

Abstract
Cancer resistance to PI3K inhibitor therapy can be in part mediated by increases in the PIM1 kinase. However, the exact mechanism by which PIM kinase promotes tumor cell resistance is unknown. Our study unveils the pivotal control of redox signaling by PIM kinases as a driver of this resistance mechanism. PIM1 kinase functions to decrease cellular ROS levels by enhancing NRF2/ARE activity. PIM prevents cell death induced by PI3K-AKT inhibitory drugs through a non-canonical mechanism of NRF2 ubiquitination and degradation and translational control of NRF2 protein levels through modulation of eIF4B and mTORC1 activity. Importantly, PIM also controls NAD(P)H production by increasing glucose flux through the pentose phosphate shunt decreasing ROS production, and thereby diminishing the cytotoxicity of PI3K-AKT inhibitors. Treatment with PIM kinase inhibitors reverses this resistance phenotype making tumors increasingly susceptible to small molecule therapeutics which block the PI3K-AKT pathway.
Keywords: PIM kinase, PI3K, NRF2, oxidative stress, redox metabolism
INTRODUCTION
Intrinsic and acquired resistance to PI3K inhibitors has been poorly understood, recent studies in breast cancer identified the PIM (Proviral Integration site for Moloney murine leukemic virus) protein kinase as additional therapeutic targets that sensitizes cancer cells to anti-cancer therapeutics that inhibit this pathway (1). These findings are highly relevant to prostate cancer where PIM is highly expressed and aberrations in the phosphatidyl inositol-3 kinase (PI3K) pathway and deletion of PTEN are detected in nearly 70% of patients with metastatic castration-resistant prostate cancer (mCRPC) (2). The prevalence of PI3K pathway activation in mCRPC cells makes this pathway an attractive target to inhibit tumor growth. However, in clinical trials PI3K inhibitor therapy induced a partial response in only 12% of patients whose tumors harbor mutations in PTEN or PIK3C isoforms (3), suggesting a possible role for PIM kinase in blocking this pathway.
PIM kinases, a family comprised of PIM1, PIM2, and PIM3, are oncogenic serine/threonine kinases that are overexpressed in multiple malignancies (4,5) that promote tumor progression by regulating cell cycle progression, proliferation, and survival (6). PIM1 expression is elevated in ~50% of human prostate cancer specimens, particularly in high Gleason grade and aggressive metastatic prostate cancer cases (5,7). These protein kinases share significant mechanistic similarities to the AKT family which might account for their ability to act as part of a resistance mechanism. For example, both PIM and AKT families are known to regulate protein translation by affecting activity of mTORC1, a nutrient/energy/redox sensor and regulator of protein synthesis, through an ability to modify identical proteins. PIM and AKT overlap in substrate modification; PIM phosphorylates TSC2, PRAS40, and eIF4B on serine (S) 406 (8).
Activation of the AKT pathway produces reactive oxygen species (ROS). Because excessive oxidative stress can lead to senescence or cell death, proliferating cancer cells limit cellular ROS levels by inducing an array of antioxidants and ROS scavengers (9). AKT-stimulated mitogenic and survival programs (10) control ROS levels through nuclear factor erythroid 2-related factor 2 (NRF2), a basic leucine zipper transcription factor, that binds to anti-oxidant response elements (AREs) to activate a battery of cytoprotective and anti-oxidant gene targets including heme oxygenase (HMOX1), NAD(P)H quinone dehydrogenase (NQO1), glutamate-cysteine ligases (GCLC and GCLM; the first rate-limiting enzymes of glutathione synthesis), glutathione peroxidases (GPX), and superoxide dismutases (SOD) (11). The activated NRF2, in addition to cytoprotective function, also promotes metabolic reprogramming by activating metabolic genes (12). Recently, it was reported that NRF2 promotes epidermal growth factor receptor (EGFR) autocrine signaling to facilitate eIF4F complex formation for cap-dependent translation initiation in KRAS mutant pancreatic adenocarcinomas (13). The activated NRF2 promotes the further activation of the PI3K-AKT pathway to accelerate aggressive cancer progression. Thus, therapeutic agents targeting NRF2 signaling may render cellular vulnerability to PI3K-AKT inhibitory drugs.
Previously, we reported that PIM kinase mediates an important adaptive survival response upon inhibition of AKT through feedback activation of receptor tyrosine kinase (RTK) signaling, leading to cancer cell resistance (14). Recently, a genome scale shRNA screening identified the PIM kinase as conferring resistance to the PI3K inhibitor alpelisib (BYL719) (1). Concurrent treatment with a PIM inhibitor and PI3K inhibitor overcame this resistance. Moreover, PIM1 was identified as a pivotal therapeutic target in triple negative breast cancer (15,16), inhibiting PIM sensitized these cells to chemotherapy.
In this study, we have defined the potential mechanism underlying the PIM1-mediated inhibition of PI3K-AKT inhibitor in both tissue culture and mouse models harboring PI3K-AKT pathway aberrations.
MATERIALS AND METHODS
Chemicals
PIM447 (17) was provided by Novartis Oncology. Buparlisib (BKM120) (18), AZD1208 (19), AZD5363 (20), and Torin1 (21) were purchased from AdooQ Bioscience. CM-H2DCF-DA (22) was from Molecular Probes (Thermo Scientific). All other chemicals were purchased from Sigma-Aldrich.
Cell Culture, Stable Cell Lines, Transfection, and CRISPR-Cas9
The human prostate cancer cell lines LNCaP, PC-3, PC3-LN4 were cultured as previously described (23,24). Mouse prostate epithelial cells were grown in DMEM medium supplemented with 2.5% charcoal stripped FCS, 5 μg/mL of insulin/transferring/selenium, 10 μg/mL of bovine pituitary extract, 10 μg/mL of epidermal growth factor, 1 μg/mL of cholera toxin as described previously (25). All cell lines were maintained at 37 °C in 5 % CO2 and were authenticated by short tandem repeat DNA profiling performed by the University of Arizona
Genetics Core Facility. The cell lines were routinely tested for mycoplasma and used for fewer than 50 passages. Cells were transfected with RNAi or transduced with lentiviruses as previously described (23,26). eIF4B sgRNA CRISPR-Cas9 system was purchased from ABM Inc. Lentiviruses were produced in 293T cells co-expressing the packaging vectors (psPAX2 and VSV-G), concentrated by ultracentrifugation (20,000 g for 2 hr at 4 °C), and resuspended with culture media for an infection (48 hr). Alt-R® CRISPR-Cas9 System are used for genome editing of the eIF4B Serine 406 site. A detailed description is provided in the Supplementary methods.
Immunoblotting, Immunofluorescence and Immunohistochemistry
For immunoblotting, total lysates were prepared, resolved by SDS-PAGE, and transferred to immobilon membranes. The following primary antibodies were utilized from the indicated vendors: PIM1, p-IRS1 (S1101), γ-H2AX (S139), p-AKT (S473), AKT, p-p70S6K (T389), p70S6K, and pS6 (S240/244) were from Cell Signaling Technology; S6, NRF2, HMOX1, GCLC, GCLM, and PRDX3 were from Santa Cruz Biotechnology; SOD2 was from Abcam; ACTIN, FLAG, HA and ubiquitin antibodies were purchased from Sigma. Immunohistochemistry and immunofluorescence staining were performed as described in detail in the Supplementary methods. Staining with H2DCF-DA followed by flow cytometry was as previously described (26).
Metabolic Analysis
Lactate dehydrogenase, NADPH and GSH production was assessed using an Amplite™ Colorimetric L-Lactate Dehydrogenase (LDH) Assay Kit (AAT Bioquest), bioluminescent cell-based NAD(P)/NAD(P)H assay, and GSH-Glo kits (Promega), respectively, and were carried out as per the manufacturer’s instructions. Measurement of oxygen consumption rate and extracellular acidification were performed using a Seahorse Bioscience XF24 Extracellular Flux Analyzer. Following exposure of LNCaP to [U-13C] glucose, carbon flux through metabolic pathways were examined at the University of Michigan Regional Comprehensive Metabolomics Resource Core (http://mrc2.umich.edu).
Animals
For xenograft studies, male SCID mice (8 weeks old) were used by approval of the Institutional Animal Care and Use Committee at the University of Arizona and described in detail in the Supplementary methods.
Prostate Cancer Patient-derived Organoids
Prostate cancer biopsies were provided by University of Arizona Cancer Center Tissues Acquisition and Cellular/Molecular Analysis Shared Resources and the study was conducted under University of Arizona Institutional Review Board approval. Prostate cancer patient-derived organoids were cultured based on previously established procedures (27,28) and are described in detail in the Supplementary methods.
Statistics
Statistical significance was determined, when appropriate, using unpaired, two-tailed Student’s t test. P-values of less than 0.05 were considered statistically significant.
RESULTS
PIM kinase and PI3K-AKT inhibitors synergize to suppress tumor growth.
Proliferation of human prostate cancer (PCa) cells, e.g., LNCaP and PC-3 lines that express constitutively active AKT, can be inhibited by the pan-PI3K inhibitor buparlisib. However, when PIM1 is overexpressed, these cells become highly resistant to this inhibitor (Figure 1A, B). In PIM1-overexpressing LNCaP prostate cancer cells (which contain a highly activated AKT pathway secondary to deletion of PTEN), simultaneously inhibiting both PI3K and the PIM kinase enhanced growth inhibition (Figure 1C). PC3-LN4 cells are a highly metastatic variant of PC-3 cells (29,30) that have increased PIM transcript levels compared to other PCa cell lines (Supplementary Fig. S1A), and are relatively resistant to buparlisib (Figure 1D, E). Synergistic inhibition of survival and growth was demonstrated when PC3-LN4 cells were treated with both PIM and PI3K/AKT inhibitors, PIM447 and buparlisib, respectively (Supplementary Fig. S1B). This growth inhibition effect is not limited to these agents, as the AKT inhibitor AZD5363 and PIM inhibitor AZD1208 also abrogated proliferation of these PCa cells (Supplementary Fig. S1C-E). To determine whether these effects were observed in vivo, mice were xenografted with PC3-LN4 tumors and then treated with buparlisib (10 mg/kg) with or without PIM447 (30 mg/kg) for 24 days. The combination treatment significantly inhibited in vivo growth, indicated by a decrease in tumor volume and weight compared with animals treated with either buparlisib, PIM447, or vehicle control (Figure 1F, G). The combination treatment significantly reduced tumor cell proliferation compared to either agent alone (Supplementary Fig. S1F, S1G), evidenced by the marked decrease in Ki67 staining. Similar results were obtained using the AKT inhibitor AZD5363 (40 mg/kg) in place of buparlisib (Figure 1H, I). These results suggest that tumor cell resistance to PI3K/AKT inhibitors was mediated, at least in part, by the PIM kinases.
Figure 1. PIM inhibition overcomes resistance to PI3K-AKT inhibitors.
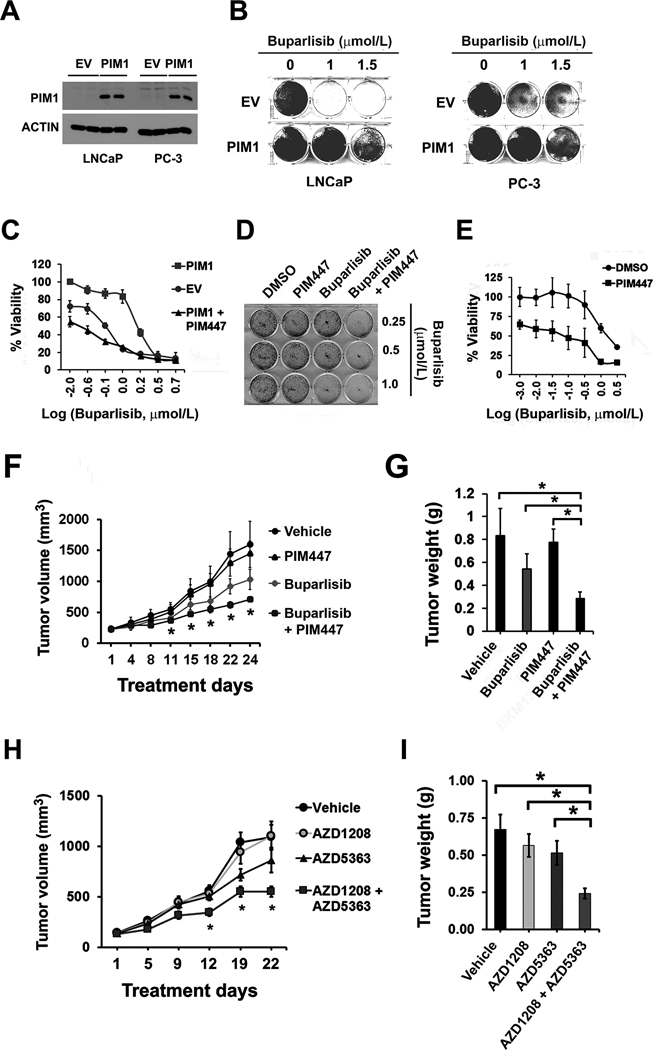
(A) Overexpression of PIM1 in LNCaP and PC-3 cells using lentivirus. Empty vector (EV) was used as a control.
(B) Representative crystal violet staining. EV and PIM1 expressing LNCaP and PC-3 cells were treated with buparlisib for 72 hr at the doses indicated.
(C) Dose-response analysis of LNCaP/PIM1 versus LNCaP/EV. LNCaP/PIM1 cells treated with 3 μmol/L PIM447 and were simultaneously exposed to varying doses of buparlisib for 72 hr. The data shown is the mean of measurements ± the standard deviation (SD, n=4). IC50 of buparlisib (μmol/L) was 0.80 for LNCaP/EV, 1.75 for LNCaP/PIM1, and 0.39 for LNCaP/PIM1 cells co-treated with PIM447.
(D) Colony focus formation visualized by crystal violet staining. Representative images are shown. PC3-LN4 cells (100 cells) were seeded and then incubated in the absence or presence of 3 μmol/L PIM447 for 7 days along with buparlisib at the doses indicated.
(E) Dose-response analysis of PC3-LN4 cells exposed to buparlisib for 72 hr in the absence or presence of 3 μmol/L PIM447. The data shown is the mean of measurements ± SD (n=4). IC50 of buparlisib (μmol/L) was 1.25 for DMSO and 0.63 for PIM447,
(F) PC3-LN4 xenografts treated with buparlisib, PIM447, or the combination. The average tumor volume ± SEM (n=5) are plotted, and the statistical comparison versus vehicle-treated control is shown using a t test (*, P<0.05).
(G) The results of tumor treatment with single or dual kinase inhibitors. The average tumor weight ± the SEM (*, P<0.05).
(H) PC3-LN4 xenografts treated with AZD5363 (40 mg/kg), AZD1208 (30 mg/kg), or the combination. The average tumor volume ± SEM (n=7) are plotted, and the statistical comparison with vehicle treated control is shown using a t test (*, P<0.05).
(I) The tumors were excised from the mice described in (H) at the end of treatment. The average weight is shown (*, P<0.05).
PIM1 kinase induces tumor cell resistance to inhibitors of PI3K/AKT signaling by increasing NRF2 levels and stimulating the production of ROS scavengers.
Small molecule PIM inhibitors inactivate the NRF2 transcription factor, reducing expression of cytoprotective genes and leading to the buildup of ROS in hypoxia (31). To understand the biochemical mechanism by which PIM kinases cause resistance to inhibitors of PI3K/AKT, the relationship between NRF2 signaling and PIM1 kinase was explored. To minimize genetic alterations affecting NRF2 regulation, we utilized mouse epithelial prostate cancer (mPrEC) cells derived from spontaneous prostate adenocarcinomas that arose in a mouse model Trp53loxP/loxP;Rb1loxP/loxP; PbCre4 (Supplementary Fig. S2A). When PIM1 was expressed in cells from these mice (mPrEC/PIM1), these cells established larger colonies in soft agar (Supplementary Fig. S2B) and had elevated levels of NRF2-ARE genes including HMOX1, NQO1, SOD2, and SOD3 (Supplementary Fig. S2C; GEO repository accession number, GSE118786). Because activated AKT was detected in mPrEC/PIM1 cells, these cells were treated with buparlisib together with PIM447. Treatment blocked AKT and PIM activity, judged by inhibition of phospho-AKT (S473) and phospho-IRS1 (S1101) expression respectively, which resulted in inhibition of mTORC1 and NRF2 activity (Figure 2A); the effects of combination treatment further resulted in DNA damage as measured by the marked induction of γ-H2AX expression. Sensitivity of mPrEC cells to buparlisib was markedly enhanced by co-treatment with PIM447 (Figure 2B). These results suggest that in prostate cell lines with few mutations, combining PI3K and PIM inhibitors may be useful in blocking tumor growth. To test activity of combination therapy to kill prostate cancer from primary patient samples, the laboratory has used organoids cultured from patient biopsies of the prostate obtained from prostatectomy specimens. These patient organoids had activated AKT and increased PSA levels (Figure 2C; F Supplementary Fig. S2 D, E). qPCR analysis confirmed the presence of PIM1, 2 and 3 transcripts in these organoids (Supplementary Fig. S2F). These organoids had detectible levels of the PIM substrate, phopho-IRS1 (S1101), indicative of PIM kinase activity (23). Organoid data resembled cell culture and animal models as combination treatment of buparlisib and PIM447 resulted in a significant reduction in organoid number and viability (Figure 2D).
Figure 2. PIM1 upregulates the NRF2-HMOX1 antioxidant response to contribute to PI3K inhibitor resistance.
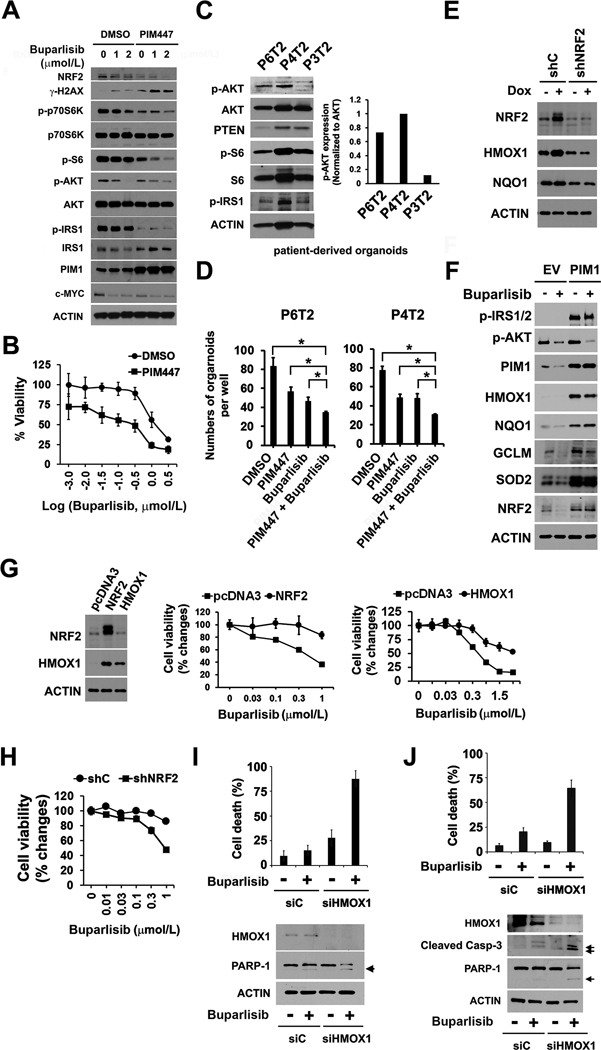
(A) Representative dose-response of mouse prostate epithelial cancer cells overexpressing PIM1 (mPrEC/PIM1) to buparlisib, PIM447 (3 μmol/L) or the combination (48 hr treatment). The data shown is the mean of measurements ± SD (n=4). IC50 of buparlisib (μmol/L) was 1.18 for DMSO and 0.35 for PIM447.
(B) Following 24 hr treatment with either buparlisib (varying doses), PIM447 (3 μmol/L) or the combination, mPrEC/PIM1 cells were lysed and immunoblotted with the indicated antibodies.
(C) Immunoblot analysis of AKT and PIM activation in prostate cancer patient-derived organoids. Total lysates of cell organoids from patients (identification number P6T2, P4T2 and P3T2) were subjected to detection of p-AKT (S473), p-S6 (S240/244), and p-IRS1 (S1101). p-AKT/AKT ratio was calculated from densitometric analysis.
(D) Organoid cells (5,000 cell density) derived from patient tissues (P6T2 and P4T2) were plated in matrigel in a 96-well plates and then treated with DMSO, 3 μmol/L PIM447, 1 μmol/L buparlisib or the combination of PIM447 plus buparlisib. Organoid number was calculated on day 7 of respective drug treatment. The data shown is the mean of measurements ± SD (n=3). *, P<0.05.
(E) Immunoblot analysis of the NRF2 and its target proteins in the Dox-inducible PIM1 LNCaP model. These cells express shRNA scrambled control (shC) or shNRF2 are treated with 20 ng/mL Dox (+ Dox) or PBS (–).
(F) Immunoblot analysis of the indicated proteins in stable PIM1-overexpressing LNCaP and control vector (EV) expressing cells exposed to 1 μmol/L buparlisib for 6 hr.
(G) Representative buparlisib dose-response curves in NRF2- or HMOX1-overexpressing LNCaP cells. The data shown is the mean of measurements ± SD (n=4). Immunoblots confirming NRF2 and HMOX1 expression compared to pcDNA3 control vector are shown.
(H) Representative buparlisib dose-responses of LNCaP/PIM1 cells expressing shNRF2, as compared to the control shC. The data shown is the mean of measurements ± SD (n=4).
(I) Immunoblots of HMOX1-depleted LNCaP/PIM1 cells exposed to 1 μmol/L buparlisib. Cell death was measured by trypan blue uptake after treatment with DMSO or buparlisib. The arrow indicates the cleavage product of PARP-1. The average number of dead cells ± SD is shown (n=3).
(J) Immunoblots of HMOX1-depleted PC3-LN4 cells exposed to 1 μmol/L buparlisib. The cleavage fragments of caspase-3 and PARP-1 are identified by arrows. Cell death is measured by trypan blue uptake in the cells treated with buparlisib or DMSO. The average number of dead cells ± SD is shown (n=3).
To examine whether NRF2 signaling induced by PIM1 kinase plays a role in resistance of human prostate cancer cell to PI3K inhibitor, a Doxycycline (Dox)-inducible vector was used to induce PIM1 overexpression in LNCaP cells; NRF2 protein expression was markedly increased (Supplementary Fig. S2G) without significant increase of NRF2 mRNA. This increase in NRF2 protein induced multiple downstream targets of NRF2, including ROS scavengers (HMOX1 and NQO1), an enzyme needed for glutathione (GSH) synthesis (GCLM), and enzymes that modulate cellular NADPH levels (i.e., ME1, IDH1 and G6PD) (Supplementary Fig. S2H). To demonstrate that the increase in ROS scavengers was secondary to PIM1-mediated NRF2 induction, and not a direct effect of elevating the PIM kinase, NRF2-targeted shRNA was expressed in the human prostate cancer cells containing Dox-inducible PIM1. PIM1-mediated induction of HMOX1 and NQO1 expression was abrogated by depletion of NRF2 (Figure 2E; Supplementary Fig. S2I). Treatment of LNCaP cells with buparlisib, a pan PI3K inhibitor, blocks AKT phosphorylation and markedly decreases NRF2 levels, leading to reduction in ROS scavengers, NQO1, HMOX1, SOD2, and in the GCLM enzyme. In contrast, when the expression of PIM1 was increased even in the presence of buparlisib treatment and blocked AKT phosphorylation, there was no change in the ROS scavenger protein levels (Figure 2F). Over a broad range of doses, buparlisib failed to downregulate HMOX1 in PIM1-overexpressing PCa cells (Supplementary Fig. S2J).
Modulation of ROS scavengers is important for buparlisib-mediated LNCaP tumor cell killing as evidenced by the fact that forced NRF2 or HMOX1 expression conferred buparlisib resistance (Figure 2G). Conversely, depletion of NRF2 in the resistant PIM1 PCa lines rendered these cells susceptible to buparlisib-induced killing (Figure 2H). Depletion of HMOX1 expression in the PIM1-overexpressing cells also restored cell sensitivity to buparlisib, as evidenced by cleavage of PARP-1, a marker of apoptosis, and markedly increased cell death (Figure 2I). In PC3-LN4 cells, siRNA-mediated depletion of HMOX1 markedly increased buparlisib-induced cell death as characterized by caspase-3 and PARP-1 cleavage (Figure 2J). Therefore, PIM1 prevents small molecule PI3K inhibitor-induced cell death by regulating the levels of NRF2 and ROS scavenger proteins.
Redox regulation of glutathione is a critical factor in PIM-mediated resistance to PI3K-AKT inhibitor.
Luminescence-based assays demonstrate that increased expression of PIM1 blocks the ability of buparlisib treatment to decrease the cellular levels of both NADPH and GSH (Figure 3A). To further examine the role of ROS in the regulation of cell death induced by PI3K inhibitors, LNCaP/PIM1 were treated with buthionine sulfoximine (BSO), an inhibitor of GSH synthesis. PIM1 transduction made these cells less sensitive to BSO killing (Figure 3B), in comparison to control cells (LNCaP expressing empty vector, LNCaP/EV). To test whether the addition of BSO significantly suppressed in vivo tumor growth, LNCaP/PIM1 cells were grown subcutaneously in immunocompromised mice, and animals were treated with BSO (400 mg/kg), buparlisib, or the combination. Combination treatment with these agents inhibited tumor growth significantly better than either agent alone (Figure 3C). To document ROS stimulation by these agents, prostate cells were stained with H2DCF-DA. In LNCaP/EV, buparlisib increased ROS accumulation, which was blocked by the addition of either the ROS scavenger N-acetylcysteine (NAC) or Trolox (Figure 3D). In contrast, buparlisib alone did not increase ROS accumulation in LNCaP/PIM1 (Figure 3E). Combined BSO/buparlisib treatment of these cells produced robust ROS accumulation, paralleling the results of animal experiments. These data indicate that increased expression of PIM1 induces resistance to buparlisib in part by stimulating GSH production and suppressing ROS accumulation.
Figure 3. Inhibition of glutathione synthesis abrogates resistance to PI3K inhibitor.
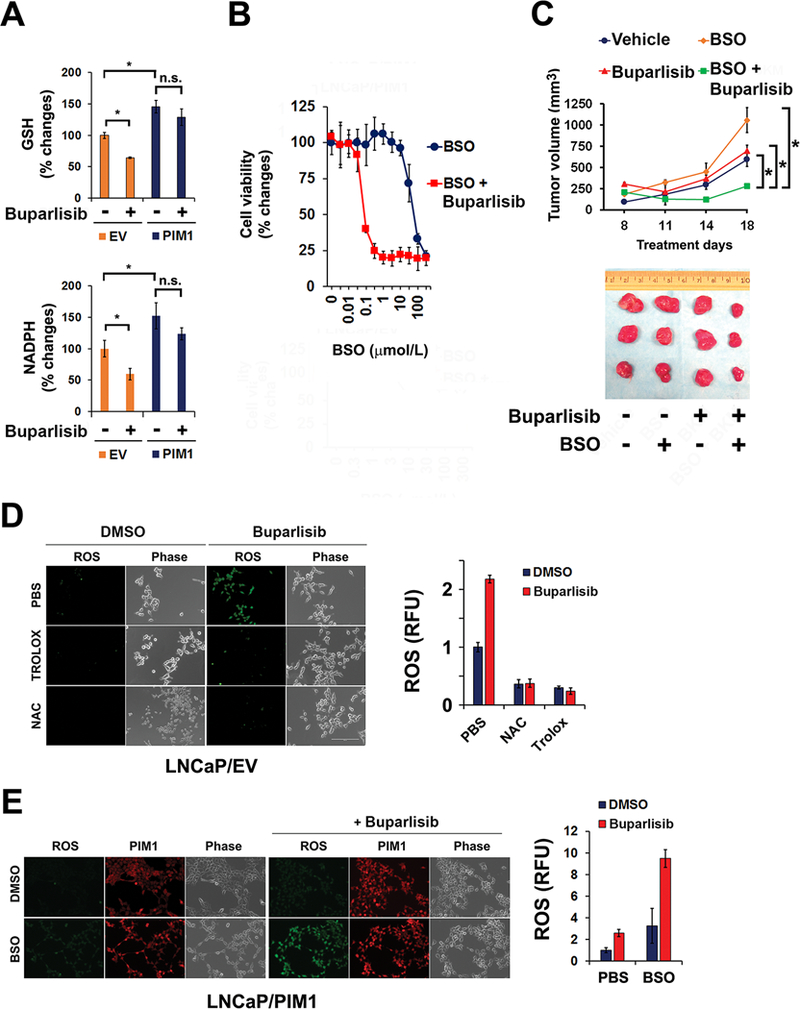
(A) The cellular levels of GSH and NADPH after treatment with buparlisib. EV and PIM1 expressing LNCaP cells were exposed to 1 μmol/L buparlisib for 48 hr. The data shown is the mean of measurements ± SD (n=4). *, P<0.05; n.s., not significant.
(B) Impact of buthionine sulfoxide (BSO) pretreatment on the sensitivity of LNCaP/PIM1 versus LNCaP/EV cells to 1 μmol/L buparlisib (72 hr). The data shown is the mean of measurements ± SD (n=4).
(C) Tumor volume of implanted LNCaP/PIM1 cells treated with the indicated agents for varying periods of time. The average volume ± SEM (n=6), and statistical comparison to the vehicle treated control is shown (*, P<0.05). The tumors shown were excised from the mice at the end of treatment.
(D) Fluorescence imaging of ROS in LNCaP/EV cells treated with buparlisib for 16 hr in presence of PBS, Trolox (1 mmol/L) or NAC (3 mmol/L) (scale bar, 20 μm). Quantitation of DCF green intensity following treatment with 1 μmol/L buparlisib. The average of these values ± the SD is shown.
(E) Fluorescence imaging of ROS in LNCaP/PIM1 cells treated with 1 μmol/L buparlisib for 16 hr in presence or absence of BSO (scale bar, 20 μm). The average value of ROS determined by DCF green florescence intensity and SD of these measurements is shown.
Given the ability of PIM1 to regulate NRF2 levels, potentially controlling the activity of multiple genes which modulate energy production, the effect of PIM1 overexpression on the metabolism of PCa cells was studied. LNCaP/EV or LNCaP/PIM1 cells were labeled with 13C-glucose and this labelling was chased at 0, 1 and 3 hours. LNCaP/PIM1 had increased carbon flux through both glycolysis and the pentose phosphate shunt. Specifically, there was an increased rate of labelling of glucose 6-phosphate (G6P)/fructose 6-phosphate (F6P) (combined), phosphoenol pyruvate (PEP), 2-phosphogluconate (2PG)/3-phosphogluconcate (3PG) (combined), 6-phosphogluconate (6PG), ribulose 5-phosphate (R5P)/xylulose 5-phosphate (X5P) (combined), and sedoheptulose-7-phosphate (S7P) (Supplementary Fig. S3). This increased flux enhanced the mass isotope abundance of NAD(P)H and NADP+ (Figure 4A). As predicted, 13C abundance in GSH was increased (Figure 4B). The Seahorse assay was then used to assess oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). Consistent with the increased flux through the glycolytic pathway, decreased OCR and increased ECAR was evident in LNCaP/PIM1 (Figure 4C). Moreover, induction of PIM1 resulted in restoration of L-lactate dehydrogenase (LDH) activity (which was decreased by buparlisib (Figure 4D). Change in mitochondrial energetics (i.e., OCR) was inhibited by buparlisib treatment but was maintained in LNCaP/PIM1 cells (Figure 4E). Thus, increased PIM1 appears to stimulate glucose carbon flux, increasing GSH synthesis, potentially through the pentose phosphate pathway, and NAD(P)H production.
Figure 4. PIM1 overexpression alters metabolic activity.
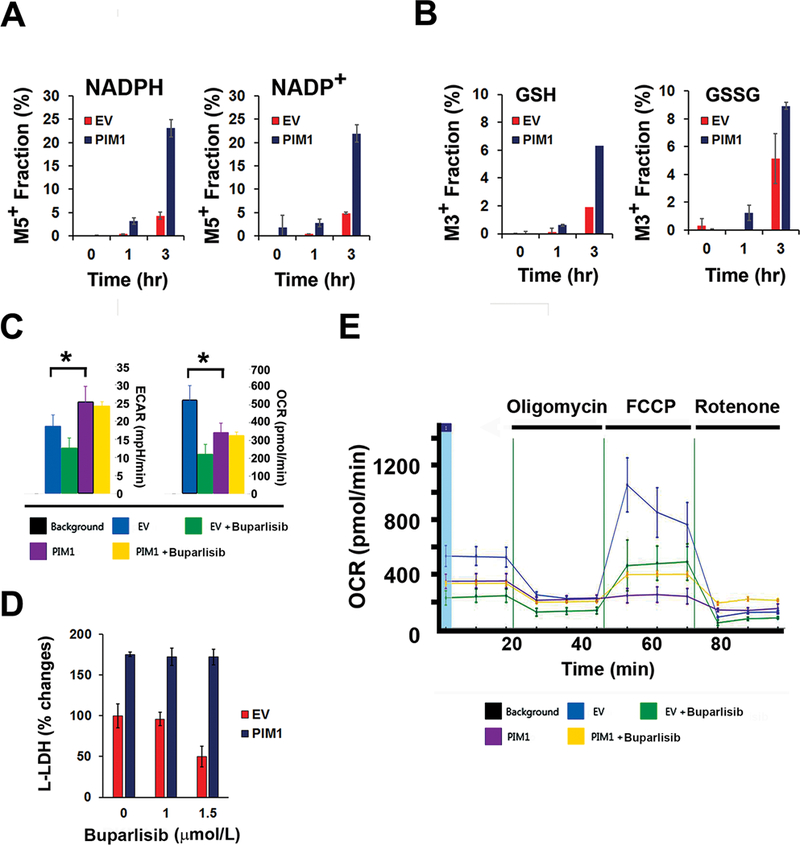
(A) (B) Flux analysis of LNCaP/PIM1 versus LNCaP/EV cells treated with 13C-glucose at the times indicated. The bars demonstrate the relative average abundance ± the SD of differing mass species.
(C) Differences of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of LNCaP/EV and LNCaP/PIM1 cells treated with buparlisib (1 μmol/L) or vehicle (DMSO). The average ± SD is shown (n=5). Statistical comparison to EV control was determined by t test (*, P<0.05).
(D) L-lactate dehydrogenase (LDH) levels in LNCaP/PIM1 or LNCaP/EV treated with varying doses of buparlisib.
(E) Representative OCR data after buparlisib treatment. LNCaP/PIM1 or LNCaP/EV cells were exposed to 1 μmol/L buparlisib or DMSO for 16 hr and then subjected to Seahorse analysis. Oligomycin (2.5 μmol/L), FCCP (2.5 μmol/L), antimycin A (1 μmol/L) and rotenone (1 μmol/L) were sequentially added.
Combined inhibition of PIM and AKT blocks the survival and proliferation of prostate cancer cells by inhibiting NRF2 expression through the regulation of mTORC1 activity.
Both PC-3 and PC3-LN4 cells with aberrant activation of AKT signaling are relatively insensitive to either buparlisib or AZD5363, but buparlisib co-treatment with PIM447 or AZD1208 (two structurally different PIM inhibitors) blocked their proliferation (Supplementary Fig. S4A, S1). Genetic inhibition of the AKT pathway by expression of PTEN in PC-3 cells downregulated NRF2 expression (Figure 5A) and resulted in a mild inhibition of PCa viability. However, this effect was blunted by PIM1 overexpression. Conversely, treatment with PIM447 enhanced PTEN-mediated suppression of PCa viability even in PIM1-overexpressing cells (Figure 5B). Treatment with the combination of PIM447 and buparlisib increased the accumulation of ROS in the PC3-LN4 cells (Figure 5C, D; Supplementary Fig. S4B). Consistent with the induction of ROS, NRF2 protein expression was markedly reduced by combination treatment (Figure 5E). Addition of tert-Butyl hydroperoxide further increased ROS and enhanced the cytotoxicity of buparlisib as well as PIM447 (Supplementary Fig. S4C). In parallel, decreases of cellular GSH and NAD(P)H levels induced by PIM447 were potentiated by buparlisib treatment (Figure 5F). Forced depletion of all three isoforms of PIM kinases with siRNAs directed at PIM1, 2 and 3 resulted in reduced GSH, NADPH, as well as HMOX1 levels (Supplementary Fig. S4 D-F), validating that these changes were secondary to PIM inhibition. The increased toxicity of ROS in these cells was reflected in induction of γ-H2AX, a DNA damage marker (Supplementary Fig. S4G). Thus, the combination of PIM and PI3K/AKT inhibitor killed PCa cells through increasing ROS levels.
Figure 5. NRF2 activity is inhibited by both PIM1 and PI3K/AKT inhibitors via inhibition of the mTORC1 pathway.
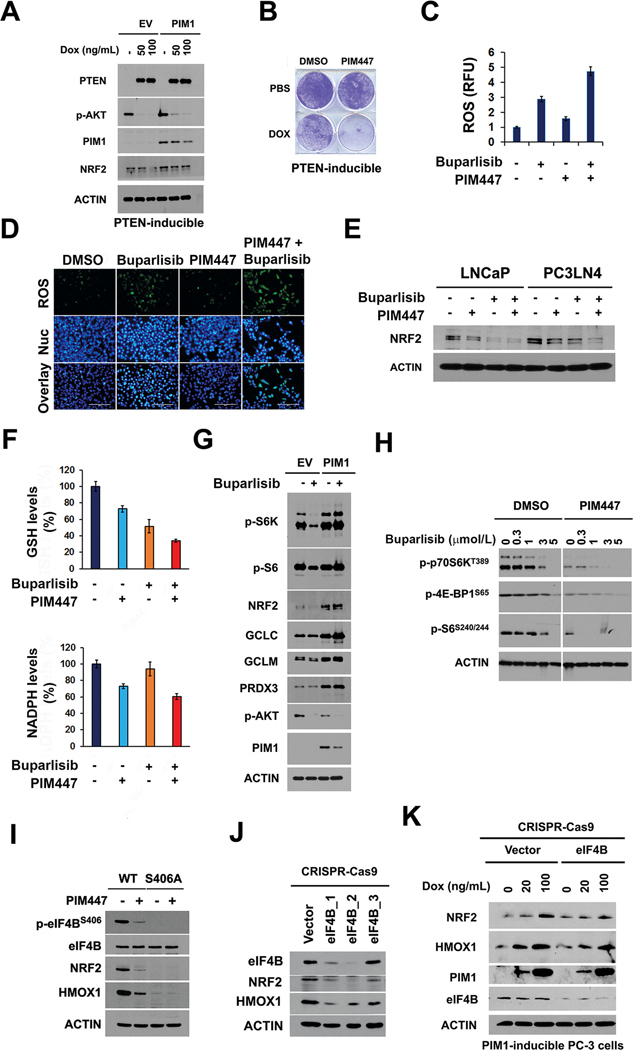
(A) PTEN was induced in PC-3 cells by varying doses of Doxycycline (Dox), and immunoblotting performed for varying proteins using actin as a control.
(B) Representative images of crystal violet-stained PTEN-inducible cells overexpressing PIM1 treated with or without Dox and either DMSO or 3 μmol/L PIM447 for 7 days.
(C) (D) Following treatment of PC3-LN4 cells with buparlisib (1 μmol/L) with or without PIM447 (3 μmol/L), ROS was imaged by quantitation of DCF green intensity (Fig 5C). The average DCF values ± SD are shown. PC3-LN4 cells treated with these inhibitors individually or in combination were labeled with DCF and Hoechst 33342 to measure ROS and document nuclei (Nuc) (Fig. 5D).
(E) Immunoblot analysis of NRF2 expression. LNCaP and PC3-LN4 cells were treated with buparlisib (1 μmol/L), PIM447 (3 μmol/L), or the combination for 24 hr.
(F) Following treatments (E), In PC3-LN4 cells GSH and NADPH levels were measured using the procedures outlined in Materials and Methods.
(G) Immunoblots of LNCaP/EV and LNCaP/PIM1 cells following 24 hr treatment with buparlisib (1 μmol/L) or DMSO.
(H) PC3-LN4 cells were treated with varying does of buparlisib with or without PIM447 for 24 hr, followed by immunoblotting with the antibodies specified.
(I) Immunoblots of wild type or eIF4B S406A mutant PC3-LN4 cells treated with PIM447 (1 μmol/L, 72 hr). PC3-LN4 eIF4B S406A knock-in cells were generated using CRISPR-Cas9 gene editing.
(J) Immunoblots of eIF4B, NRF2 and HMOX1 expression in LNCaP cells after lentiviral transduction of eIF4B CRISPR-Cas9 carrying three different sgRNA target sequences. Vector control was the scrambled sgRNA CRISPR lentivector.
(K) Immunoblots of eIF4B, NRF2 and HMOX1 expression in PIM1-inducible PC-3 (TRIPZ-PIM1) cells transduced with eIF4B CRISPR-Cas9 (carrying sgRNA eIF4B_2) or scrambled sgRNA CRISPR lentivector (Vector). After 72 hr transduction, Doxycycline (Dox) at the indicated dose was added for 48 hr to induce PIM1 overexpression.
While buparlisib treatment of LNCaP cells effectively inhibited mTORC1 activity as judged by phosphorylation of p70S6K, an mTORC1 substrate, PIM1 overexpression abrogated the mTORC1 inhibitory activity of buparlisib (Figure 5G). When PC-3 or PC3-LN4 cells were treated with buparlisib and PIM447, phosphorylation levels of p70S6K, S6 and 4E-BP1 protein were more strongly inhibited compared with buparlisib alone. Together, these data validate that both the AKT/PI3K pathway and PIM control mTORC1 signaling (Figure 5H). In LNCaP cells, AZD5363, buparlisib, or Torin1 (a mTOR kinase inhibitor) all blocked mTORC1 signaling and decreased HMOX1 levels, but in PIM1 overexpressing cells only Torin1 suppressed mTORC1 and HMOX1 expression (Supplementary Fig. S4H). The NQO1-ARE luciferase vector measures the stimulation of transcription by NRF2. When PC3-LN4 cells expressing NQO1-ARE luciferase were treated with Torin1, luciferase activity was inhibited to a similar extent as PIM447 plus buparlisib (Supplementary Fig. S4I). PIM kinase in part regulates mRNA translation by phosphorylating eIF4B on S406 (8). Using CRISPR-Cas9 technology, a knock in of serine to alanine (eIF4B S406A) mutation was produced in human prostate cancer line PC3-LN4. Similar to PIM inhibitor treatment, blocking this eIF4B S406 phosphorylation via a genetic approach markedly inhibited NRF2 and HMOX1 protein expression (Figure 5I). To examine whether eIF4B is needed for PIM-mediated induction of NRF2 signaling, PIM1-inducible PC-3 cells were transduced with lentivirus of CRISPR-Cas9 eIF4B (sgRNA eIF4B_2) and control vector. Compared to the increased expression of NRF2 and HMOX1 upon Doxycycline-stimulated PIM1 induction in control vector expressing cells, increases of HMOX1 and NRF2 was blunted in eIF4B-deficient cells even with Doxycycline stimulation (Figure 5J, K), indicating critical role of eIF4B in PIM1 regulation of NRF2 signaling. Mouse embryonic fibroblasts (MEFs) obtained from mice that are genetically deleted in all PIM kinases (triple knockout; TKO) do not phosphorylate eIF4B. These results suggest that inhibition of mTORC1 by PI3K and PIM inhibitors functions to block NRF2 activation, and thus increase ROS production. It is noteworthy that when PIM triple knockout MEF cells were engineered to express either PIM1, PIM2, or PIM3 (23,26) expression of each PIM isoform was capable of increasing NRF2 expression (Supplementary Fig. S4J), indicating the overlapping effects of these isoforms on NRF2 signaling.
Because mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system (32), we explored whether increased expression of PIM changes NRF2 levels by partially blocking ubiquitination and subsequent proteasome-mediated protein degradation. To test this possibility, LNCaP cells were co-transfected with PIM1, hemagglutinin (HA)-tagged NRF2, and Flag-tagged ubiquitin plasmids. NRF2 protein was immunoprecipitated using anti-HA agarose beads, and ubiquitylated NRF2 was further detected by immunoblot analysis with an anti-Flag antibody. In LNCaP cells co-transfected with PIM1, a decrease in the level of ubiquitylated NRF2 protein was detected (Figure 6A), indicating that PIM1 prevents NRF2 ubiquitination. To further examine the role of PIM1, PCa cells were co-transfected with HA-NRF2 and Flag-Ubiquitin followed by the treatment with PIM447 in presence of MG132, a compound that blocks the degradation of ubiquitylated NRF2. PIM447 treatment induced accumulation of ubiquitinated NRF2 protein (Figure 6B). Reduction of NRF2 protein levels by PIM447 was blocked by the application of MG132 (Figure 6C). MG132 also blocked the decrease in NRF2 induced by genetic deletion of PIM (Supplementary Fig. S4K). The half-life of NRF2 protein was decreased by treatment with PIM447 compared to untreated PC3-LN4 control cells from 32.8 min to 21.9 min (Figure 6D). This demonstrates the potential ability of PIM kinases to maintain NRF2 protein stability and its function by controlling both the translation and degradation pathways that regulate the levels of NRF2. Therefore, PIM and PI3K-AKT inhibitors synergize to block NRF2 activity, augmenting oxidative stress to suppress tumor growth (Figure 6E).
Figure 6. NRF2 ubiquitination and protein stability is regulated by PIM1 kinase.
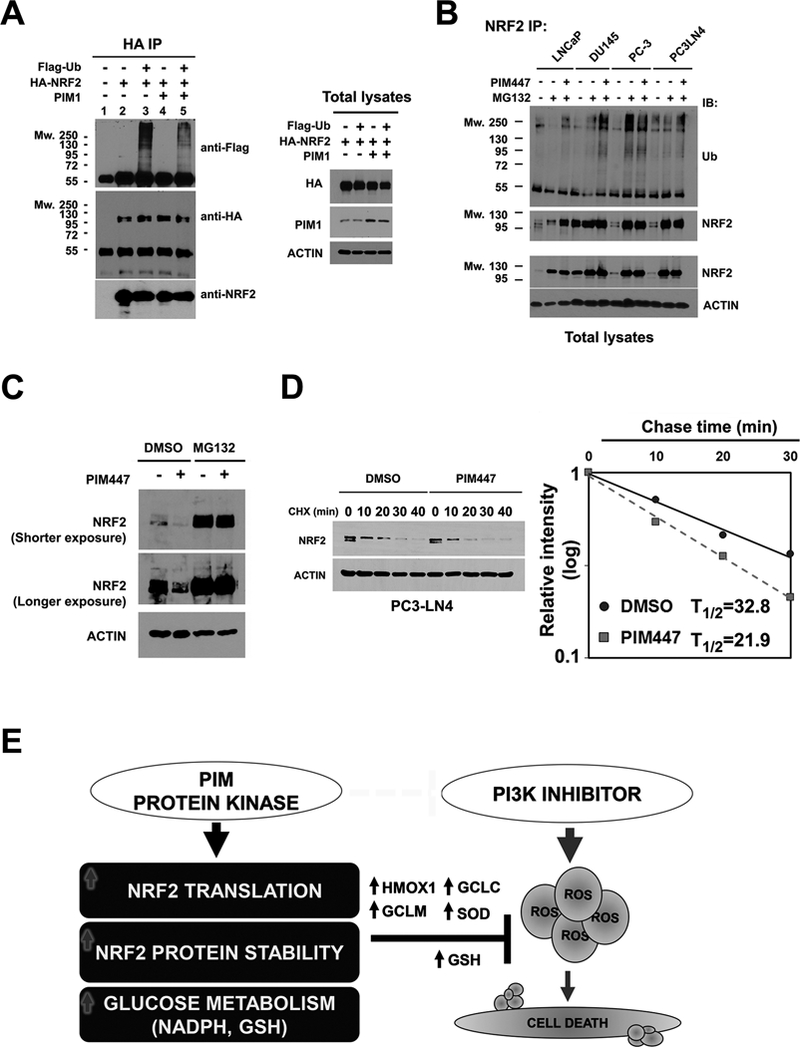
(A) Immunoblot analysis of ubiquitinated NRF2 protein. LNCaP cells were transfected with Flag-Ub, HA-NRF2, and PIM1 as indicated. After 48 hr, these transfectants were exposed to 10 μmol/L MG132 for 4 hr. NRF2 was immunoprecipitated with an anti-HA antibody, and was subjected to immunoblot analysis with an anti-Flag antibody used for detection of ubiquitylated NRF2. An aliquot of the total lysate was evaluated for immunoblot analysis.
(B) Immunoblot analysis of endogenous NRF2 ubiquitination. Following exposure to MG132 total extracts from LNCaP, DU145, PC-3 and PC3-LN4 cells were immunoprecipitated with an anti-NRF2 antibody. Immunoprecipitated NRF2 was subjected to immunoblot analysis with an anti-ubiquitin (anti-Ub) antibody for detection of endogenous ubiquitylated NRF2. An aliquot of the total cell lysate was used for immunoblot analysis.
(C) Immunoblot analysis of NRF2 accumulation after MG132 treatment. PC3-LN4 cells were treated with 3 μmol/L PIM447 for 24 hr in the presence or absence of 10 μmol/L MG132.
(D) The half-life of NRF2 in PC3-LN4 cells following treatment with DMSO or 3 μmol/L PIM447. After 24 hr treatment, cells were exposed to 10 μg/ml Cycloheximide for the indicated times. Total lysates were subjected to immunoblot analysis. NRF2 and Actin levels were determined by densitometry, and the level of NRF2 relative to that of Actin was plotted as a function of time to determine the decrease in NRF2 protein.
(E) Schematic outline of the resistance mechanism induced by PIM kinase. Expression of the PIM protein kinase induces tumor resistance to PI3K inhibitors. PIM promotes NRF2 synthesis through phosphorylation of eIF4B (S406) and increases NRF2 activity by inhibiting ubiquitination-mediated proteasome degradation. PIM modulates glycolytic metabolism to favor a decrease in ROS accumulation by producing NADPH and GSH. Genetic depletion of ROS scavenger proteins overcomes PIM-mediated drug resistance. Combining PIM and PI3K inhibitors promotes cancer death by inhibition of NRF2 signaling.
DISCUSSION
The PIM protein kinases have been shown to confer resistance to PI3K and AKT inhibitors in part by activating PI3K downstream effectors in an AKT-independent manner (1,14,33). PIM inhibition enhances sensitivity to AKT inhibitors, and cancers with mutation in PIK3CA cells often develop increased levels of the PIM kinases (1). Treatment of tumors with agents that inhibit cell surface tyrosine kinases, e.g., MET inhibitors, blocks AKT activation, elevates PIM levels and ultimately, induces drug resistance (34). Experiments reported here demonstrate the mechanism by which PIM functions to induce resistance to these agents, namely, 1) by increasing NRF2 levels and consequently augmenting the level of ROS scavengers, and 2) by stimulating cellular metabolism to drive reduced Glutathione levels.
The NRF2 transcription factor is an important regulator of cancer survival and proliferation. Redox homeostasis is essential to maintain cellular equilibrium and the survival of cancer cells that have abnormal metabolism (12). Approximately 70% of tumors in biopsies from patients with metastatic prostate cancer harbor aberrant PI3K pathway signaling. This leads to AKT activation, and in this tumor type, HMOX1 plays a pivotal role in antioxidant defense and the regulation of resistance to PI3K inhibition. In prostate cancer cell lines harboring PTEN deletion or mutation, PI3K inhibitor treatment increases ROS levels while decreasing scavenger enzymes. Here, the importance of ROS generation as a means by which these compounds kill prostate cancer was demonstrated by the observation that the expression of HMOX1 blocked the cytotoxicity of the pan-PI3K inhibitor buparlisib, while the depletion of ROS scavengers enhanced the activity of this compound. Importantly, even in buparlisib-treated tumor cells, expression of PIM1 elevated NRF2 levels and increased ROS scavengers, e.g., HMOX1, preventing PI3K inhibitor killing of prostate cancer. The combination of a PIM and an AKT inhibitor markedly decreased NRF2 and increased ROS production. Importantly, further increasing ROS generation with tert-Butyl Hydroperoxide stimulated the death of these prostate cancer cells.
Elevated GSH biosynthesis is known to be required for PI3K/AKT-driven resistance to oxidative stress, initiation of tumor spheroids, and anchorage independent growth (35). The addition of buparlisib to prostate cancer treatment lowered GSH levels along with lowering NAD(P)H, a molecule essential for keeping GSH reduced and inducing oxidative stress. In contrast to buparlisib, PIM1 overexpression enhanced GSH synthesis, abrogating the inhibitory effects of buparlisib. Recent studies have shown that NRF2 regulates genes that are involved in anabolic metabolism. Shown here, in prostate cancer cells, PIM1 facilitated glucose flux into substrates for glutathione production and the recycling process. Genes involved in glutathione synthesis were not induced by PIM1 when NRF2 was depleted, suggesting that PIM regulation of NRF2 is coordinating the high GSH content in PIM1-overexpressing tumor cells. Synthetic lethal interaction between NRF2 loss and mutant k-Ras was detected in pancreatic ductal adenocarcinomas (13). Cells that lack PIM kinase or are treated with a PIM inhibitor were not tolerant of mutant KRAS expression due to lethal ROS accumulation (26), again suggesting that PIM kinases are important regulators of cellular redox signaling. In agreement with other reports (13,35), AKT inhibitor efficacy was markedly enhanced by co-treatment with the glutathione synthesis inhibitor BSO. Here, it is shown that tumor suppression is enhanced by combining BSO and buparlisib. BSO re-sensitized PIM1-overexpressing tumors to buparlisib both in vitro and in vivo. This suggests that the maintenance of GSH is important to the mechanism by which PIM protects cells from death. Clearly, the regulation of redox homeostasis by PIM1 overlaps with the action of AKT and controls cellular homeostasis.
PIM1 regulates translation by phosphorylating eIF4B, a protein associated with mRNA unwinding, on S406 (8). Shown here, mutation of the PIM phosphorylation site in eIF4B (S406A) markedly decreases NRF2 protein levels, suggesting a mechanism by which PIM controls NRF2 translation. NRF2 exists in complex with KEAP1 via direct protein-protein interactions between the KEAP1 Kelch domain and the ETGE and DLG motifs on the Neh2 domain of NRF2 (36). We further show that PIM kinases appear to partially block the ubiquitination of NRF2, enhancing its half-life, accounting in part for the increased levels of NRF2 when PIM is expressed. However, the mechanism by which PIM regulates NRF2 degradation is not clear. Although there is variability in the level of KEAP1 in prostate cancer cell lines, experiments did not find that PIM1 regulated the levels of NRF2 by modifying either the interaction of NRF2 with KEAP1 or the degradation of KEAP1. NRF2 degradation could be regulated in these cells by the phosphorylation on Neh6 domain of NRF2 by glycogen synthase kinase-3 (GSK3) can be recognized by β-TrCP, which acts as a receptor for the SKP1-CUL1-RBX1/ROC1 ubiquitin ligase complex (37) and leads to the degradation of NRF2 in a KEAP1-independent fashion. In human lung cancer A549 cells, inhibition of the PI3K/AKT pathway markedly reduced endogenous NRF2 protein through this mechanism (38).
The mechanism by which PIM regulates NRF2 can have broader impact for molecularly targeted therapy in varied tumor types. Targeting the PI3K signaling pathway has resulted in the development of novel PI3K inhibitors including buparlisib, pictilisib, ZSTK474, dactolisib, apitolisib and omipalisib. A PI3K-α-selective inhibitor alpelisib is currently in clinical development for breast cancer therapy in a Phase III clinical trial (NCT02437318), and Phase II study for treatment of head and neck squamous cell carcinoma (NCT02051751). In 2014, the p110δ specific inhibitor idelaisib was approved in the US and Europe as the first-in-class PI3K inhibitor for use in the treatment of chronic lymphocytic leukemia and follicular lymphoma. As well, copanlisib with activity against the PI3K-α and -δ isoforms was recently approved by the US Food and Drug Administration for treatment of patients with relapsed follicular lymphoma. However, in prostate cancer although mutations activate the PI3K pathway inhibitors targeting this pathway have not shown activity. Our findings in prostate cancer models that combining PI3K inhibitors with a PIM inhibitor enhances the induction of cell death by markedly increasing ROS generation may provide molecular basis to overcome the limited efficacy of PI3K-targeted therapies. The therapeutic vulnerabilities caused by redox regulation in cancer suggest the use of PIM inhibitors as an additional treatment modality.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by University of Arizona Cancer Center support grant NIH P30CA023074, NIH award R01CA173200, and DOD award W81XWH-12–1-0560 (to A.S. Kraft) and American Lung Association Award LCD-504131 (to N.A. Warfel). The authors thank Novartis Oncology for providing PIM447. The UACC Shared Resources provided support for histological and tissue staining, microarray, and flow cytometry analysis.
Footnotes
Disclosure of Potential Conflicts of Interest Research.
The authors disclose no competing interest.
REFERENCES
- 1.Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, et al. Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer . Cancer Discov 2016;6(10):1134–47 doi 10.1158/2159-8290.CD-16-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161(5):1215–28 doi 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crumbaker M, Khoja L, Joshua AM. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers (Basel) 2017;9(4) doi 10.3390/cancers9040034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu HT, Wang N, Wang X, Li SL. Overexpression of Pim-1 is associated with poor prognosis in patients with esophageal squamous cell carcinoma. J Surg Oncol 2010;102(6):683–8 doi 10.1002/jso.21627. [DOI] [PubMed] [Google Scholar]
- 5.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature 2001;412(6849):822–6 doi 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 6.Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer 2011;11(1):23–34 doi 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- 7.Xie Y, Xu K, Dai B, Guo Z, Jiang T, Chen H, et al. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene 2006;25(1):70–8 doi 10.1038/sj.onc.1209058. [DOI] [PubMed] [Google Scholar]
- 8.Cen B, Xiong Y, Song JH, Mahajan S, DuPont R, McEachern K, et al. The Pim-1 protein kinase is an important regulator of MET receptor tyrosine kinase levels and signaling. Mol Cell Biol 2014;34(13):2517–32 doi 10.1128/MCB.00147-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res 2010;44(5):479–96 doi 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z, et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc Natl Acad Sci U S A 2014;111(12):4472–7 doi 10.1073/pnas.1324136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol 2003;43:233–60 doi 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 12.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012;22(1):66–79 doi 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 13.Chio II, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016;166(4):963–76 doi 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cen B, Mahajan S, Wang W, Kraft AS. Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1. Cancer Res 2013;73(11):3402–11 doi 10.1158/0008-5472.CAN-12-4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, et al. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat Med 2016;22(11):1321–9 doi 10.1038/nm.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braso-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med 2016;22(11):1303–13 doi 10.1038/nm.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burger MT, Nishiguchi G, Han W, Lan J, Simmons R, Atallah G, et al. Identification of N-(4-((1R,3S,5S)-3-Amino-5-methylcyclohexyl)pyridin-3-yl)-6-(2,6-difluorophenyl)- 5-fluoropicolinamide (PIM447), a Potent and Selective Proviral Insertion Site of Moloney Murine Leukemia (PIM) 1, 2, and 3 Kinase Inhibitor in Clinical Trials for Hematological Malignancies. J Med Chem 2015;58(21):8373–86 doi 10.1021/acs.jmedchem.5b01275. [DOI] [PubMed] [Google Scholar]
- 18.Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer . ACS Med Chem Lett 2011;2(10):774–9 doi 10.1021/ml200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keeton EK, McEachern K, Dillman KS, Palakurthi S, Cao Y, Grondine MR, et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014;123(6):905–13 doi 10.1182/blood-2013-04-495366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 2012;11(4):873–87 doi 10.1158/1535-7163.MCT-11-0824-T. [DOI] [PubMed] [Google Scholar]
- 21.Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benz o[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem 2010;53(19):7146–55 doi 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2’,7’-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 1992;5(2):227–31. [DOI] [PubMed] [Google Scholar]
- 23.Song JH, Padi SK, Luevano LA, Minden MD, DeAngelo DJ, Hardiman G, et al. Insulin receptor substrate 1 is a substrate of the Pim protein kinases. Oncotarget 2016;7(15):20152–65 doi 10.18632/oncotarget.7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song JH, Kraft AS. Pim kinase inhibitors sensitize prostate cancer cells to apoptosis triggered by Bcl-2 family inhibitor ABT-737. Cancer Res 2012;72(1):294–303 doi 10.1158/0008-5472.CAN-11-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun H, Wang Y, Chinnam M, Zhang X, Hayward SW, Foster BA, et al. E2f binding-deficient Rb1 protein suppresses prostate tumor progression in vivo. Proc Natl Acad Sci U S A 2011;108(2):704–9 doi 10.1073/pnas.1015027108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song JH, An N, Chatterjee S, Kistner-Griffin E, Mahajan S, Mehrotra S, et al. Deletion of Pim kinases elevates the cellular levels of reactive oxygen species and sensitizes to K-Ras-induced cell killing. Oncogene 2015;34(28):3728–36 doi 10.1038/onc.2014.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014;159(1):163–75 doi 10.1016/j.cell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, et al. Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc 2016;11(2):347–58 doi 10.1038/nprot.2016.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pettaway CA, Pathak S, Greene G, Ramirez E, Wilson MR, Killion JJ, et al. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin Cancer Res 1996;2(9):1627–36. [PubMed] [Google Scholar]
- 30.Gravina GL, Ranieri G, Muzi P, Marampon F, Mancini A, Di Pasquale B, et al. Increased levels of DNA methyltransferases are associated with the tumorigenic capacity of prostate cancer cells. Oncol Rep 2013;29(3):1189–95 doi 10.3892/or.2012.2192. [DOI] [PubMed] [Google Scholar]
- 31.Warfel NA, Sainz AG, Song JH, Kraft AS. PIM Kinase Inhibitors Kill Hypoxic Tumor Cells by Reducing Nrf2 Signaling and Increasing Reactive Oxygen Species. Mol Cancer Ther 2016;15(7):1637–47 doi 10.1158/1535-7163.MCT-15-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U S A 2015;112(52):15790–7 doi 10.1073/pnas.1521919112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zwang Y, Jonas O, Chen C, Rinne ML, Doench JG, Piccioni F, et al. Synergistic interactions with PI3K inhibition that induce apoptosis. Elife 2017;6 doi 10.7554/eLife.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.An N, Xiong Y, LaRue AC, Kraft AS, Cen B. Activation of Pim Kinases Is Sufficient to Promote Resistance to MET Small-Molecule Inhibitors. Cancer Res 2015;75(24):5318–28 doi 10.1158/0008-5472.CAN-15-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lien EC, Lyssiotis CA, Juvekar A, Hu H, Asara JM, Cantley LC, et al. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat Cell Biol 2016;18(5):572–8 doi 10.1038/ncb3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol 2006;26(8):2887–900 doi 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol 2011;31(6):1121–33 doi 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013;32(32):3765–81 doi 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.