

Abstract
Purpose:
MET inhibitors can be effective therapies in patients with MET exon 14 (METex14) mutant non-small cell lung cancer (NSCLC). However, long term efficacy is limited by the development of drug resistance. In this study, we characterize acquired amplification of wild type (WT) KRAS as a molecular mechanism behind crizotinib resistance in three cases of METex14 mutant NSCLC and propose a combination therapy to target it.
Experimental Design:
The patient-derived cell line and xenograft (PDX) DFCI358 were established from a crizotinib-resistant METex14 mutant patient tumor with massive focal amplification of WT KRAS. To characterize the mechanism of KRAS-mediated resistance, molecular signaling was analyzed in the parental cell line and its KRAS siRNA-transfected derivative. Sensitivity of the cell line to ligand stimulation was assessed and KRAS-dependent expression of EGFR ligands was quantified. Drug combinations were screened for efficacy in vivo and in vitro using viability and apoptotic assays.
Results:
KRAS amplification is a recurrent genetic event in crizotinib-resistant METex14 mutant NSCLC. The key characteristics of this genetic signature include uncoupling MET from downstream effectors, relative insensitivity to dual MET/MEK inhibition due to compensatory induction of PI3K signaling, KRAS-induced expression of EGFR ligands and hypersensitivity to ligand-dependent and independent activation, and reliance on PI3K signaling upon MET inhibition.
Conclusions:
Using patient-derived cell line and xenografts, we characterize the mechanism of crizotinib resistance mediated by KRAS amplification in METex14 mutant NSCLC and demonstrate the superior efficacy of the dual MET/PI3K inhibition as a therapeutic strategy addressing this resistance mechanism.
Keywords: Lung cancer, drug resistance, amplification, MET exon 14, KRAS
Introduction
Genotype directed therapy is the standard of care for patients with advanced non-small cell lung cancer (1–3). However, the vast majority of patients ultimately develop acquired drug resistance to targeted therapies (4–7). The mechanistic understanding of drug resistance has been instrumental in the development of therapeutic strategies to overcome or prevent acquired resistance (8–11).
MET encodes the tyrosine kinase proto-oncogene c-MET, the receptor for hepatocyte growth factor (HGF). MET exon 14 mutations (METex14) have only recently been appreciated as relatively frequent alterations in NSCLC (3–6% of all NSCLC) with sensitivity to tyrosine kinase inhibitors (TKIs) (12–15). Exon 14 carries the tyrosine 1003 (Y1003) residue necessary for the binding of the E3 ubiquitin ligase CBL. Amino acid substitution at Y1003 or mutations or deletions in METex14 or its flanking introns that cause in-frame skipping of METex14, eliminate the CBL binding site and lead to increased MET protein stability and effector signaling (13,16). Despite dramatic responses seen in MET amplified or METex14 mutant NSCLC patients treated with MET-directed TKIs, acquired resistance inevitably develops. To date, only a handful of studies have characterized the mechanisms of resistance to MET inhibitors, all of which are attributed to different secondary mutations in the MET tyrosine kinase domain, believed to interfere with adequate binding of the drug to the kinase (4,17–19).
In the current study, we identify acquired wild-type (WT) KRAS amplification in three cases of crizotinib resistant METex14 mutant NSCLC. Using a cell line and xenograft derived from one of these patients at the time of crizotinib resistance, we demonstrate the unique mechanisms by which KRAS amplification leads to drug resistance, and develop a therapeutic approach to treat these resistant cancers.
Materials and Methods
Patients
All patient studies were conducted in accordance with the Declaration of Helsinki and the Dana-Farber/Harvard Cancer Center institutional review board (IRB)-approved clinical research correlative protocols which allow for the collection and reporting of clinical, pathologic, and genomic characteristics from participants. All patients included in the study provided a written informed consent.
Next generation sequencing, plasma genotyping and identification of MET exon 14 skipping mutation
Methods for targeted NGS were previously reported (15). Cell-free circulating tumor DNA was isolated from blood and analyzed using the Guardant360 assay (Guardant Health, Inc, Redwood City, CA) as previously reported (45). The skipping of exon 14 in MET was initially investigated by RT-PCR, carried out using the QuanTitec Reverse Transcription kit (Qiagen) (see Supplementary Methods).
Fluorescence in situ hybridization (FISH) analysis
FISH analysis was performed on formalin-fixed paraffin-embedded (FFPE) tissue using Chromosome 7 centromere (CEN7), chromosome 8 centromere (CEN8), and chromosome 12 centromere (CEN12), KRAS, EGFR, BRAF, and MYC bacterial artificial chromosomes labeled as FISH probes (Chromosomescience laboratory, Sappro, Japan) (see Supplementary Methods).
Antibodies, compounds, oligonucleotides, and other reagents
Immunohistochemical (IHC) studies
FFPE tissue was subjected to IHC for KRAS (1:200, ab55391, Abcam) or phospho-ERK (1:400, 4370S, Cell Signaling Technology) (see Supplementary Methods).
KRAS copy number and mRNA quantification
RNA and DNA from the frozen cell pellet of processed DFCI358 pleural fluid and DFCI358 cell line were extracted using Trizol according to manufacturer’s instructions. Real Time quantitative PCR was performed using inventoried or Made to Order Assays-on-DemandTM provided by Applied Biosystems (see Supplementary Methods).
Cell Line generation and maintenance
DFCI358 cell line was derived from a pleural effusion harvested from a crizotinib-resistant patient (case #1) (see Supplementary Methods) and maintained in complete RPMI1640. PDX-derived DFCI358 cell line (DFCI358 PDX) was established from a freshly resected tumor expanded in a mouse according to the described PDX generation protocol (see Supplementary Methods).
Hs746T and H596 were a generous gift from the Belfer Institute for Advanced Science (2016). Calu-6 cells were purchased from ATCC (2015). Hs746T were maintained in DMEM supplemented with 10% FBS, streptomycin and penicillin. Remaining cell lines were maintained in RPMI1640 supplemented with 10% FBS, streptomycin and penicillin. Hs746T were authenticated before banking in 2012 and used for this study immediately after thawing in 2016; Calu-6 were authenticated within 6 months of being used for this study, using the Promega GenePrint 10 System at the RTSF Genomics Core at Michigan State University in 2016. All cell lines used in the study were routinely tested for mycoplasma, using the Mycoplasma Plus PCR Primer Set (Agilent).
PC9 (EGFR del19), A549 (KRAS G12S), HCC827 (EGFR del19) and H1975 (EGFR L858R) were maintained in complete RPMI1640. The PC9 cells were obtained from Dr. Nishio (Kinki University, Osaka, Japan) (2005), the others from ATCC. PC9, HCC827 and H1975 were authenticated in 2014; A549 in 2016, and used within 6 months of being authenticated.
MTS growth and viability assay
Growth and inhibition of growth was assessed by MTS assay according to previously established methods (46). All experimental points were set up in 6 to 12 wells and all presented data are representative of several replicates (see Supplementary Methods).
Growth and apoptosis assay
Cells undergoing drug treatments were continuously monitored for growth and apoptosis using the CellEvent™ Caspase-3/7 Green Detection Reagent (Life Technologies) and Incucyte ZOOM (Essen BioScience) (see Supplementary Methods).
Drug treatment and Western blotting
Cells were treated with drugs at concentrations and for durations specific to each experiment as indicated in figure legends. Western blotting and immunoblotting were done as described previously (10). Blots were developed on Amersham Imager 600 (GE Healthcare Life Sciences) (see Supplementary Methods for details).
siRNA knock-down studies
The ON-TARGETplus Human KRAS SMARTpool siRNA, ON-TARGETplus Non-targeting Pool control and ON-TARGETplus Human MET SMARTpool siRNA (Dharmacon) were used for all siRNA experiments. Drug treatments or IncuCyte assays were conducted 72 hours after transfection or 96 hours after transfection where overnight serum starvation was required (see Supplementary Methods for details).
KRAS overexpression
For stable KRAS overexpression, Hs746T cells were transduced with a KRAS or empty vector lentiviral construct according to standard procedures (47) (see Supplementary Methods for details).
Co-immunoprecipitation (co-IP) and active KRAS studies
Cells were lysed in the MB lysis buffer (50 mM HEPES pH 7.5; 50mM NaCl; 1% glycerol, 0.3% NP-40; 1.5 nM MgCl), containing phosphatase and protease inhibitors. Lysates for the detection of GTP-bound RAS/KRAS were further processed using 300 μg lysate and the Active Ras Detection Kit (Cell Signaling Technologies) adhering to the manufacturer’s protocol. Mouse anti-p110 (SCBT) was used for co-IP. Mouse anti-p110 (SCBT); rabbit anti-KRAS (SCBT); rabbit anti-cRaf (CST); mouse anti-RAS (CST) were used for immunoblotting (see Supplementary Methods for details).
Human phospho-RTK array
For experiments in Fig. 2J and S3B, cells were seeded at 4 × 106 cells/100 mm2 dish, let attach overnight and treated with 0.1 μM of DMSO, each drug alone, or in combination for 6 hours. Using the Human Phospho-RTK Array kit (R&D) and following the manufacturer’s protocol, cells were lysed and 1mg of protein used to hybridize to antibody membranes (see Supplementary Methods for details).
Figure 2.
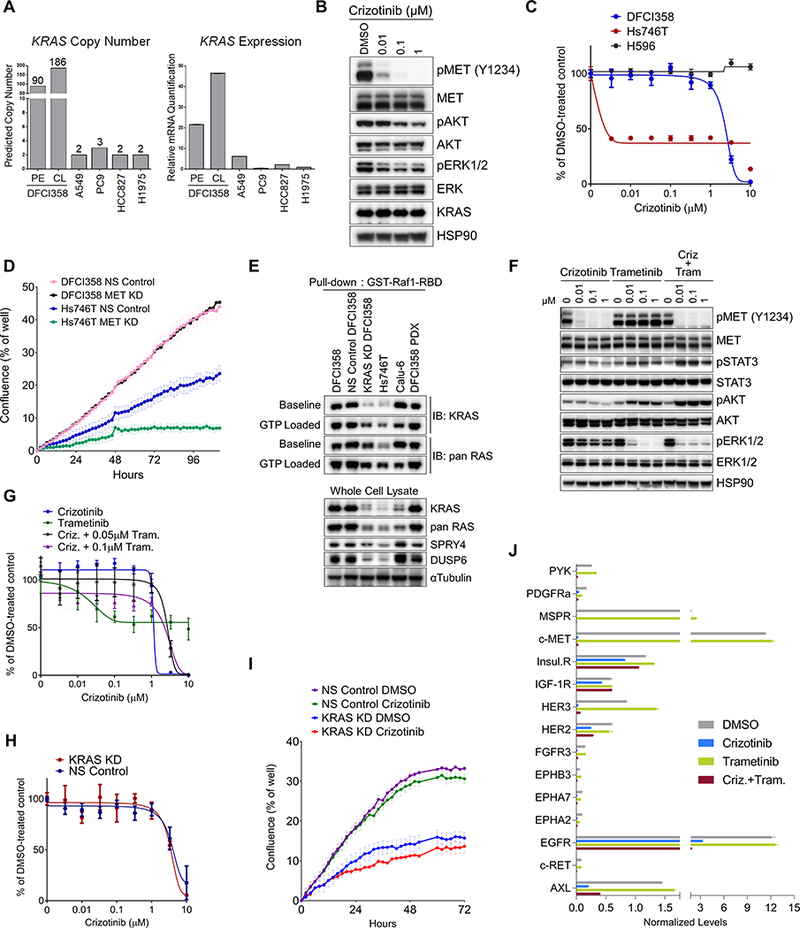
KRAS amplified crizotinib resistant cells exhibit enhanced KRAS/MAPK/ERK signaling. A, Real time qPCR quantification of wild type KRAS copy number and transcript in DFCI358 tumor tissue, patient derived DFCI358 cell line, and non-amplified lung cancer cell lines for comparison. B, Western blot of MET inhibition and sustained downstream signaling in DFCI358 cell line following 6-hour crizotinib treatment. C, Dose-response curves of crizotinib resistant DFCI358, H596 and crizotinib sensitive Hs746T METex14 mutant cell lines treated with crizotinib for 3 days. D, IncuCyte analysis of proliferation as measured by confluence per well of DFCI358 or Hs746T cells, each transduced with either non-silencing siRNA (NS Control) or MET siRNA pool (MET KD), each at 50nM siRNA. Error bars represent standard error of the mean of 9 technical and 3 biological replicates in a 12-well format. E, Western blot of GTP-bound KRAS or pan RAS following pull-down with active RAS-reactive GST-Raf1-RBD fusion protein in parental, non-silencing control siRNA (NS Control), or KRAS siRNA transduced DFCI358 (KRAS KD), DFCI358 PDX cell line, Hs746T and Calu-6 cell lines. GTP loaded controls illustrate the maximum achievable GTP-bound KRAS levels. Baseline shows GTP-RAS levels without GTP incubation. αTubulin was used as a loading control. F, Western blot of DFCI358 cells treated with dose escalated crizotinib (criz), trametinib (tram), or crizotinib combined with 0.1 μM trametinib, for 6 hours. HSP90 was used as a loading control. G, Dose-response growth and viability curve of DFCI358 cells treated with dose-escalated crizotinib alone or in combination with trametinib for 3 days, assessed by MTS assay. H, Dose-response curve of DFCI358 transduced with non-silencing control (NS Control) or KRAS siRNA (KRAS KD), treated with dose-escalated crizotinib for 3 days and assessed by MTS assay. I, IncuCyte analysis of proliferation as measured by confluence per well of vehicle or crizotinib treated DFCI358 NS Control or KRAS KD cells. Error bars represent standard error of the mean of 9 technical and 3 biological replicates. J, Quantification of phospho-RTK array signals (shown in Supplementary Fig. S3B) following a 6-hour treatment with DMSO, crizotinib (criz), trametinib (tram) or the combination. The mean signal (n=2) was calculated and normalized to the mean reference signal (n=6). Error bars represent standard deviation of the mean.
Ligand stimulation studies
72 hours post-transfection, cells were starved for 24 hours in serum-free RPMI1640. Cells in Fig. 4A were treated for 30 minutes at 37°C with FBS, recombinant EGF, insulin, or IGF-1 prepared in serum-free RPMI1640 at the indicated concentrations. Cells in Figs. 4B,C were treated with EGF or FBS at the indicated concentrations for 10 minutes at 37°C. After the 10-minute treatment, EGF/FBS was washed off with PBS and cells were returned to 37°C for another 20 minutes, for a total of 30 minutes. Cells in Fig. 4H were starved and treated with supernatants (normalized to cell number) collected from DFCI358 NS Control or KRAS KD for 30 minutes. Following ligand treatment, cells were washed in ice-cold PBS, lysed in the MB buffer and processed as outlined elsewhere.
Figure 4.
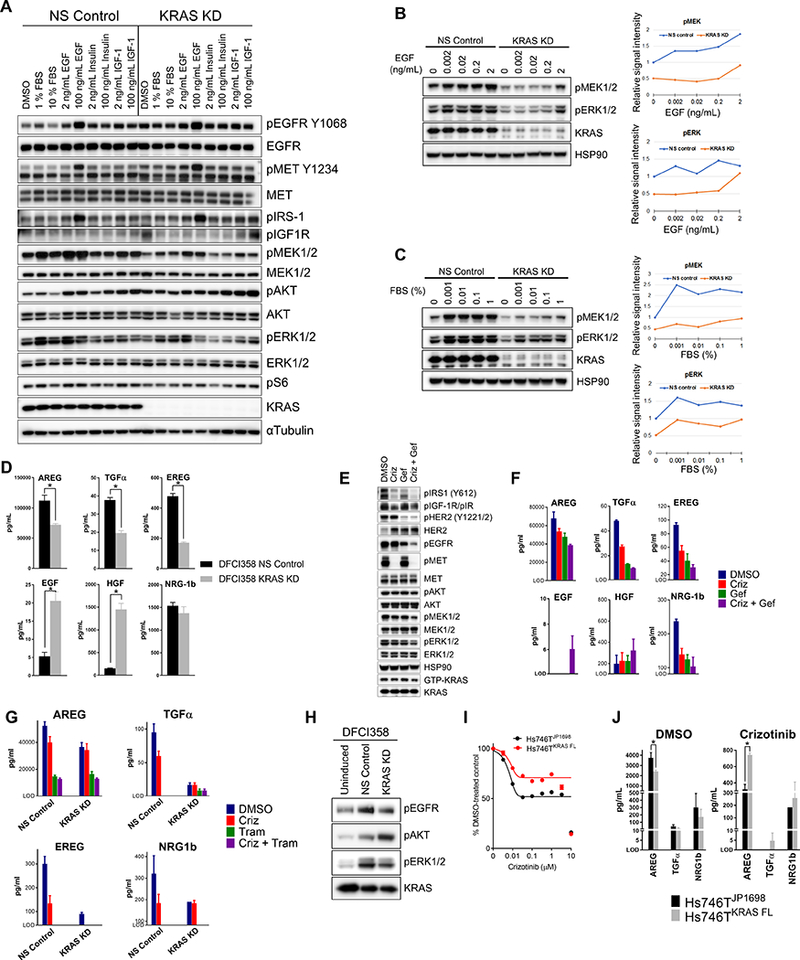
KRAS amplified cells are hypersensitive to growth factor stimulation. A, Western blot of serum-starved growth factor induced DFCI358 transduced with either non-silencing control (NS Control) or KRAS siRNA (KRAS KD). αTubulin was used as a loading control. B, Western blot of serum-starved DFCI358 transduced with either non-silencing control (NS Control) or KRAS siRNA (KRAS KD) and induced with EGF at increasing concentrations. HSP90 was used as a loading control. p-MEK (top panel) and pERK1/2 (bottom panel) Western blot signals were quantified and plotted relative to untreated NS Control values. C, Western blot of serum-starved DFCI358 transduced with either non-silencing control (NS Control) or KRAS siRNA (KRAS KD) and induced with fetal bovine serum (FBS) at increasing concentrations. HSP90 was used as a loading control. p-MEK (lower left panel) and pERK1/2 (lower right panel) Western blot signals were quantified in Image Studio Lite and plotted relative to the value of untreated NS Control. D, AMMP™ based quantification of ligands in cell culture media conditioned by NS Control or KRAS KD DFCI358 cells, expressed as mean concentration of 3 biological and 3 technical replicates each. Error bars represent standard error of the mean. Asterisks denote statistically significant difference. E, Western blot of DFCI358 cells treated with crizotinib (criz), gefitinib (gef), or crizotinib/gefitinib combination, each at 1 μM, for 6 hours. GTP-KRAS levels were determined from a GST-Raf1-RBD pull-down, using the same lysates. HSP90 was used as a loading control. F, AMMP™ based quantification of ligands in cell culture media conditioned by DFCI358 cells treated with each drug at 1 μM, for 6 hours, expressed as mean concentration of 3 technical replicates. Error bars represent standard error of the mean. Only ligands and conditions with values above level of detection (LOD) are shown. G, AMMP™ based quantification of ligands in cell culture media conditioned by DFCI358 cells treated with each drug at 0.5 μM, for 24 hours, expressed as mean concentration of 2–3 technical replicates. Error bars represent standard error of the mean. Only ligands and conditions with values above level of detection (LOD) are shown. H, Starved DFCI358 cells were treated with PBS (Uninduced) or supernatants collected from either DFCI358 NS Control or DFCI358 KRAS KD. Western blot shows induction of pEGFR and pERK1/2 in DFCI358 treated with supernatant from NS Control, while pAKT is induced by KRAS KD supernatant. I, Dose-response growth and viability curve of empty vector-transduced (JP1698) and KRAS-FLAG overexpressing (KRAS FL) Hs746T derivatives treated with dose-escalated crizotinib for 3 days, assessed by MTS assay. J, AMMP™ based quantification of ligands in cell culture media conditioned by empty vector-transduced (JP1698) or KRAS-FLAG overexpressing (KRAS FL) Hs746T derivatives treated with crizotinib at 0.5 μM, for 24 hours, expressed as mean concentration of 2–3 technical replicates. Error bars represent standard error of the mean. Only ligands and conditions with values above level of detection (LOD) are shown. Asterisks denote statistically significant difference.
EGFR ligand quantification
For experiments in Figs. 4D and 4G, 0.75 or 1.25 × 106 cells/dish were plated in 60 mm2 dishes for transfection with control or KRAS siRNA, respectively, and transfected as previously described. 72 hours post-transfection, media were replaced with 3.5 mL fresh complete RPMI1640. Supernatants were collected, filtered and snap-frozen 24 hours later. Each experiment was done in biological triplicates. For experiments in Figs. 4F, G, J, cells were treated with 1 μM drug in 3.5 mL complete RPMI1640 for 24 hours. Ligand concentrations were measured in cell culture media using an immunoassay based on the acoustic membrane microparticles (AMMP™) technology (48). Cells were trypsinized and counted for the purposes of normalization (see Supplementary Methods for details).
DFCI358 PDX In vivo efficacy and PD studies
All in vivo mouse studies were performed at the Dana-Farber Cancer Institute, Boston, USA in accordance with the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of Dana-Farber Cancer Institute. All mice were housed in a pathogen-free environment at DFCI animal facility and handled in strict accordance with good animal practice as defined by the office of laboratory animal welfare. Ten-week-old female NOD.Cg-Prkdc< scid>IL2rg< tm1Wjl>/Szj (NSG) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). DFCI358 PDX was built from patient derived core biopsy specimen obtained with patient consent under an IRB approved protocol.
Expanded DFCI358 PDX tumors were implanted in the right flank of each mouse. When tumors reached 70–150 mm3 or 100–200 mm3 for the copanlisib or alpelisib study, respectively, mice were randomized to groups of n=6 mice/arm (copanlisib study) or n=5 mice/arm (alpelisib study) and treated for 2–3 weeks. Tumor growth was monitored until the average tumor volume (TV) reached 2000 mm3. Tumor size and body weight (BW) were monitored twice weekly. TV was calculated as follows: TV (mm3)=length×width×width×0.5. Statistical analysis was performed using the Kruskal–Wallis/Dunn’s post-hoc test. P≤0.05 was considered significant. PD study was performed following a 3-day treatment (see Supplementary Methods and Supplementary Table S2 for details on drug dosing).
Results
Recurrent amplification of WT KRAS in three patients with METex14 mutant NSCLC who developed acquired resistance to crizotinib
We identified three METex14 patients treated with crizotinib, all of whom developed amplification of WT KRAS in their drug resistant tumor (Supplementary Figs. S1, S2). Patient #1 (MET c.2903_3028+67del193insA) had an initial marked response to crizotinib (Fig. 1A) followed by disease progression within 4 months (Fig. 1A) of crizotinib treatment. Targeted next generation sequencing (NGS) on thoracentesis and a repeat biopsy of an enlarging left cervical lymph node revealed no secondary mutations in MET but revealed marked focal WT KRAS amplification (estimated 55 copies by NGS), which was not present in either of the pre-crizotinib lung or adrenal samples (Fig. 1B). Acquired KRAS amplification was further confirmed by fluorescence in situ hybridization (FISH), and a marked acquired increase in KRAS and phospho-ERK expression was demonstrated by immunohistochemistry (IHC) (Fig. 1B). Plasma-based cell-free DNA NGS at the time of crizotinib resistance also demonstrated high-level focal KRAS amplification with no other significant copy number gains or genomic mutations in a targeted 73-gene panel (Fig. 1C). MET expression was detected in both the pre-crizotinib and post-crizotinib samples, and none of the samples displayed MET amplification by NGS or FISH (Supplementary Fig. S1).
Figure 1.
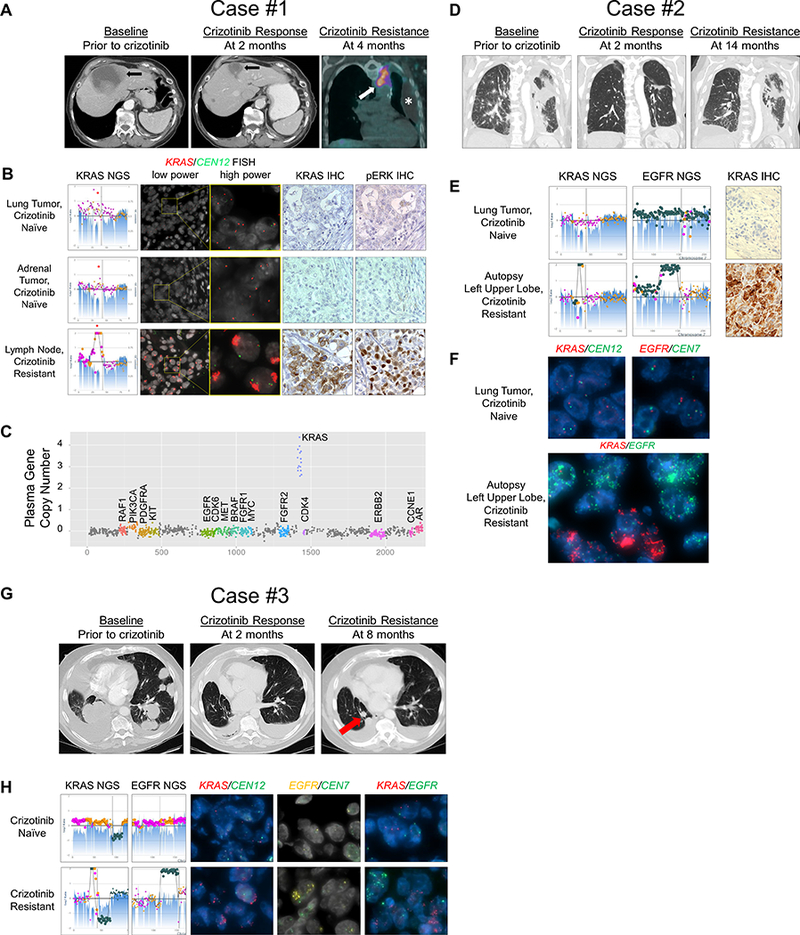
Acquired wild-type KRAS amplification in three patients with MET exon 14 mutant NSCLC at the time of crizotinib resistance. A, Axial CT images of the upper abdomen for Case #1 showing a liver metastasis at baseline (left panel) which significantly decreased in response to crizotinib at 2 months of therapy (middle panel). Fused coronal image of the PET-CT scan obtained at 4 months of therapy (right panel) demonstrated FDG-avid, enlarging mediastinal adenopathy (arrow) and a new loculated left pleural effusion (asterisk) from which the DFCI358 cell line was derived. B, Next generation sequencing (NGS, left panels) copy number plots of the KRAS locus on chromosome 12 from two crizotinib-naïve tumors from Case #1 and the crizotinib-resistant supraclavicular lymph node (bottom left panel) with estimated 55–75 copies of KRAS. Fluorescence in situ hybridization (FISH) for KRAS (red) and centromere 12 (CEN12, green) is shown at low and high power for the crizotinib naïve and resistant samples. Immunohistochemistry (IHC) for KRAS and phosphor-ERK (pERK) are also shown. C, Plasma gene copy number for several genes are displayed for Case #1 from blood taken at the time of crizotinib resistance. D, Coronal CT images for Case #2 showing a consolidated mass in the left upper lobe of the lung at baseline (left panel), which responded to crizotinib at 2 months of therapy (middle); however, progression with regrowth of left lung tumor was noted at 14 months indicative of the development of acquired resistance (right). E, NGS copy number plots of the KRAS locus on chromosome 12 and the EGFR locus on chromosome 7 are shown for the pre- and post-crizotinib samples for Case #2. Post-crizotinib sample shows estimated 25 copies of KRAS in the upper lobe. KRAS IHC before and after crizotinib treatment are also shown. F, KRAS (red) and CEN12 (green) FISH (top left) and EGFR (red) and centromere 7 (CEN7, green) FISH (top right) are shown in the crizotinib naïve tumor. Dual KRAS/EGFR (red/green, respectively) FISH in the crizotinib resistant tumor is shown (bottom panel). G, Axial CT images of the chest for Case #3 showing bilateral lung masses and a nodule at baseline (left), which partially responded to crizotinib (middle), followed by growth of one of the nodules in the right lower lobe (right, arrow), which was biopsied at the time of acquired resistance. H, NGS copy number plots of the KRAS and EGFR loci are shown for the pre- and post-crizotinib samples for Case #3. Post-crizotinib sample shows estimated 21 copies of KRAS. KRAS IHC before and after crizotinib treatment are also shown. FISH for KRAS/CEN12 (red/green, respectively), EGFR/CEN7 (yellow/green, respectively) and dual KRAS/EGFR (red/green) are shown in the crizotinib naïve and crizotinib resistant tumor samples.
Patient #2 (MET c.2888–20_2888–10del) experienced a dramatic initial response to crizotinib but developed disease progression after 14 months and died shortly thereafter (Fig. 1D). At autopsy three different crizotinib resistant lesions were analyzed by NGS (Figs. 1E and S2), none of which contained a MET secondary mutation (data not shown). All three demonstrated evidence of WT KRAS amplification not present in the pre-crizotinib treated tumor, and increased KRAS expression by IHC (Figs. 1E, S2). In addition, they contained variable degree of WT EGFR amplification (Supplementary Fig. S2). Dual color FISH detecting the KRAS and EGFR amplifications further revealed that they were located in different cells within the drug resistant tumor (Fig. 1F).
Patient #3 (MET c.3028 +1G>T) showed a partial response after 2 months of treatment with crizotinib but progressed 4 months later with a growth of a lung lesion (Fig. 1G). Targeted NGS of a growing lesion revealed no secondary mutations in MET, but compared to the pre-crizotinib tumor specimen, there was evidence of amplification of WT KRAS and EGFR (Fig. 1H). Genomic amplification of KRAS and EGFR was confirmed by FISH and detected in non-overlapping cells (Fig. 1H).
KRAS amplification uncouples MET from downstream effectors in crizotinib resistant cells
A pleural effusion (PE) collected from patient #1 following disease progression was used to generate the DFCI358 cell line (CL) and at the same time lymph node biopsy was used to also establish the DFCI358 patient derived xenograft (PDX) model. The cell line retained the high level of KRAS amplification and expression observed in the pleural effusion (Fig. 2A). We analyzed the pMET status and downstream signaling in the DFCI358 cell line upon crizotinib treatment and observed high levels of activated MET at baseline and complete MET inhibition with as little as 10 nM of crizotinib, implying a MET-independent mechanism of resistance (Fig. 2B). In contrast, MAPK/ERK and AKT pathway activation was only minimally inhibited at well above the clinically achievable concentration of 57 nM free drug (20) (Fig. 2B), demonstrating that the activation of these pathways was partially uncoupled from that of MET. This was consistent with the failure of crizotinib to inhibit cell proliferation even at micromolar concentrations (Fig. 2C), similar to the crizotinib resistant METex14/PI3K mutant H596 (homozygous MET c.3251spl+1 G>T (21); PIK3CA E545K mutation) and in contrast to the gastric crizotinib sensitive METex14 mutant Hs746T (MET c.3082+1G>T) cell lines (Fig. 2C). MET siRNA further inhibited proliferation of Hs746T, but not of DFCI358 (Fig. 2D). To investigate the functional involvement of KRAS in crizotinib resistance, we downregulated KRAS using a siRNA (knockdown; KD) and compared these cells to those expressing non-silencing siRNA control (NS Control). KRAS siRNA treatment was effective in downregulating KRAS expression and activation in the DFCI358 cells to levels observed in the crizotinib-sensitive METex14 mutant Hs746T cells (22,23)(Fig. 2E). We demonstrated that parental DFCI358, NS Control DFCI358 and the DFCI358 PDX cell line exhibited high baseline KRAS activation, as evidenced by the levels of GTP-bound KRAS precipitating with the fusion protein GST-Raf1-RBD and that the levels were comparable to those seen in the KRAS G12C mutant cell line Calu-6 (Fig. 2E). In addition, the amplified KRAS DFCI358 cells exhibited upregulation of negative regulators of the RAS and RTK signaling, DUSP6 and SPRY4, respectively, a feature shared with mutant KRAS cell lines, including Calu-6. In contrast, the levels of both negative regulators were low in the Hs746T cells and in the KRAS KD DFCI358 derivative. The high GTP-bound state of KRAS at baseline is further indicative of functional involvement of amplified KRAS in the signaling of DFCI358.
MEK inhibition or KRAS knockdown fail to restore sensitivity to crizotinib due to activation of PI3K/AKT signaling
Given that the MAPK/ERK pathway is one of the major KRAS effectors, we assessed whether inhibiting MEK alone, using trametinib, could restore sensitivity to crizotinib. Although trametinib completely inhibited pERK1/2 at a concentration of 10 nM (Fig. 2F), treatment with trametinib alone and in combination with crizotinib prominently induced PI3K/AKT, and to a lesser extent JAK/STAT3 pathway signaling (Fig. 2F), but didn’t increase pS6 levels, the target of the mTOR pathway (Supplementary Fig. S3A). A growth viability assay demonstrated that the sensitivity of the DFCI358 cells to crizotinib in the presence of trametinib was limited (Fig. 2G). Next, we examined whether the knock-down of KRAS, used as a surrogate for KRAS inhibition, could overcome crizotinib resistance. The viability assay showed a comparable IC50 between the control and the KD cells with crizotinib treatment (Fig. 2H), however, the KRAS KD cells exhibited a substantially reduced growth rate at baseline (Fig. 2I), suggesting strong dependence on amplified KRAS. To identify potential mechanism(s) leading to PI3K/AKT activation following drug treatment in the parental or KRAS KD cell lines, we performed a phospho-receptor tyrosine kinase (RTK) array following crizotinib and/or trametinib treatment, and identified the epidermal growth factor receptor (EGFR), receptor tyrosine-protein kinase erbB-2 (HER2), insulin-like growth factor 1 receptor (IGF1R) and insulin receptor (IR) as RTKs that remained activated following drug treatment (Figs. 2J and S3B).
To further differentiate between the relative contributions of amplified KRAS and the above identified RTKs to PI3K/AKT pathway induction upon pharmaceutical or siRNA-mediated MAPK/ERK inhibition, and by so doing implicate amplified KRAS in resistance to crizotinib +/− trametinib, we compared the signaling responses of DFCI358 NS Control and KRAS KD following pharmaceutical inhibition of MET, MEK, EGFR/HER2, IGF1R/IR and the combinations thereof using crizotinib, trametinib, lapatinib and linsitinib, respectively (Fig. S3C, 3A-G). Comparing the levels of RTK induction between crizotinib-treated NS Control or KRAS KD as determined by quantification of the immunoblot in Fig. 3A, we observe that KRAS knock-down rescues from MET inhibition via PI3K/AKT by inducing stimuli upstream of PI3K, namely IRS1 and HER2, (Fig. 3B-D). Despite the relatively higher contribution of these stimuli in pro-survival signaling in KRAS KD cells than in the NS Control, the NS Control cells still exhibit a substantially higher induction of AKT upon the inhibition of MET and MAPK/ERK (Fig. 3E), implicating amplified KRAS in the molecular rewiring and maintenance of pro-survival signaling via PI3K/AKT in these cells. Consistent with this conclusion is the finding that pShc, an adaptor upstream of MAPK/ERK, is induced to higher levels in the NS Control than in the KRAS KD cells (Fig. 3F). Finally, both DFCI358 derivatives induce JAK/STAT signaling in response to MET/MAPK/ERK inhibition (Fig. 3G), however, the cause and effect of this induction remains unclear.
Figure 3.
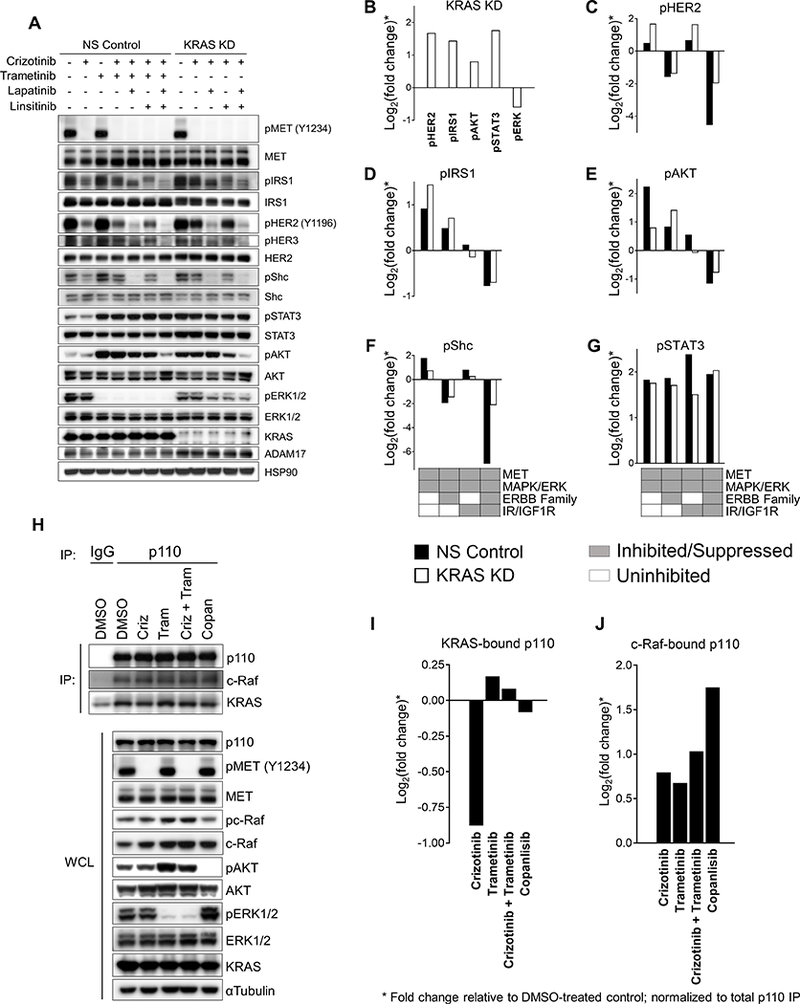
Crizotinib/trametinib-treated NS Control DFCI358 and crizotinib-treated KRAS KD DFCI358 induce PI3K/AKT via distinct mechanisms. A, Western blot of DFCI358 transduced with non-silencing control (NS) or KRAS siRNA (KRAS KD) treated with each drug at 0.5 μM, for 6 hours. HSP90 was used as a loading control. B, Quantification of Western blot signals shown in Fig. 3A for indicated markers in crizotinib-treated KRAS KD DFCI358 cells. Ratios of crizotinib-treated KRAS KD to crizotinib-treated NS control values were calculated to determine fold-change (induction of each marker) upon KRAS knock-down, which was ploted as Log2 for each marker. C-G, Quantification of Western blot signals shown in Fig. 3A for pHER2 (C), pIRS1 (D), pAKT (E), pShc (F) and pSTAT3 (G). The induction of each marker in response to MET/MAPK/ERK inhibition by pharmaceutical (NS Control) and/or knock-down (KRAS KD) methods relative to MET inhibition only (NS Control) was assessed by calculating the ratios of crizotinib-treated KRAS KD or crizotinib/trametinib-treated NS Control relative to crizotinib-treated NS Control values for each marker. Additional inhibition of the ERBB family (lapatinib) and/or insulin family (linsitinib) receptors was assessed for the relative involvement of these pathways in resistance to MET/MAPK/ERK inhibition. The fold change was expressed as Log2 for each marker and treatment condition. H, Co-IP study using an anti-pan-p110 antibody or IgG control in DFCI358 treated with DMSO, crizotinib (criz), trametinib (tram), crizotinib + trametinib (criz+tram), or copanlisib (copan), each at 0.5 μM for 6 hours. Interactions of p110 with KRAS or c-Raf were assessed by immunoblotting the immunoprecipitate against KRAS or c-Raf antibodies, respectively. Immunoblot against p110 was used to assess total levels of immunoprecipitated p110. Whole cell lysates (WCL) were blotted against targets of interest. I, J, Quantification of IP Western blot signals shown in Fig. 3H, expressed as Log2 of fold change, where fold change is the ratio of each value relative to DMSO-treated control, normalized to total immunoprecipitated p110 levels and corrected for IgG background.
The catalytic subunit of PI3K, p110, can be directly engaged and activated by KRAS (24). To further study the relationship of amplified KRAS and PI3K/AKT signaling, we evaluated the levels of KRAS-bound p110 (pan) using co-immunoprecipitation (IP) studies following drug treatments (Fig. 3H). An immunoblot quantification revealed that upon crizotinib treatment, the interaction between KRAS and p110 decreased, consistent with our previous data implicating the MAPK/ERK pathway in crizotinib resistance. In contrast, MEK +/− MET inhibition led to enhanced interaction between KRAS and p110, suggesting that the relatively low efficacy of MEK inhibition can be attributed to a compensatory induction of p110 by KRAS. Inhibiting PI3K with copanlisib slightly diminished the p110/KRAS complex levels, possibly accounting for the compensatory increase in MAPK/ERK signaling (Fig. 3I). In addition, p110 was found in complex, directly or indirectly, with c-Raf regardless of drug treatment (Fig. 3J). Our findings suggest that MEK inhibition or KRAS KD lead to compensatory activation of PI3K/AKT signaling, suggesting that this pathway may need to be inhibited to restore crizotinib sensitivity.
KRAS amplified cells are hypersensitive to growth factor stimulation
In order to understand how amplified KRAS may govern downstream signaling in the context of MET inhibition, we exposed serum-starved NS Control and KRAS KD DFCI358 to EGF, insulin, or IGF1, the stimulatory ligands for RTKs whose activation was maintained following crizotinib treatment in Fig. 2J (Fig. 4A). NS Control cells exhibited marked activation of pMEK, pERK1/2, and pAKT in response to both low and high concentrations of EGF and fetal bovine serum (FBS), while KRAS KD cells demonstrated a substantially attenuated response under the same conditions. Interestingly, EGF at 100 ng/mL not only activated EGFR but also led to massive induction of insulin receptor substrate 1 (IRS1), independently of IGF1R activation. EGF further increased the levels of phosphorylated MET precursor. Finally, although we observed no RTK induction in response to insulin, KRAS KD DFCI358 exhibited enhanced reliance on the PI3K/AKT pathway compared to the NS control, as evidenced by increased pAKT following IGF1 treatment. In light of these results, we sought to determine the lowest concentration of EGF or fetal bovine serum (FBS) sufficient for activation of signaling downstream of KRAS (Fig. 4B, C). Starved NS Control cells exhibited prominent activation of MEK and ERK1/2 with EGF concentrations as low as 0.002 ng/mL, while the KRAS KD equivalent required a minimum of 2 ng/mL EGF, a 1000-fold difference, to fully activate its downstream effectors (Fig. 4B). Similar trends were observed with pMEK following treatment with serially diluted FBS, even though the dynamics of ERK1/2 activation was similar between the NS and KD cells, after accounting for the difference in the absolute pERK1/2 levels (Fig. 4C). These studies suggest that amplified KRAS imparts resistance, in part, by rendering cells hyper-sensitive to ligand stimulation. In light of previously published reports demonstrating that ERK1/2 transcriptionally induces the expression of EGFR ligands, including transforming growth factor alpha (TGF-α), amphiregulin (AREG), and heparin-binding epidermal growth factor–like growth factor (HB-EGF) (25,26), we also sought to determine whether amplified KRAS may enhance autocrine signaling in DFCI358. Supernatants from cultured NS control and KRAS KD DFCI358 cells were subjected to quantification of secreted EGFR ligands AREG, TGF-α, epiregulin (EREG), EGF; the MET specific ligand hepatocyte growth factor (HGF); the HER3/4 ligand neuregulin 1 (NRG1) (Fig. 4D). NS Control cells expressed high levels of all of these ligands, and knockdown of KRAS resulted in a significant decrease in AREG (p<0.011), TGFα (p<0.0006) and EREG (p<0.0001), respectively, which are EGFR ligands shown to promote EGFR recycling, unlike EGF known to induce degradation (27,28). In contrast, KRAS KD DFCI358 exhibited an increase in secreted EGF (p<0.005) and HGF (p<0.0007), respectively, although the absolute EGF levels remained low. While the mechanistic basis and biological implications of this increase remain unclear, it may represent a compensatory response to the attenuated MAPK/ERK signaling in KRAS KD cells. NRG1 expression remained unchanged in KRAS KD cells.
To further examine the role of EGFR activation in crizotinib resistance, we subjected DFCI358 to the combination of the MET inhibitor crizotinib and/or the EGFR inhibitor gefitinib. The combination acutely reduced the levels of all, active MET, EGFR, HER2 (an EGFR dimerization partner) and IRS1, suggesting crosstalk and interdependence between the receptors (Fig. 4E). More importantly, even though all tested RTKs were strongly inhibited by the combination, the levels of GTP-bound KRAS, pMEK and pERK1/2 remained high, further supporting our observations that KRAS amplification renders cells hypersensitive to upstream stimuli and/or leads to a partial uncoupling of the MAPK/ERK pathway from activated RTKs, akin to mutant KRAS signaling (Fig. 4E). In addition, the quantification of EGFR ligands expressed by DFCI358 following crizotinib/gefitinib treatment demonstrated that both, EGFR and to a lesser extent MET, cooperate with amplified KRAS to promote autocrine signaling by sustaining EGFR ligand production, as evidenced by a decrease in EGFR ligand levels upon inhibition of EGFR and/or MET (Fig. 4F) and upon KRAS knock-down and/or MET/MEK inhibition (Fig. 4G). Supernatant collected from NS Control cells induced pEGFR/pERK1/2, while that from KRAS KD cells induced pAKT signaling in starved DFCI358 as expected (Fig. 4H). Together these data suggest that KRAS amplification partially uncouples RTKs from MAPK/ERK signaling and brings about heightened expression of three EGFR agonists that promote receptor recycling. Moreover, we speculate that following downregulation of KRAS expression, these cells increase HGF and EGF production via an unknown mechanism to activate MET and EGFR/IRS1, as a compensation for the loss of KRAS expression. Finally, a GST-Raf1-RBD fusion protein pull-down showed that in the context of estimated ~186-copy KRAS amplification and long-term drug treatment (Supplementary Fig. S4A), especially when compared to an acute drug treatment (Supplementary Fig. S4B), none of the tested drug combinations appreciably decreased the levels of GTP-bound KRAS, despite a modest short-term decrease in pERK1/2 achieved by the gefitinib combinations (Supplementary Fig. S4B). A kinetics analysis of the GTP-KRAS levels in the DFCI358 cell line undergoing a combined crizotinib/gefitinib treatment over time further illustrates that the inhibition of KRAS is modest and transient, limited to ~1–6 hours of drug exposure, and followed by compensatory KRAS reactivation at longer time points (Supplementary Fig. S4C). These data suggest a rapid and dynamic rebound in GTP-KRAS in response to drug pressure, in order to sustain signaling via MEK or PI3K.
To further study the role of KRAS amplification in crizotinib resistance in METex14 mutant models, we engineered the crizotinib sensitive Hs746T cell line to overexpress WT KRAS (Supplementary Fig. S4D) and evaluated its crizotinib sensitivity. As assessed by the growth and viability assay, KRAS overexpression imparted a degree of resistance to crizotinib in this cell line (Fig. 4I). In addition, quantification of EGFR ligands revealed that upon crizotinib treatment, Hs746TKRAS cells sustained higher expression of AREG as compared to the vector-transduced Hs746T (Fig. 4J). The effect of KRAS overexpression in Hs746T is likely less pronounced due to the cellular context and lower relative KRAS expression as compared to KRAS amplified DFCI358.
Inhibition of PI3K/AKT signaling restores sensitivity to crizotinib in METex14 KRAS amplified models in vitro and in vivo
A model of crizotinib and crizotinib/trametinib resistance in parental, NS Control and KRAS KD DFCI358 derivatives proposes possible mechanisms and hints at alternative approaches to target them (Fig. 5). In search of a therapeutic combination to overcome KRAS amplification-mediated crizotinib resistance, we evaluated proliferation and apoptosis of DFCI358 cells during treatment with MET, ERBB family, MEK, PI3K, and IGF1R/IR inhibitors, either alone or in combination. Among the tested treatments, combining crizotinib with the pan PI3K inhibitor copanlisib even at low concentrations (100 nM) potently induced cellular apoptosis (Fig. 6A). Strikingly, even copanlisib alone showed modest pro-apoptotic activity. In comparison, no other crizotinib combination was effective at inducing apoptosis in vitro (Fig. 6A), including with trametinib, which showed only an anti-proliferative effect (Fig. 6B). Of note is that alpelisib, a p110α selective inhibitor, was ineffective at this concentration in vitro (Fig. 6A), although some efficacy was observed at higher concentrations when combined with crizotinib in vitro (data not shown). Both DFCI358, and Hs746TKRAS cells responded modestly to single-agent copanlisib and were strongly inhibited by the crizotinib/copanlisib combination (Fig. 6C). The inhibitory effects of the combination were somewhat less pronounced in the Hs746TKRAS, presumably because of our limited ability to overexpress KRAS. We further show that p110 inhibition alone leads to transient suppression of the MAPK/ERK pathway, as evidenced by the temporary decreases in active c-Raf, MEK1/2 and ERK1/2 (Fig. 6D, E).
Figure 5.

Model of resistance to MET and MET/MAPK/ERK inhibition in DFCI358. A, At baseline, mutant MET in parental DFCI358 cells activates KRAS, which in turn amplifies downstream MAPK/ERK signaling resulting in enhanced transcription and release of EGFR ligands. B, Upon MET inhibition in parental DFCI358 cells, amplified KRAS sustains MAPK/ERK signaling via EGFR-ligand stimulated EGFR and other activated RTKs. C, Upon dual MET/MEK inhibition in KRAS amplified NS Control DFCI358 cells, cell viability is sustained by the PI3K/AKT pathway activated directly by amplified KRAS. D, With KRAS downregulation and MET inhibition in KRAS KD DFCI358 cells, resulting in diminished MAPK/ERK signaling, cell viability is sustained by the PI3K/AKT pathway activated by RTKs, such as IR/IGF1R, HER2 and EGFR.
Figure 6.
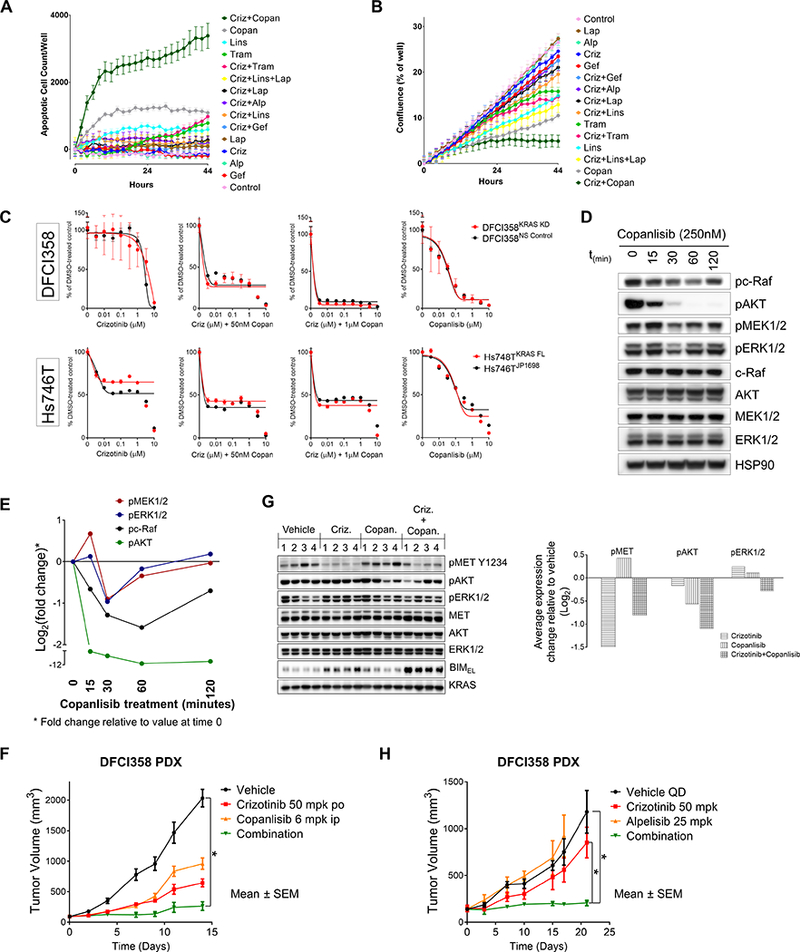
Inhibition of PI3K/AKT signaling restores sensitivity to crizotinib in METdel14 KRAS amplified cells. A, IncuCyte analysis of rate of apoptosis normalized to confluence, expressed as the mean apoptosis reached per condition and timepoint in DFCI358 cells. Error bars represent standard error of the mean. B, IncuCyte analysis of proliferation as measured by confluence per well of TKI-treated DFCI358 cells. Error bars represent standard error of the mean (n=6 or 12). Abbreviations: crizotinib, criz; gefitinib, gef; trametinib, tram; linsitinib, lins; alpelisib, alp; lapatinib, lap; copanlisib, copan. C, Dose-response growth and viability curve of DFCI358 NS Control or KRAS KD and Hs746TJP1698 or Hs746TKRAS FL derivatives treated with dose-escalated crizotinib +/− 0.05μM or 1μM copanlisib or dose-escalated copanlisib alone for 3 days, assessed by MTS assay. D, Western blot showing a time-course of copanlisib treatment in DFCI358 (250nM) with respect to the inhibition of downstream effectors. E, Quantification of Western blot signals shown in Fig. 6D for indicated markers. Ratios of phospho signals at each timepoint relative to those at time=0 were calculated to determine fold-change (suppression of each marker) upon copanlisib treatment, which was plotted as Log2 for each marker. F, DFCI358 PDX study comparing the efficacy of each indicated single agent and drug combination. Asterisks denote statistically significant difference. G, Pharmacodynamics of the DFCI358 PDX study evaluating target inhibition following a 3-day treatment. Graph shows quantification of average Western blot signals for indicated markers. Inhibition relative to vehicle average value is expressed as Log2 for each marker. HSP90 or KRAS was used as a loading control. H, DFCI358 PDX study comparing the efficacy of each indicated single agent and drug combination. Asterisks denote statistically significant difference.
Informed by our in vitro data, we performed an in vivo study using the DFCI358 PDX and evaluated both efficacy and pharmacodynamics (PD) of the crizotinib/copanlisib combination co-targeting MET and PI3K. The crizotinib/copanlisib combination was well tolerated. While neither of the single agents achieved tumor regression within the course of the study (14 days), the combination of crizotinib and copanlisib was more efficacious than crizotinib alone at inhibiting tumor growth (%TGI of 90.2 vs. 69.4, respectively) (Fig. 6F). In vivo efficacy of the combination was lower than expected, we believe, due to suboptimal availability of copanlisib when administered as a mannitol suspension intraperitoneally, a conclusion further supported by the incomplete pAKT inhibition achieved within the PD study (Fig. 6G). In a separate study, the combination of crizotinib and the selective PI3Kα inhibitor alpelisib maintained tumor stasis (%TGI of 93.68 for the combination vs. 31.62 for crizotinib; p≤0.0249) for the duration of the study (21 days) (Fig. 6H), further strengthening our conclusions, despite this combination being less efficacious in vitro at low concentrations than crizotinib/copanlisib (Fig. 6A, B and data not shown). We confirmed that PDX tumors retained KRAS amplification and overexpression (Supplementary Fig. S5).
Discussion
METex14 expressing NSCLCs are clinically sensitive to MET TKIs (12–15). Unfortunately, as with many other genotype-directed TKI therapies, the benefit of a single-agent approach is almost always limited by the acquisition of secondary kinase domain resistance mutations and/or activation of compensatory pathways. Here we describe three cases of crizotinib-resistant METex14 mutant NSCLC where resistant tumors presented with a high copy number gain of the WT KRAS allele. We developed and characterized, both in vitro and in vivo, a unique patient derived model from one of those patients, and implicated amplified KRAS in crizotinib resistance in this patient. We define 3 hallmarks of KRAS-mediated resistance in this context: 1) relative insensitivity to dual MET/MEK inhibition due to strong compensatory induction of PI3K, 2) KRAS-induced expression of EGFR ligands and hypersensitivity to ligand-dependent and independent activation, 3) reliance on PI3K signaling upon MET inhibition.
Both de novo and acquired KRAS mutations have been shown to impart resistance to single-agent EGFR targeted therapies in colorectal cancers (CRC) (29–31) by constitutively activating downstream signaling despite complete inhibition of the driver RTK. Amplified WT KRAS can impart resistance to cetuximab in CRC (31) and dual KRAS/MET amplification can confer resistance in MET amplified cell lines chronically exposed to MET TKIs (32). WT KRAS amplification has been implicated in the resistance of the ALK-rearranged H3122 cell line chronically treated with ALK TKIs in vitro, leading to MAPK pathway reactivation (33).
Both the DFCI358 pleural effusion and primary cell line contain a massive amplification in KRAS. Intriguingly, the levels of GTP-bound KRAS in DFCI358 were not linearly proportional to the gene copy number and were like those observed in a KRAS mutant model. Given that overexpression/overactivation of KRAS may be toxic, the RTK/RAS/ERK signaling must be tightly regulated by feedback and feedforward loops (34). Indeed, we observed heightened expression of negative ERK1/2 and RTK regulators, DUSP6 and SPRY4, respectively, in KRAS amplified DFCI358, DFCI358 PDX and KRAS mutant Calu-6, but not in KRAS KD DFCI358 or crizotinib sensitive Hs746T. This finding, in part, explains the non-linear relationship between the KRAS copy number and the levels of activated KRAS, MEK and ERK1/2 in DFCI358 seen throughout the study. It is likely that the MAPK/ERK baseline activation in DFCI358 is regulated to a tolerable level that allows cells to bypass RTK inhibition with the minimum amount of upstream stimulus necessary. This is evident in the relatively lower levels of activated RTKs in DFCI358 NS Control, compared to the KRAS KD cells, following serum starvation, as well as upregulation of SPRY4 in KRAS amplified cells, dampening RTK activation and/or GTP-KRAS formation. Nevertheless, given the large reservoir of inactive KRAS available for compensatory activation in response to RTK inhibition, kinase-based strategies to address KRAS-mediated resistance are likely to pose a challenge. As illustrated by the sustained GTP-KRAS levels following long-term exposure to an array of inhibitor combinations, the compensatory activation of copious KRAS reserves interferes with the effectiveness of most tested drug combinations. Our conclusion is further strengthened by the ability of the crizotinib/gefitinib combination to better inhibit GTP-RAS and ERK1/2 when administered acutely (3 hours), before KRAS rebound thwarts its effectiveness (Supplementary Fig. S4).
Our study provides a line of evidence implicating amplified WT KRAS in crizotinib resistance, in part, by rendering cells hyper-sensitive to ligand stimulation (Figs. 4A-C,H). This quality of amplified KRAS signaling could, in and of itself, confer resistance to crizotinib by transducing ligand-dependent activation of expressed RTKs even in the absence of appreciable amounts of stimulating ligands. Indeed, the upregulation of EGFR ligands by the MAPK/ERK pathway has long been recognized (26,35,36). In CRC, both TGFα and AREG were overexpressed and shown to promote crosstalk between EGFR and MET, conferring EGFR blockade resistance (37,38). Conversely, EGF and TGFα were implicated in EGFR-mediated phosphorylation of MET (39,40). In DFCI358, augmented EGFR ligand production is of particular significance, given the intricate crosstalk involving RTKs of the ERBB family and MET (34,41,42). Intriguingly, 2 of the 3 patients (Figs. 1E, F, H, S2) showed evidence of WT EGFR amplification, in non-overlapping cells with KRAS amplification, suggesting this may be a complementary mechanism of activating EGFR signaling leading to drug resistance, where KRAS amplified cells activate EGFR amplified cells via paracrine signaling. Morgillo et al. reported that in NSCLC, erlotinib increased the levels of EGFR/IGF1R heterodimer, activating IGF1R and its downstream effectors, thereby compromising the antitumor activity of erlotinib (43). This or a similar mechanism may be partially responsible for the relative long-term ineffectiveness of the crizotinib/gefitinib combination seen in vitro (Supplementary Figs. S4A, C).
Our studies implicate KRAS/MAPK/ERK signaling as the main pathway mediating crizotinib resistance in the presence of KRAS amplification. However, since MEK inhibition strongly induces pro-survival PI3K/AKT signaling, in contrast to PI3K inhibition which triggers apoptosis presumably via temporary suppression of MAPK/ERK (Fig. 6D-E) (44), and since all of the ligand-activated RTKs signal through PI3K, concomitant inhibition of MET and PI3K emerged as the most effective treatment strategy both in vitro and in vivo (Figs. 6). Although the number of resistant METex14 mutant cases studied to date is still small requiring a larger study of drug resistant patients, our findings suggest that acquired WT KRAS amplification may emerge as one of the more common mechanisms of resistance, in addition to MET secondary mutations. Whether the recurrent nature of KRAS amplification observed in METex14 patients is due to an inherent predisposition of these cancers to developing such amplification remains to be determined. Continued systematic genomic studies of tumors from patients with METex14 mutant NSCLC will be necessary to establish the frequency of different acquired resistance mechanisms and to determine what proportion of patients may benefit from an alternative MET inhibitor (in case of a MET secondary mutation) or require a combination treatment approach.
Supplementary Material
Translational Relevance.
MET exon 14 mutations (METex14) have recently been recognized as a targetable oncogenic driver in non-small cell lung cancer (NSCLC), and these cancers inevitably acquire resistance to MET inhibitors. While secondary mutations in the MET kinase domain that confer resistance to MET inhibitors have been reported, characterization of mechanisms involving parallel signaling in METex14 mutant NSCLC have not yet been described. Our study describes one such mechanism and proposes a therapeutic strategy to target it.
Acknowledgments
This work was supported by The American Cancer Society (CRP-17–111-01-CDD to PAJ), the National Cancer Institute (R01CA135257; R01CA222823 to PAJ), the Conquer Cancer Foundation of the American Society of Clinical Oncology (to MMA), the Mugar Family Fund (to PAJ) and the Goldstein Family Fund (to PAJ).
Footnotes
Conflicts of interest
PAJ has received consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Merrimack Pharmaceuticals, Chugai Pharmaceuticals, Ariad Pharmaceuticals, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics and LOXO Oncology; receives post-marketing royalties from DFCI owned intellectual property on EGFR mutations licensed to Lab Corp; has sponsored research agreements with AstraZeneca, Daichi-Sankyo, PUMA, Eli Lilly and Company and Astellas Pharmaceuticals; and has stock ownership in Gatekeeper Pharmaceuticals. MMA has received consulting fees from AbbVie, Ariad, Clovis, Bristol-Myers Squibb, Nektar, AstraZeneca, Genentech/Roche, Boehringer Ingelheim, and Merck. LMS has received consulting fees from Genentech and speaker fees from Research to Practice. CPP has received honoraria from BioRad and is on the scientific advisory board for Digital Bioanalytics. MN Consults for Bristol-Myers Squibb, Toshiba Medical Systems, WorldCare Clinical; received a research grant from Merck Investigator Studies Program, Toshiba Medical Systems; Honorarium from Bayer. AJR has received consulting fees from Medtronic, Boehringer-Ingelheim, Ariad, Roche, and AstraZeneca. DBC has received consulting fees and honoraria from Pfizer, Boehringer Ingelheim and Ariad pharmaceuticals. RJN and RBL are employees of Guardant Health and have stock ownership. PAV has received consulting fees from Gala Therapeutics. All remaining authors have no conflicts of interest.
References
- 1.Paweletz CP, Sacher AG, Raymond CK, Alden RS, O’Connell A, Mach SL, et al. Bias-Corrected Targeted Next-Generation Sequencing for Rapid, Multiplexed Detection of Actionable Alterations in Cell-Free DNA from Advanced Lung Cancer Patients. Clin Cancer Res 2016;22(4):915–22 doi 10.1158/1078-0432.CCR-15-1627-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol 2016. doi 10.1200/JCO.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drilon A, Wang L, Arcila ME, Balasubramanian S, Greenbowe JR, Ross JS, et al. Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin Cancer Res 2015;21(16):3631–9 doi 10.1158/1078-0432.CCR-14-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS, Kuang Y, et al. Acquired METD1228V Mutation and Resistance to MET Inhibition in Lung Cancer. Cancer Discov 2016;6(12):1334–41 doi 10.1158/2159-8290.CD-16-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316(5827):1039–43 doi 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 6.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015;21(6):560–2 doi 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 2011;71(18):6051–60 doi 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatcher JM, Bahcall M, Choi HG, Gao Y, Sim T, George R, et al. Discovery of Inhibitors That Overcome the G1202R Anaplastic Lymphoma Kinase Resistance Mutation. J Med Chem 2015;58(23):9296–308 doi 10.1021/acs.jmedchem.5b01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 2015;372(18):1689–99 doi 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- 10.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009;462(7276):1070–4 doi 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer Discov 2015;5(9):960–71 doi 10.1158/2159-8290.CD-15-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50 doi 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 2015;5(8):850–9 doi 10.1158/2159-8290.CD-15-0285. [DOI] [PubMed] [Google Scholar]
- 14.Paik PK, Drilon A, Fan PD, Yu H, Rekhtman N, Ginsberg MS, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5(8):842–9 doi 10.1158/2159-8290.CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol 2016;34(7):721–30 doi 10.1200/JCO.2015.63.4600. [DOI] [PubMed] [Google Scholar]
- 16.Drilon A MET Exon 14 Alterations in Lung Cancer: Exon Skipping Extends Half-Life. Clin Cancer Res 2016;22(12):2832–4 doi 10.1158/1078-0432.CCR-16-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heist RS, Sequist LV, Borger D, Gainor JF, Arellano RS, Le LP, et al. Acquired Resistance to Crizotinib in NSCLC with MET Exon 14 Skipping. J Thorac Oncol 2016;11(8):1242–5 doi 10.1016/j.jtho.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 18.Ou SI, Young L, Schrock AB, Johnson A, Klempner SJ, Zhu VW, et al. Emergence of Preexisting MET Y1230C Mutation as a Resistance Mechanism to Crizotinib in NSCLC with MET Exon 14 Skipping. J Thorac Oncol 2017;12(1):137–40 doi 10.1016/j.jtho.2016.09.119. [DOI] [PubMed] [Google Scholar]
- 19.Dong HJ, Li P, Wu CL, Zhou XY, Lu HJ, Zhou T. Response and acquired resistance to crizotinib in Chinese patients with lung adenocarcinomas harboring MET Exon 14 splicing alternations. Lung Cancer 2016;102:118–21 doi 10.1016/j.lungcan.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Ou SH. Crizotinib: a novel and first-in-class multitargeted tyrosine kinase inhibitor for the treatment of anaplastic lymphoma kinase rearranged non-small cell lung cancer and beyond. Drug Des Devel Ther 2011;5:471–85 doi 10.2147/DDDT.S19045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res 2006;66(1):283–9 doi 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- 22.Asaoka Y, Tada M, Ikenoue T, Seto M, Imai M, Miyabayashi K, et al. Gastric cancer cell line Hs746T harbors a splice site mutation of c-Met causing juxtamembrane domain deletion. Biochem Biophys Res Commun 2010;394(4):1042–6 doi 10.1016/j.bbrc.2010.03.120. [DOI] [PubMed] [Google Scholar]
- 23.Park CH, Cho SY, Ha JD, Jung H, Kim HR, Lee CO, et al. Novel c-Met inhibitor suppresses the growth of c-Met-addicted gastric cancer cells. BMC Cancer 2016;16:35 doi 10.1186/s12885-016-2058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007;129(5):957–68 doi 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 25.Reznik TE, Sang Y, Ma Y, Abounader R, Rosen EM, Xia S, et al. Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor. Mol Cancer Res 2008;6(1):139–50 doi 10.1158/1541-7786.MCR-07-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev 2001;15(8):981–94 doi 10.1101/gad.191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldys A, Gooz M, Morinelli TA, Lee MH, Raymond JR Jr., Luttrell LM, et al. Essential role of c-Cbl in amphiregulin-induced recycling and signaling of the endogenous epidermal growth factor receptor. Biochemistry 2009;48(7):1462–73 doi 10.1021/bi801771g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stern KA, Place TL, Lill NL. EGF and amphiregulin differentially regulate Cbl recruitment to endosomes and EGF receptor fate. Biochem J 2008;410(3):585–94 doi 10.1042/BJ20071505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010;304(16):1812–20 doi 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 30.Valtorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer 2013;133(5):1259–65 doi 10.1002/ijc.28106. [DOI] [PubMed] [Google Scholar]
- 31.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012;486(7404):532–6 doi 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cepero V, Sierra JR, Corso S, Ghiso E, Casorzo L, Perera T, et al. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res 2010;70(19):7580–90 doi 10.1158/0008-5472.CAN-10-0436. [DOI] [PubMed] [Google Scholar]
- 33.Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med 2015;21(9):1038–47 doi 10.1038/nm.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volinsky N, Kholodenko BN. Complexity of receptor tyrosine kinase signal processing. Cold Spring Harb Perspect Biol 2013;5(8):a009043 doi 10.1101/cshperspect.a009043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarthy SA, Samuels ML, Pritchard CA, Abraham JA, McMahon M. Rapid induction of heparin-binding epidermal growth factor/diphtheria toxin receptor expression by Raf and Ras oncogenes. Genes Dev 1995;9(16):1953–64. [DOI] [PubMed] [Google Scholar]
- 36.Gangarosa LM, Sizemore N, Graves-Deal R, Oldham SM, Der CJ, Coffey RJ. A raf-independent epidermal growth factor receptor autocrine loop is necessary for Ras transformation of rat intestinal epithelial cells. J Biol Chem 1997;272(30):18926–31. [DOI] [PubMed] [Google Scholar]
- 37.Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res 2013;19(24):6751–65 doi 10.1158/1078-0432.CCR-13-0423. [DOI] [PubMed] [Google Scholar]
- 38.Hobor S, Van Emburgh BO, Crowley E, Misale S, Di Nicolantonio F, Bardelli A. TGFalpha and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin Cancer Res 2014;20(24):6429–38 doi 10.1158/1078-0432.CCR-14-0774. [DOI] [PubMed] [Google Scholar]
- 39.Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem 2000;275(12):8806–11. [DOI] [PubMed] [Google Scholar]
- 40.Breindel JL, Haskins JW, Cowell EP, Zhao M, Nguyen DX, Stern DF. EGF receptor activates MET through MAPK to enhance non-small cell lung carcinoma invasion and brain metastasis. Cancer Res 2013;73(16):5053–65 doi 10.1158/0008-5472.CAN-12-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai AZ, Abella JV, Park M. Crosstalk in Met receptor oncogenesis. Trends Cell Biol 2009;19(10):542–51 doi 10.1016/j.tcb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 42.Shepard HM, Brdlik CM, Schreiber H. Signal integration: a framework for understanding the efficacy of therapeutics targeting the human EGFR family. J Clin Invest 2008;118:3574–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res 2006;66(20):10100–11 doi 10.1158/0008-5472.CAN-06-1684. [DOI] [PubMed] [Google Scholar]
- 44.Will M, Qin AC, Toy W, Yao Z, Rodrik-Outmezguine V, Schneider C, et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov 2014;4(3):334–47 doi 10.1158/2159-8290.CD-13-0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One 2015;10(10):e0140712 doi 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calles A, Kwiatkowski N, Cammarata BK, Ercan D, Gray NS, Janne PA. Tivantinib (ARQ 197) efficacy is independent of MET inhibition in non-small-cell lung cancer cell lines. Mol Oncol 2015;9(1):260–9 doi 10.1016/j.molonc.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A, et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010;29(16):2346–56 doi onc2009526[pii]10.1038/onc.2009.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dickerson WM, Saab A, Leong K, Miller M, Latterich M, Beausang LA, et al. Measurement of downstream kinase activity modulation as indicator of epidermal growth factor receptor inhibitor efficacy. Anal Biochem 2014;448:65–7 doi 10.1016/j.ab.2013.11.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.