGT CCA G, respectively (18), while the forward and reverse primers for GAPDH were: GAG TCA ACG GAT TTG GTC GT and TTG ATT TTG GAG GGA TCT CG, respectively.
ROS Measurement
Cells were incubated 30 min with 500 nM DCF-DA (Invitrogen) at 37°C in phenol red-free media, trypsinized, washed with cold PBS, and analyzed by flow cytometry (CytoFlex).
Gel-free Affinity Purification for MS
Immunoprecipitates were washed 4× in lysis buffer, 5× in rinse buffer (50 mM ammonium bicarbonate [pH 8–8.5] and 75 mM KCl), and eluted 3× with 100 μL of 0.5 M ammonium hydroxide (pH≥11) at RT with constant rotation. Combined eluates were dried in a SpeedVac, resuspended in 100 μL 50 mM ammonium bicarbonate, incubated in 4.5 mM DTT at 56°C for 30 min, and then treated with 10 mM IAM 30 min at RT in the dark. Eluted proteins were digested with trypsin and cleaned through StageTips before MS analysis.
Statistical Analysis
Data were presented as the mean ± SEM. Statistical significance was determined using unpaired Student’s t-test or one-way ANOVA (comparing all the experimental groups to the same basal control group), as appropriate. If ANOVA was significant, individual differences were evaluated using the Dunnett post-test. Statistical analyses were performed using Graph-Pad Prism 7, with p < 0.05 considered significant.
Results and Discussion
Refined q-oxPTPome for detecting oxidation of classical PTPs
For q-oxPTPome, reduced PTPs (PTP-S−) are alkylated with N-ethylmaleimide (NEM; PTP-S-NEM) to render them unreactive to other modifications, while reversibly oxidized PTPs (PTP-SOH or other rearrangements) are converted to their sulfonic acid forms (PTP-SO3H) via the sequential use of dithiothreitol (DTT) and pervanadate (PV). Consequently, q-oxPTPome detects both reversibly oxidized PTPs and PTPs that are basally in the sulfonic acid state. This approach also can be modified to quantify total PTPs (qPTPome) or those basally in the sulfonic acid (PTP-SO3H) state, but it does not measure PTPs in the sulfinic acid (PTP-SO2H) state (Fig. 1A).
Figure 1.

Refined q-oxPTPome can sensitively monitor H2O2-induced classical PTP oxidation. A, Scheme for q-oxPTPome, qPTPome and PTP-SO3H approaches; see text for details. NEM: N-ethylmaleimide, DTT: dithiothreitol, PV: pervanadate. FASP: filter-aided sample preparation. B-D, Rat1 cells were treated with H2O2 at the indicated concentrations for 4 min and then processed for q-oxPTPome. B, Samples were directly analyzed by immunoblotting using oxPTP Ab. C, Samples were immunoprecipitated using oxPTP Ab and immunoblotted for PTP1B (PTPN1). D, Samples were analyzed by mass spectrometry (MS) and quantified by label-free quantification. Data represent mean ± SEM (n=4–8; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; ANOVA with Dunnett post-test).
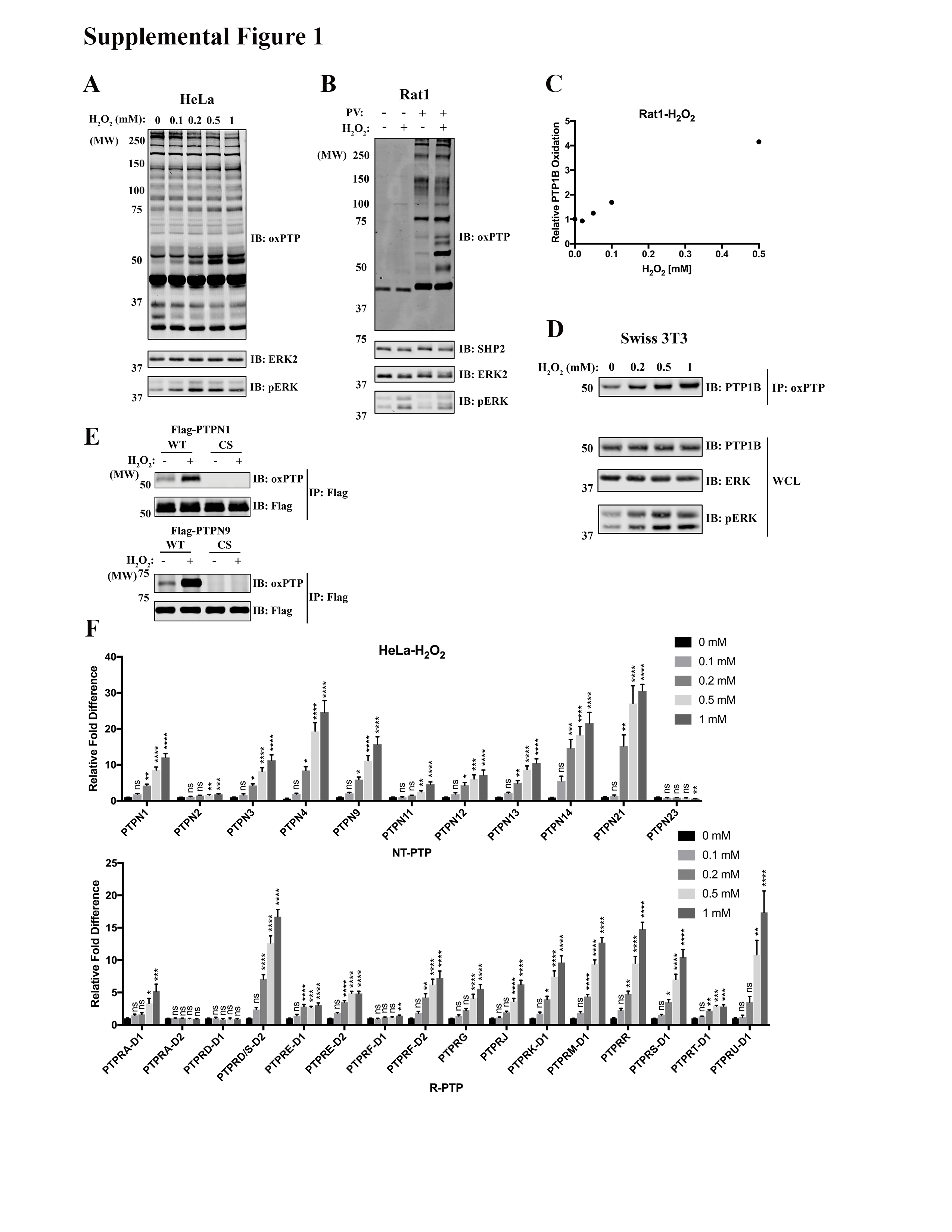
Our original protocol was only capable of reproducibly quantifying PTP oxidation in cells exposed to extracellular H2O2 concentrations ≥1 mM (16). We further optimized q-oxPTPome, incorporating FASP purification (17) to increase its sensitivity (see Materials and Methods). To test our refined protocol, we treated Rat1 and HeLa cells with 0.1–1 mM of H2O2, subjected cell lysates to q-oxPTPome, and resolved the treated lysates by SDS-PAGE, followed by immunoblotting with oxPTP antibody (Ab). We observed multiple bands, whose intensities increased with increasing H2O2 levels, consistent with increased oxidation (Fig. 1B and Supplementary Fig. S1). To confirm that PTPs were oxidized, treated lysates were immunoprecipitated with oxPTP Ab, followed by immunoblotting for PTP1B (encoded by Ptpn1). Indeed, PTP1B showed an H2O2 dose-dependent increase in oxidation, with levels as low as 50 nM evoking detectable signals (Fig. 1C and Supplementary Fig. S1). Because q-oxPTPome detects reversible oxidation and the sulfonic acid state, we compared PTP-SO3H levels with q-oxPTPome (Supplementary Fig. S1). As anticipated, nearly all of the bands were present in the q-oxPTPome lanes, consistent with reversible oxidation. We also transfected HEK293 cells with WT or catalytic cysteine-mutated (CS) Flag-PTPN1 or Flag-PTPN9 expression constructs and assessed oxidation. After immunoprecipitation with anti-Flag Ab and immunoblotting with oxPTP Ab, only the cognate WT PTPs were detected under basal or H2O2-treated conditions (Supplementary Fig. S1). These data confirm that our improved q-oxPTPome approach only detects catalytic cysteine oxidation, comporting with our earlier report (16).
To assess oxidation of all classical PTPs, Rat1 or HeLa cells were exposed to 0.1–1 mM H2O2 treatment and oxidation was monitored by MS (Fig. 1D and Supplementary Fig. S1). We detected >20 classical PTPs, including the D1 and D2 domains of some R-PTPs, in each cell line. Most PTPs showed differential, gradually higher levels of oxidation in response to increasing H2O2, and many exhibited statistically significant increases in oxidation at 0.1 or 0.2 mM. This level of sensitivity is at least 10-fold higher than our original approach (16). Consistent with our previous results, some PTPs (e.g., PTPN23, PTPRA-D2) did not show increased oxidation or even appeared to have decreased oxidation at high concentrations of H2O2. These findings reflect the oxidation of these PTPs to the sulfinic acid state, which cannot be detected by our assay (16). The refined q-oxPTPome assay detects exogenous ROS-induced PTP oxidation at levels comparable to the most sensitive approaches in the literature (15). Furthermore, its ability to simultaneously monitor oxidation of all classical PTPs makes q-oxPTPome a useful tool to analyze physiological and pathological states.
Multiple PTPs are highly oxidized in FH-deficient cells
FH-deficient cells have enhanced ABL1 tyrosyl phosphorylation, which is indirectly the consequence of increased ROS (14), leading us to suspect ROS-induced oxidation/inactivation of an ABL-directed PTP. To test this possibility, we compared UOK262 cells, which derive from an FH-mutant HLRCC patient (UOK262 FH-deficient), to UOK262 cells with wild-type FH re-introduced (UOK262 FH-WT) (19), as well as YUNK1 (Yale University Normal Kidney) cells, an SV40-immortalized kidney cell line with or without FH knockdown (YUNK1 FH-WT and YUNK1 FH-KD). In accord with the previous studies, ROS levels were increased and ABL1 was hyper-phosphorylated in FH-deficient cells (Supplementary Fig. S2 and Fig. 2A). Using refined q-oxPTPome and qPTPome, we observed several proteins with increased oxidation in UOK262 FH-deficient, compared with FH-reconstituted, cells (q-oxPTPome; Fig. 2B left panels), without a change in their expression levels (qPTPome; Fig. 2B right panel). MS demonstrated a global increase in classical PTP oxidation, normalized to the expression level of each PTP, with PTPN7, PTPN9, PTPN12, and PTPN13 being the top four oxidized PTPs (Fig. 2C). Increased PTPN12 oxidation was confirmed by immunoblotting (Fig. 2D and Supplementary Fig. S2). Although most PTPs were expressed at similar levels in both cell lines, PTPN13 and PTPN18 appeared to be overexpressed in UOK262 FH-deficient, compared with FH-reconstituted, cells (Supplementary Fig. S2). This global increase in PTP oxidation is consistent with high oxidative stress in FH-deficient cells, suggesting that PTP catalytic cysteines are sensitive ROS targets. In addition, PTPs from UOK262 FH-WT cells showed variable sensitivity to H2O2-evoked oxidation (Supplementary Fig. S2). Notably, PTPN7, PTPN9, and PTPN12, the top three PTPs oxidized in response to H2O2, were also the most highly oxidized in FH-deficient cells (Fig. 2C and Supplementary Fig. S2), implying that PTPs that are more sensitive to ROS might be more vulnerable to FH deficiency-induced oxidative stress.
Figure 2.

Multiple classical PTPs are oxidized in FH-deficient PRCC cell lines. A, UOK262 (FH-WT and FH-deficient) cells (upper panels) and YUNK1 (FH-WT and FH-KD) cells (lower panels) were lysed, and the levels of ABL1 and pABL-Y245 were assessed by immunoblotting. B, UOK262 (FH-WT or FH-deficient) cells were processed by q-oxPTPome (left panels) or qPTPome (right panel) and immunoblotted with oxPTP Ab. Arrows indicate potentially highly oxidized proteins in FH-deficient cells. SHP2 serves as a loading control. C, Samples from Fig. 2B were analyzed by MS and label-free quantification. The q-oxPTPome signal was normalized to the qPTPome signal (to adjust for PTP expression). Data represent mean ± SEM (n=3; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; unpaired two-tailed t-test). D, UOK262 (FH-WT or FH-deficient) were processed through q-oxPTPome. Samples were immunoprecipitated with oxPTP Ab and immunoblotted for PTPN12. H2O2, 1 mM for 4 min.
Because q-oxPTPome detects reversible oxidation and the PTP-sulfonic acid state, we asked how much of the observed oxidation reflected basal PTP-SO3H. Indeed, several proteins had increased SO3H hyper-oxidation in FH-deficient cells, which was also seen when we treated cells with 10 mM H2O2 (Supplementary Fig. S2). We only loaded 1/3 of the q-oxPTPome samples compared with other samples (Supplementary Fig. S2), but most bands in the q-oxPTPome lane had much higher intensity than in the SO3H hyper-oxidation lanes, indicating that most PTP (or protein-thiol) oxidation in FH-deficient cells was reversible. This conclusion was supported by MS quantification, shown as the percentage of SO3H hyper-oxidation, relative to q-oxPTPome signal, from FH-deficient cells (Supplementary Fig. S2). Most PTPs had low or undetectable SO3H hyper-oxidation (Supplementary Fig. S2), except for PTPN4 and PTPN13, which exhibited 30% and 40% oxidation to the SO3H state, respectively. Presumably, these PTPs are either intrinsically more vulnerable to irreversible oxidation or they localize to a cellular compartment(s) with higher levels of FH deficiency-induced ROS.
PTPN12 trapping mutant interacts with phosphorylated ABL1
Because PTPN7, PTPN9, and PTPN12 showed the highest specific oxidation in FH-deficient cells (Fig. 2C), we asked whether they regulate ABL1 phosphorylation. Notably, PEST-type PTPs, including PTPN12, PTPN18, and PTPN22, were reported to dephosphorylate ABL1 (20). Because PTPN18 was expressed in all tested HLRCC cell lines and was overexpressed in UOK262 FH-deficient cells (Fig. 2D and Supplementary Fig. S2), we included PTPN18 in our analysis.
PTP substrate-trapping mutants, including mutants of the catalytic cysteine to serine (CS) or the aspartate in the WPD motif to alanine (DA), are used widely for substrate identification; such mutants retain substrate binding capacity without catalyzing dephosphorylation (21). We generated HEK293 cells with tetracycline-inducible expression of Flag-tagged PTPN7, PTPN9, PTPN12, PTPN18, or their cognate CSDA mutants. For some experiments, cells were pre-treated with pervanadate, which can boost the intracellular phosphotyrosine by inactivating all cysteine-based PTPs. After pervanadate pre-treatment, only cells expressing PTPN12-CSDA interacted with a 140 kDa protein (consistent with the expected size of ABL1) on phosphotyrosine (pY) blots (Supplementary Fig. S3). Using both in-gel and gel-free MS affinity purification, we found that only PTPN12-CSDA co-immunoprecipitated (co-IPed) with ABL1, along with other known PTPN12 substrates, such as p130Cas and PSTPIP2 (Fig. 3A and Supplementary Fig. S3). Immunoblotting confirmed that PTPN12-CSDA interacted with ABL1 under non-denaturing detergent conditions but not in an SDS-containing buffer (Supplementary Fig. S3), consistent with a typical trapping mutant/substrate interaction. Similar results were obtained using HeLa cells (Fig. 3B).
Figure 3.

PTPN12, but not PTPN18, can trap tyrosyl phosphorylated ABL1. A, HEK293 T-REx cells, treated with tetracycline to induce expression of Flag vector (V), Flag-PTPN12-WT, or Flag-PTPN12-CSDA, were exposed to 50 μM PV or buffer control for 30 min before lysis. PTP-interacting proteins were co-immunoprecipitated with anti-Flag beads and identified by MS (combining results of both gel-free affinity purification and in-gel trypsin digestion). Heat maps show the relative amounts of each protein identified (compared with Flag vector control samples), as measured by label-free quantification. B, HeLa T-REx cells, treated with tetracycline to induced expression of Flag vector, Flag-PTPN12-WT, Flag-PTPN12-CSDA, Flag-PTPN18-WT, or Flag-PTPN18-CSDA, were exposed to 50 μM PV or buffer control for 30 min before lysis. Samples were immunoprecipitated with anti-Flag beads and immunoblotted with anti-ABL1, -pABL-Y245, -phosphotyrosine (pY) and -Flag Abs. ERK2 levels serve as a control for loading.
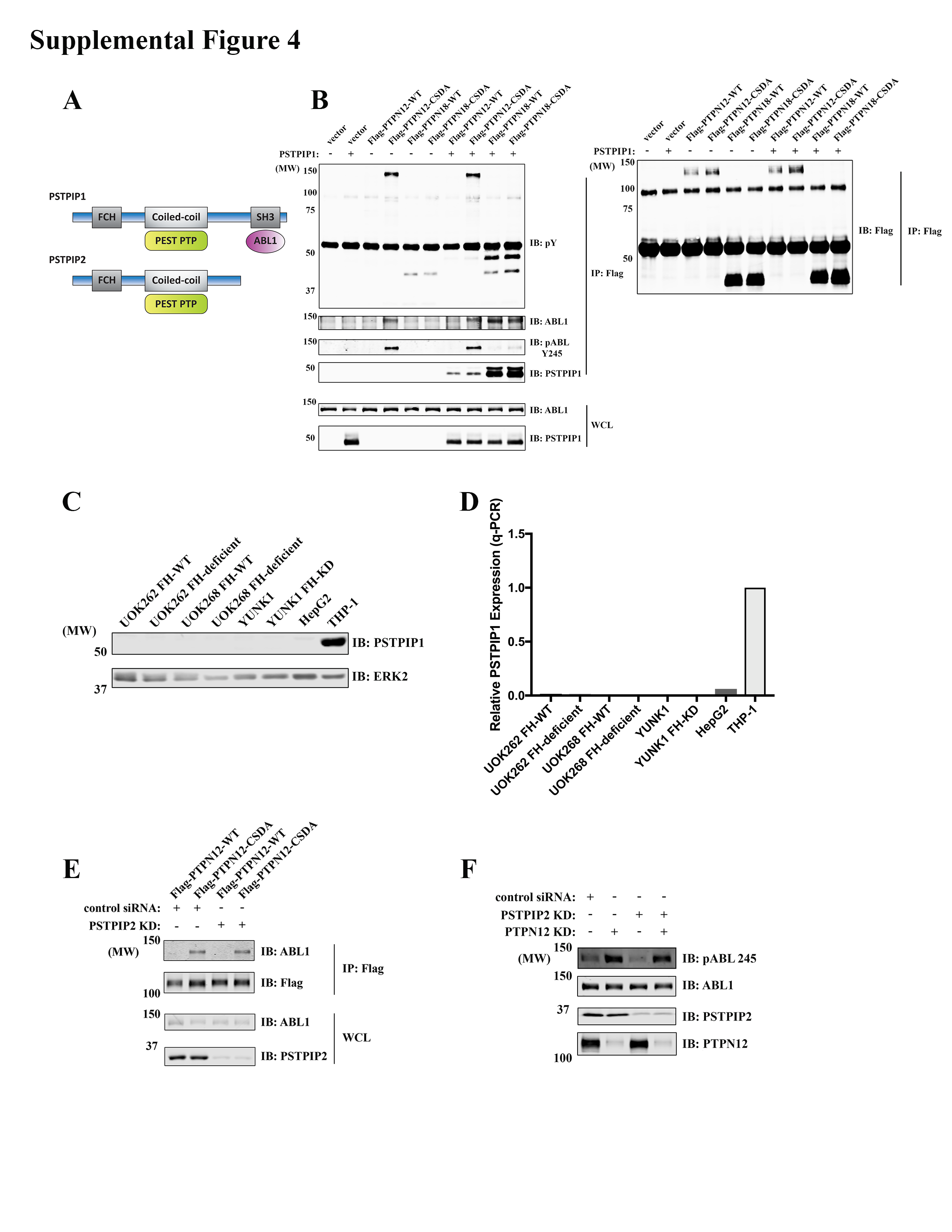
PEST-type PTPs reportedly dephosphorylate ABL1 via the adaptor PSTPIP1 (20). Surprisingly, we only observed the related adaptor PSTPIP2 in PTPN12 or PTPN18 immunoprecipitates (Fig. 3A and Supplementary Fig. S3). PSTPIP2 lacks the critical SH3 domain that interacts with ABL1 to bridge it to PEST-type PTPs (Supplementary Fig. S4), so we hypothesized that failure of PTPN18 to interact with ABL1 might be due to low (or no) expression of PSTPIP1 in these cell lines. To test this hypothesis, we overexpressed PSTPIP1 in HeLa cells, and consistent with previous results, PSTPIP1 co-IPed with PTPN12 and PTPN18 (Supplementary Fig. S4). Furthermore, PSTPIP1 expression helped PTPN18 to interact with ABL1. However, unlike ABL1 recovered in PTPN12-CSDA immunoprecipitates, ABL1 that co-IPed with PTPN18 was not heavily tyrosine-phosphorylated (Supplementary Fig. S4), suggesting that PTPN12 might be a better ABL1 phosphatase, regardless whether or not PSTPIP1 is expressed. We also knocked down PSTPIP2 in HeLa cells; as expected, there was no difference in the ability of PTPN12-CSDA to trap ABL1 (Supplementary Fig. S4).
PTPN12 regulates ABL1 phosphorylation in FH-deficient PRCC cell lines
To investigate the function of PEST-type PTPs in FH-deficient PRCC, we knocked down PTPN12 or PTPN18 in UOK262 FH-WT or FH-deficient lines. PTPN12-KD, but not PTPN18-KD, evoked increased phosphorylation of a 140 kDa protein in total pY immunoblots, as well as increased tyrosine-phosphorylated ABL1, as detected by a pY-specific antibody (Fig. 4A left panel and Supplementary Fig. S5). Similar results were obtained using another patient-derived FH-deficient PRCC cell line, UOK268 (Fig. 4A right panel). ABL1-KD led to a markedly reduced intensity of the hyper-phosphorylated 140 kDa band in PTPN12-KD cells (Supplementary Fig. S5), indicating that most, if not all, of the hyper-phosphorylated 140 kDa species was ABL1. PSTPIP2 knockdown did not affect PTPN12 KD-induced ABL1 phosphorylation (Supplementary Fig. S4), confirming that PTPN12 does not require PSTPIP2 to regulate ABL1 in these cells.
Figure 4.

PTPN12 regulates ABL1 phosphorylation, downstream signaling, and cell proliferation in FH-deficient PRCC cell lines. A, UOK262 (FH-WT and FH-deficient) and UOK268 (FH-WT and FH-deficient) cells were transfected with control, PTPN12 or PTPN18 siRNAs (SMARTpool) before lysis and immunoblotting with anti-pY and -pABL Y245 Abs. B, UOK262 FH-WT, UOK268 FH-WT or YUNK1 cells were transfected with control, PTPN12 or PTPN18 siRNAs (SMARTpool), and then were treated with or without 2 μM Imatinib for 1 hr before lysis and immunoblotting with the indicated antibodies. C, UOK262 (FH-WT and FH-deficient) cells were infected with pLX304 lentivirus to overexpress GFP, PTPN12-WT, or PTPN12-R237M. The same number of newly transduced cells were allowed to proliferate for seven days and then counted by using a Countess II FL Automated Cell Counter. Data represent means ± SEMs (n=3; *p<0.05, **p<0.01, ***p<0.001; unpaired two-tailed t-test). D, Schematic showing how FH deficiency-induced ROS oxidize/inhibit PTPN12, which leads to ABL1 hyper-phosphorylation and FH-deficient PRCC tumor growth and survival. GSH: glutathione.
We also assessed ABL1-dependent downstream signaling. CRKII was hyper-phosphorylated in UOK268 and YUNK1 cells after PTPN12 KD, but there was not much difference in UOK262 cells (Fig. 4B). Increased CRKII phosphorylation was reversed by Imatinib, consistent with ABL1 being the major CRKII kinase. However, after PTPN18-KD, there was a slight increase in ABL1 and CRKII phosphorylation in YUNK1 cells (Fig. 4B right panel); this difference cannot be explained by PSTPIP1 because all of the cell lines have undetectable levels of PSTPIP1 by IB and qPCR (Supplementary Fig. S4). Therefore, whereas PTPN12 is the major PTP regulating ABL1 phosphorylation, other PEST-type PTPs might play a minor role. Because multiple other classical PTPs were oxidized in FH-deficient cells (Fig. 2C), we cannot rule out the possibility that they might also control ABL1 phosphorylation. PTPN12-KD did not confer a proliferative advantage on FH-WT or FH-deficient cells in vitro (Supplementary Fig. S5). Conceivably, PTPN12 oxidation-induced ABL1 activation mainly allows FH-deficient cells to survive fumarate accumulation-induced oxidative stress (14). FH-WT cells are not under such stress, whereas FH-deficient cells already activate ABL1 by PTPN12 oxidation, potentially explaining why PTPN12-KD does not give either cell type a proliferative advantage. We also overexpressed PTPN12-WT, PTPN12-R237M (phosphatase-dead mutant) or GFP (control) in UOK262 cells. Only PTPN12-WT decreased ABL1 phosphorylation (Supplementary Fig. S5) and specifically inhibited the growth of FH-deficient cells (Fig. 4C), implying a tumor suppressor role of PTPN12 in FH-deficient HLRCC.
In conclusion, our refined q-oxPTPome approach sensitively and globally quantifies classical PTP oxidation, allowing us to discover that PTPN12 is highly oxidized in FH-deficient HLRCC-associated PRCC cell lines as a consequence of their elevated oxidative stress. We found that PTPN12 can trap ABL1 in the absence of the adaptors PSTPIP1 or PSTPIP2 and is the major PTP regulating ABL1 phosphorylation in HLRCC cell lines. Because ABL1 phosphorylation/activation is critical for FH-deficient PRCC cells to proliferate and survive (14), PTPN12 appears to be an important, although “dynamically regulated,” tumor suppressor gene in HLRCC (Fig. 4D). Previous studies showed that PTPN12 can suppress mammary epithelial cell proliferation and transformation and is an important tumor suppressor in triple-negative breast cancer (TNBC). In TNBC, however, PTPN12 is mainly inactivated through deletion or loss-of-function mutations (22). Our data demonstrate that oxidative stress-induced PTPN12 oxidation can inhibit PTPN12 without deletion/mutation, and suggest that activation of (a) PTK(s) via ROS-mediated inactivation of cognate PTPs is a novel mechanism for cancer pathogenesis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Ann Marie Pendergast (Duke University Medical School) for alerting us to the hyper-activation of, and the requirement for, ABL1 in HLRCC. We thank Drs. Anne-Claude Gingras (Lunenfeld-Tanenbaum Research Institute) and Nicole St-Denis (University of Toronto) for providing several WT PTP constructs. We thank Drs. Yubao Wang and Kwan-Ho Tang for help with flow cytometry, Dr. Jieyu Jen for assistance with qPCR, and Dr. Carmine Fedele (all at NYU Langone) for providing the GFP pLX304 construct. This work was supported by R01CA49152 (to BGN), the Proteomics Shared Resource of the Perlmutter Cancer Center (P30 CA16087), and the Princess Margaret Cancer Foundation. MFM was supported by a Canada Research Chair, the Leukemia and Lymphoma Society of Canada, the Canadian Institutes of Health Research, and the Canadian Cancer Society. MS experiments were supported by Cancer Center Support grant P30CA016087 and an NIH shared instrumentation grant (1S10OD010582–01A1) for the purchase of an Orbitrap Fusion Lumos.
Financial support: This work was supported by R01CA49152 (BGN), the Proteomics Shared Resource of the Perlmutter Cancer Center (P30 CA16087), and the Princess Margaret Cancer Foundation. MFM is supported by Canada Research Chairs, Leukemia and Lymphoma Society of Canada, Canadian Institutes of Health Research, and Canadian Cancer Society. The mass spectrometric experiments were in part supported by Cancer Center Support grant P30CA016087 from the National Cancer Institute and an NIH shared instrumentation grant from the NIH (1S10OD010582) for purchase of an Orbitrap Fusion Lumos.
Footnotes
Conflict of interest: BGN is a co-founder, member of the Board of Directors, has equity in, and receives consulting funds from, Northern Biologics, and is a co-founder, chair of the Scientific Advisory Board, has equity in, and receives consulting funds from, Navire Pharmaceuticals. A member of his immediate family has equity in Pfizer, Inc. RSB holds equity in, and receives consulting income and research funding from and Cybrexa Therapeutics. He also receives research funding from Astra-Zeneca, Inc. and has received speakers’ honoraria from Beigene Corporation, Merck KgA.
References
- 1.Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol. 2015;33:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tonks NK. Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013;280(2):346–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.St-Denis N, Gupta GD, Lin ZY, Gonzalez-Badillo B, Veri AO, Knight JDR, et al. Phenotypic and Interaction Profiling of the Human Phosphatases Identifies Diverse Mitotic Regulators. Cell Rep. 2016;17(9):2488–501. [DOI] [PubMed] [Google Scholar]
- 4.Ostman A, Frijhoff J, Sandin A, Bohmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150(4):345–56. [DOI] [PubMed] [Google Scholar]
- 5.Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001;98(6):3387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grubb RL 3rd, Franks ME, Toro J, Middelton L, Choyke L, Fowler S, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007;177(6):2074–9; discussion 9–80. [DOI] [PubMed] [Google Scholar]
- 7.Merino MJ, Torres-Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31(10):1578–85. [DOI] [PubMed] [Google Scholar]
- 8.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30(4):406–10. [DOI] [PubMed] [Google Scholar]
- 9.Zheng L, Cardaci S, Jerby L, MacKenzie ED, Sciacovelli M, Johnson TI, et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat Commun. 2015;6:6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51(2):236–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20(4):524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20(4):511–23. [DOI] [PubMed] [Google Scholar]
- 13.Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, Yang Y, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29(15):4080–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sourbier C, Ricketts CJ, Matsumoto S, Crooks DR, Liao PJ, Mannes PZ, et al. Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell. 2014;26(6):840–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karisch R, Neel BG. Methods to monitor classical protein-tyrosine phosphatase oxidation. FEBS J. 2013;280(2):459–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karisch R, Fernandez M, Taylor P, Virtanen C, St-Germain JR, Jin LL, et al. Global proteomic assessment of the classical protein-tyrosine phosphatome and “Redoxome”. Cell. 2011;146(5):826–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–62. [DOI] [PubMed] [Google Scholar]
- 18.Akkaya-Ulum YZ, Balci-Peynircioglu B, Purali N, Yilmaz E. Pyrin-PSTPIP1 colocalises at the leading edge during cell migration. Cell Biol Int. 2015;39(12):1384–94. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet. 2010;196(1):45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cong F, Spencer S, Cote JF, Wu Y, Tremblay ML, Lasky LA, et al. Cytoskeletal protein PSTPIP1 directs the PEST-type protein tyrosine phosphatase to the c-Abl kinase to mediate Abl dephosphorylation. Mol Cell. 2000;6(6):1413–23. [DOI] [PubMed] [Google Scholar]
- 21.Blanchetot C, Chagnon M, Dube N, Halle M, Tremblay ML. Substrate-trapping techniques in the identification of cellular PTP targets. Methods. 2005;35(1):44–53. [DOI] [PubMed] [Google Scholar]
- 22.Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144(5):703–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.