

Abstract
Mismatch repair (MMR) deficiency through MSH6 inactivation has been identified in up to 30% of recurrent high-grade gliomas, and represents a key molecular mechanism underlying the acquired resistance to the alkylating agent temozolomide (TMZ). To develop a therapeutic strategy that could be effective in these TMZ-refractory gliomas, we first screened 13 DNA damage response modulators for their ability to suppress viability of MSH6-inactivated, TMZ-resistant glioma cells. We identified a PLK1 selective inhibitor, Volasertib, as the most potent in inhibiting proliferation of glioblastoma cells. PLK1 inhibition induced mitotic catastrophe, G2/M cell cycle arrest, and DNA damage, leading to caspase-mediated apoptosis in glioblastoma cells. Importantly, therapeutic effects of PLK1 inhibitors were not influenced by MSH6 knockdown, indicating that their action is independent of MMR status of the cells. Systemic treatment with Volasertib potently inhibited tumor growth in an MMR-deficient, TMZ-resistant glioblastoma xenograft model. Further in vitro testing in established and patient-derived cell line panels revealed an association of PLK1 inhibitor efficacy with cellular Myc expression status. We found that cells with deregulated Myc are vulnerable to PLK1 inhibition, as Myc overexpression sensitizes, while its silencing desensitizes, glioblastoma cells to PLK1 inhibitors. This discovery is clinically relevant as glioma progression post-TMZ treatment is frequently accompanied by MYC genomic amplification and/or pathway activation. In conclusion, PLK inhibitor represents a novel therapeutic option for recurrent gliomas, including those TMZ-resistant from MMR deficiency. Genomic MYC alteration may serve as a biomarker for PLK inhibitor sensitivity, as Myc-driven tumors demonstrated pronounced responses.
Introduction
As a heterogenous group of tumors characterized by different molecular and clinical features, diffuse gliomas invariably recur after multidisciplinary treatments that include surgical resection, radiation therapy and alkylating chemotherapeutic agents. Temozolomide (TMZ) is the most commonly used alkylator for gliomas, with clinical activity in both lower-grade tumors carrying isocitrate dehydrogenase 1 (IDH1) mutations (1, 2), as well as primary glioblastoma exhibiting methylation of the MGMT (O6-methyguanine DNA methyltransferase) promoter (3, 4). Unfortunately, prolonged treatment with TMZ typically leads to development of acquired resistance to TMZ, contributing to malignant progression, tumor recurrence and mortality. Inactivation of mismatch repair (MMR) genes, i.e., MSH2, MSH6, MLH1 and PMS2, has been identified in recurrent malignant gliomas (5, 6) that have progressed from lower-grade lesions and in previously treated glioblastoma, whereas these alterations are extremely rare in primary tumors (7). In tumors with deficient MMR, TMZ-induced base mispairing of O6-methylguanine with thymine is not recognized by the MMR machinery and escapes the “futile cycling” of repair otherwise mediated by proficient MMR (8), the mechanism by which TMZ induces apoptosis in intact MMR cells. Therefore, MMR inactivation represents a key molecular mechanism of acquired TMZ resistance in gliomas (9). Previous studies showed strong association between TMZ treatment and the development of MMR deficiency, and MMR alterations after TMZ treatment of low grade gliomas are considered a strong driver for malignant progression and a post-TMZ hypermutator phenotype (7, 10). There is an urgent need for new treatment strategies that target MMR-deficient, TMZ-resistant recurrent gliomas.
Genetic alterations involved in glioma progression or recurrence are diverse and not limited to MMR genes. An integrated genomic characterization study of paired (i.e, pre- and post-progression) glioma samples identified amplification of MYC locus as one of the most frequent genetic events that occur during glioma malignant progression (11). Deletions affecting the gene encoding FBW7, a Myc (c-Myc) suppressor, were also found in a subset of progressed gliomas. These genetic alterations resulted in significant upregulation of Myc target genes and signaling activation during the evolution of gliomas. A key oncoprotein contributing to malignancy by regulating diverse cellular functions including cell proliferation, metabolism and programmed cell death (12, 13), Myc plays a major role in the maintenance of glioma stem cells. Previous studies have shown that inhibition of Myc decreases cell proliferation, induces apoptosis and impairs self-renewal of glioma stem cells, revealing their dependency on Myc (14, 15). Since glioma stem cells are considered the cellular reservoir from which tumor resistance and recurrence emerges, Myc therefore serves as a critical driver of glioma evolution and thus an important therapeutic target in recurrent, progressed glioma. However, pharmacological direct targeting of Myc-mediated transcriptional regulation remains a challenge, and different approaches have been proposed to exploit Myc-induced downstream signaling pathways (16–19).
Here, screening of DNA damage modulators identified PLK1 inhibitor as a potent therapeutic for glioma, independent of MMR status. Furthermore, we show that deregulated Myc generates vulnerability to PLK1 inhibition in glioma cells. Thus, we propose that PLK1 inhibitor is a promising treatment strategy for recurrent gliomas, irrespective of MMR-deficiency, especially those driven by Myc.
Materials and Methods
Cells and compounds
Human glioblastoma cell lines (U87, U251, LN229, A172, U118, and LN18) were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were authenticated in 2017 by comparison of STR profiles to the ATCC public dataset. Gli36 was provided by Dr. Khalid Shah at Massachusetts General Hospital, Boston, MA, in 2014. Normal human astrocytes (NHA) were purchased from ScienCell in 2009, and used before passage 10. Glioblastoma cell lines and NHA were maintained in Dulbecco’s modified Eagle medium (DMEM) with 4.5 g/L glucose, L-glutamine and sodium pyruvate supplemented with 10% fetal bovine serum and 1% penicillin/Streptomycin/Amphotericin. Patient-derived glioma neurosphere lines (MGG4, MGG6, MGG8, MGG18, MGG23, MGG75, and MGG152) were established and cultured in serum-free neural cell medium as described previously (20–22). All the glioma cell lines were confirmed to be mycoplasma-free using LookOut Mycoplasma PCR Detection Kit from Sigma in 2016–2017. Volasertib (23), Irinotecan, KU-55933 (24), VE-821 (25), Alisertib (26), Barasertib (27), MK8776 (28), NU7441 (29), Palbociclib, Carboplatin, and Imatinib were purchased from SelleckChem. GSK461364 (30, 31) was from APExBIO and Temozolomide, Etoposide, and Ex527 (32) were from Sigma-Aldrich.
Western blot analysis
Cells were lysed in radioimmunoprecipitation (RIPA) buffer (Boston Bioproducts) with a cocktail of protease and phosphatase inhibitors (Roche). Protein (10–15 μg) was separated by 4–20% SDS-PAGE and transferred to polyvinylidene difloride membrenes by electroblotting. After blocking with 5% non-fat dry milk in TBS-T (20 mM Tris [pH, 7.5], 150 mM NaCl, 0.1% Tween20) for 1–2 hours at room temperature, membranes were incubated with primary antibody at 4C overnight. Membranes were washed in TBS-T and incubated with appropriate peroxidase conjugated secondary antibodies for 1 hour at room temperature. Signals were visualized using the enhanced chemiluminescense (ECL) kit (Amersham Bioscience). Primary antibodies used were: MSH6 (#5425), MGMT (#2739), cleaved-PARP (#5625), cleaved-caspase3 (#9661), phospho-H2Ax (#2577), Myc (#9402), N-Myc (#9405), phosphor-HistoneH3 (Ser10, #9701)(Cell Signal Technology), PLK1 (ab70697)(Abcam), and β-actin (A1978)(Sigma).
Cell viability and apoptosis assay
Cells were seeded in 96-well plates at 1,000–3,000 cells per well. After overnight incubation, compounds were serially diluted and added to wells. Cell viability was evaluated by Cell Titer-Glo (Promega) according to the manufacturer’s instruction, on day 6 for TMZ, and day 3 (72 hours) for Volasertib and GSK461364. Daily evaluation of cell viability following drug exposure was used to determine the time course of treatment effects and plotted as % cell viability relative to day 0 (treatment initiation). Caspase3/7 activities were evaluated by Caspase-Glo 3/7 assay (Promega) according to the manufacturer’s instruction at 24 hours after Volasertib treatment.
Cell cycle analysis
After 24-hour incubation with/without Volasertib, cells were fixed in chilled 70% ethanol overnight and stained with propidium iodide in the presence of 100 μg/ml RNase and 0.1% TritonX-100 in PBS. Data were acquired by flow cytometry (BD FACSAria ll or BD LSRII) and cell cycle distributions were analyzed with the FlowJo software (FlowJo, LLC).
Immunofluorescence and mitotic catastrophe detection
Gli36, U87 and LN229 cells were cultured in 4-well chamber slides with/without Volasertib for 24 hours and fixed with 4% paraformaldehyde. MGG4 and MGG75 cells were treated with and without Volasertib for 24 hours, fixed with 4% paraformaldehyde and mounted on slides by cytospin. Cells were permeabilized with 0.1%Triton X-100 in PBS. After blocking with 10% goat serum in PBS, cells were stained with phospho-Histone H3 primary antibody (CST#9701 1:200) overnight at 4°C and followed by incubation with fluorophore-conjugated secondary antibody (1:1000, Alexa Fluor 546 goat anti-rabbit IgG; Life Technologies Corporation) at 4°C for 2–4 hours. Slides were mounted with DAPI (H1200: Vector Laboratories). Staining was observed under a Nikon Eclipse Ci microscope equipped with CoolSNAP DYNO (Photometrics), and positivity for pHH3 and mitotic catastrophe were evaluated by acquiring microscopic images (NIS Elements, Nikon) and counting at least 100 cells per slide.
MSH6-shRNA, Myc overexpression and Myc-shRNA cell lines generation
Lentivirus vector plasmids containing shRNA sequences for MSH6 were TRCN0000286578 from Sigma (sh1) and V3LHS 318784 from Dharmacon (sh8). Non-targeting shRNA lentivirus vector (SHC002) was from Sigma. Myc overexpression construct, pCDH-puro-cMyc was from Addgene (#46970), and GFP control was pCDH-CMV-MCS-EF1-copGFP from System Bioscience (CD511B-1). For regulatable Myc knockdown, cMyc-shRNA sequences (No.1: CCGGAGGTAGTTATCCTTAAAAACTCGAG TTTTTAAGGATAACTACCTTTTTT, No.2: CCGGCCTGAGACAGATCAGCAACAACTCGAGTTGTTGCTGATCTGTCTCAGGTTTTT) were ligated into the pLKO-Tet-On vector (#21915, Addgene). To generate lentiviral particles, 293T cells were transfected with a lentiviral plasmid, packaging plasmid (pCMV-dR8.2), and envelope plasmid (pCMV-VSV-G) with FuGene (Promega). Cells were infected with lentivirus in the presence of polybrene (8 μg/ml) for 8 hours. Three days later, cells were selected with puromycin (0.6 μg/ml for LN229, U251, U87; 0.2 μg/ml for Gli36, MGG4, MGG75, and MGG152) for 3–4 days before use. To induce expression of Myc shRNA, infected cells were cultured with Doxycycline (1 μg/ml) for at least 72 hours. Knockdown and overexpression was confirmed by western blot.
Immunohistochemistry
Tumor tissue specimens for control (n=3) and Volasertib groups (n=3) were fixed in neutral-buffered formalin and embedded in paraffin. Five μm sections were deparaffinized, rehydrated, heated in 10mM Citrate Na buffer for antigen retrieval, and treated with 3% H2O2 in methanol. After blocking, slides were incubated with pHH3 antibody (Cell Signaling Technology #9701, 1:200) overnight at 4C, followed by biotinilated secondary antibody for 30 minutes at room temperature and ABC solution (PK6200: Vectastain) for 30 minutes at room temperature. Sections were stained with 3,3-diaminobenzine (Dako) as chromogen and counterstained with Hematoxylin. Immunopositivity for pHH3 was evaluated by acquiring microscopic images and counting at least 1,000 cells per tumor.
Animal study
Five million LN229-MSH6sh1 cells were implanted subcutaneously into the flank of 7-week-old female nude mice (NCI). When the maximum diameter of established tumors reached 5 mm, mice were randomized and treated with PBS (n=6) or Volasertib (n=5, 40mg/kg) once a week for 5 cycles. Volasertib was formulated in hydrochloric acid (0.2N), diluted with 0.9% NaCl and injected intravenously into the tail vein. Tumor diameters were measured twice a week using digital caliper. Tumor volumes were calculated using the formula: tumor volume (mm3) = tumor length x tumor width2 /2. Orthotopic model of MSH6-deficient glioblastoma was established by intracerebral implantation of 1×105 MGG4-MSH6sh1 cells or 5×105 LN229-MSH6sh1 cells in 7-week-old female nude mice (NCI) as previously described (20). Mice were randomized and treated with PBS (n=8) or Volasertib (n=8, 40mg/kg) once a week for 4 cycles. Animals were euthanized when significant deterioration of neurological or general conditions was noted. For immunohistochemistry of pHH3, tumor-bearing mice were treated with PBS (n=3), GSK461364 (n=3, 40mg/kg, intraperitoneally), BI2536 (SelleckChem, n=3, 40mg/kg, intravenously)(33) or Volasertib (n=3, 40mg/kg, intravenously) and sacrificed 24 hours after the treatment. GSK461364 was dissolved in DMSO and diluted with 5% glucose in water. BI2536 was dissolved in 0.2N HCl and diluted with 0.9% NaCl. All animal procedures were approved by Institutional Animal Care and Use Committee at Massachusetts General Hospital.
Statistical analysis
The Prism (GraphPad software) software package was used for statistical analysis. Comparisons between 2 groups were done with Student t-test (unpaired). Differences of tumor volumes were analyzed by non-parametric Mann-Whitney test. P values < 0.05 were considered statistically significant.
Results
Volasertib inhibits the proliferation of glioma cells independent of MSH6 status.
MSH6 inactivation has been identified in approximately 25–30% of recurrent gliomas and is associated with treatment with TMZ (6, 10). MSH6 inactivation renders glioblastoma cells resistant to TMZ (9). We induced shRNA suppression of MSH6 expression in glioblastoma cell lines and patient-derived glioma stem-like cell lines, and confirmed MSH6 knockdown conferred TMZ resistance (Figure 1A). The impact of MSH6 silencing on TMZ sensitivity was more pronounced in glioma cells that do not express MGMT, than MGG152 cells, which express MGMT and are relatively resistant to TMZ (Figure 1B). In order to identify a new candidate therapeutic target in MSH6-deficient, TMZ-resistant gliomas, we tested 13 compounds that modulate/inhibit different pathways of DNA damage response for potent cytoxicity in our glioma models. We identified volasertib, a selective inhibitor of Polo-like kinase-1 (PLK1), as potently cytotoxic to LN229 glioblastoma cells engineered with control and MSH6 shRNA as well as U251 cells engineered with MSH6 shRNA, reducing cell viability by 85–90% at 1 μM (Figure 1C, Supplementary Figure S1). PLK1 inhibitors Volasertib and GSK461364 exerted comparable and consistent anti-glioma activity in glioblastoma cell lines and spheres (Figure 1D and Supplementary Figure S1). Importantly, effects were independent of cellular MSH6 status in all the cell lines tested, indicating MMR signaling is not involved in glioma response to PLK1 blockade (Figure 1D and Supplementary Figure S2A). Testing of a second independent shRNA sequence targeting MSH6 showed that cellular responses to TMZ and PLK1 inhibitors were essentially identical with the two used shRNAs, supporting their on-target effects (Supplementary Figure S2B). We next assessed the in vitro efficacy of PLK1 inhibitor combination with TMZ. There were no additive or synergic effects with TMZ, and PLK1 inhibitors did not reverse TMZ resistance in MSH6-silenced cell lines (Supplementary Figure S3). These results indicate that PLK1 inhibitor effects are independent of MMR deficiency in glioma cells, and that the efficacy is not influenced by MMR status or TMZ treatment.
Figure 1. Volasertib inhibits the proliferation of glioma cells independent of MSH6 status.
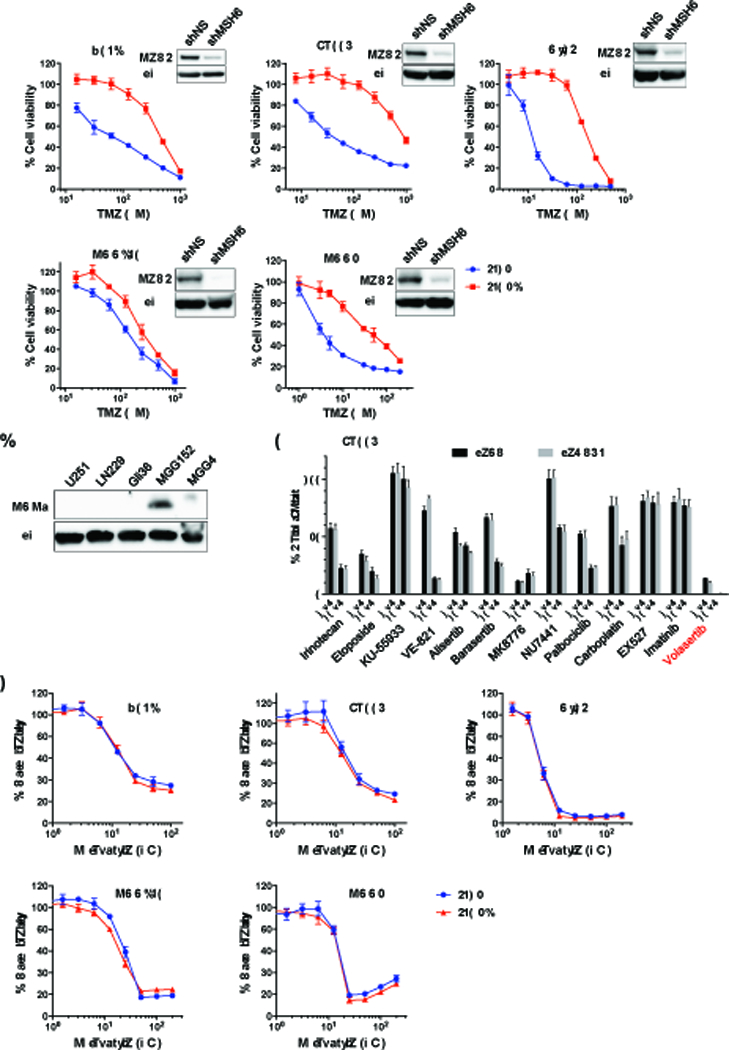
(A) Cell viability assay to determine temozolomide (TMZ) dose response in glioblastoma cells engineered with a non-targeting shRNA (shNS, blue) or MSH6-directed shRNA (shMSH6, red) lentivirus. MSH6 inactivation rendered glioma cell lines (U251, LN229 and Gli36) and patient-derived glioma sphere lines (MGG152 and MGG4) more resistant to TMZ. Immunoblot for each cell line confirmed MSH6 knockdown. Actin was used as a loading control. Cells were treated with specified concentrations of TMZ, and cell viability was evaluated by CellTiter Glo on day 6. (B) Immunoblot for MGMT in parental (unmodified) cell lines used for Figure 1A. Actin was used as a loading control. (C) A compound screening of 13 DNA damage response modulators in LN229shNS (non-silencing) and LN229shMSH6 cells at 1 and 10 μM. Cell viability was measured by CellTiter Glo on day 6. Also see Supplementary Figure S1. (D) Volasertib dose response in control (shNS, blue) and MSH6-knockdown (shMSH6, red) glioblastoma cells. Cell viability was measured by CellTiter Glo assay at 72 hours.
Volasertib induces G/2M cell cycle arrest and disrupts mitosis in both MSH6-intact and deficient glioma cells.
PLK1 plays critical roles in all aspects of mitosis and is also known as a direct or indirect regulator of the cell cycle-dependent proteins (34, 35). Since PLK1 inhibition has been shown to induce G2/M cell cycle arrest and mitotic cell death (36, 37), we evaluated cell cycle distribution after treatment of control (non-silencing shRNA, shNS) and shMSH6 cells with Volasertib. As expected, Volasertib treatment induced a large increase in G2/M population indicative of G2/M cell cycle arrest (Figure 2A). Previous studies have shown that G2/M cell cycle arrest by PLK1 inhibition results in an increase in the mitotic marker phospho-histone H3 (Ser10) (pHH3) (38), which allows monitoring the cell cycle effect of PLK1 inhibitors (39). Volasertib treatment increased pHH3-positive cells in LN229 and MGG4 cells, an effect that was not affected by cells’ MSH6 status (Figure 2B and Figure 2C). We also found post-treatment cells exhibited multi-lobular, giant nuclear and/or highly fragmented/bursted nuclei, characteristic of the type of abnormal mitosis described as “mitotic catastrophe” (Figure 2D). We investigated whether these abnormal mitoses were associated with DNA damage and apoptosis in shNS and shMSH6 glioblastoma cells. Volasertib treatment accumulated DNA damage (p-H2Ax), and induced apoptosis as shown by cleaved PARP and/or cleaved caspase3 (Figure 2E) and a dose-dependent increase in caspase3/7 activity (Figure 2F). Importantly, all of these Volasertib effects were observed to a similar degree in control shNS in addition to shMSH6 glioblastoma cells, suggesting that Volasertib efficacy is not influenced by MSH6 status.
Figure 2. Volasertib induces G/2M cell cycle arrest, apoptosis and DNA damage in both MSH6-intact and deficient glioma cells.
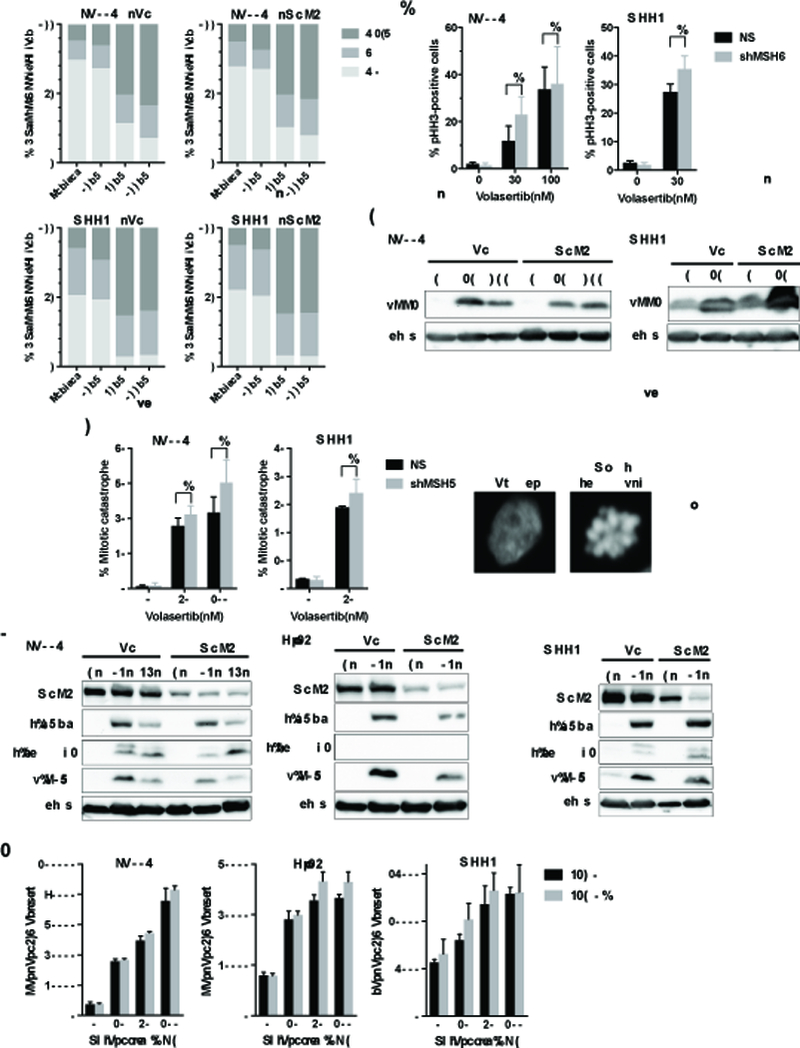
(A) Flow cytometric analysis showing cell cycle distribution of LN229 (shNS, shMSH6) and MGG4 (shNS, shMSH6) cells treated with Volasertib at indicated concentrations for 24 hours. (B) LN229 (shNS, shMSH6) and MGG4 (shNS, shMSH6) cells with/without Volasertib treatment (30 or 100 nM for 24 hours) were stained with DAPI and pHH3 antibody. % pHH3 positive cells was quantified in at least 100 cells per experiment. (C) Immunoblot of pHH3 expression in LN229 (shNS, shMSH6) and MGG4 (shNS, shMSH6) cells treated with indicated doses of Volasertib for 24 hours. Actin was used as a loading control. (D) In the same experiment as B, % cells with abnormal mitosis (mitotic catastrophe; shown right is a representative seen in MGG4, along with control healthy nucleus) was evaluated in at least 100 cells per experiment. (E) Immunoblot of MSH6, cleaved-PARP (c-PARP), cleaved-caspase3 (c-caspase3) and phospho-H2Ax (p-H2Ax) expression in shNS and shMSH6 cell lines treated with/without Volasertib (30 nM, 24 or 48 hours). Actin blot was used as a loading control. (F) shNS and shMSH6 glioblastoma cells were treated with indicated concentrations of Volasertib for 24 hours. Caspase3/7 activity was measured by Caspase-Glo 3/7 assay. In B and D, NS, not statistically significant (student t-test).
PLK1 inhibitor suppresses MMR-deficient TMZ resistant glioma tumor growth in vivo.
We next asked the question whether Volasertib is therapeutically active in vivo against MSH6-deficient, TMZ-resistant xenografts. Cycles of systemic intravenous administration of Volasertib or PBS were initiated in nude mice when their subcutaneously established LN229shMSH6 xenografts reached 5 mm in diameters. Volasertib completely abrogated tumor growth for the entire duration of the experiment (28 days after treatment initiation) whereas xenograft tumors treated with PBS continuously grew and needed animal euthanasia (Figure 3A). Immunohistochemical analysis revealed that pHH3 positive cells in xenografts treated with Volasertib were about 5 times of those seen in control tumors, indicating Volasertib induction of mitotic arrest (Figure 3B and Figure 3C). All mice maintained good general health status and stable body weight during treatment. However, similar treatment of Volasertib (4 injections instead of 5) in animals bearing orthotopic MGG4shMSH6 xenografts did not yield a significant therapeutic benefit of survival (Supplementary Figure S4A). Pharmacodynamic studies testing 3 currently available PLK1 inhibitors, Volasertib, GSK461364 and BI2536 in mice bearing LN229shMSH6 tumors in the brain revealed no significant increase in pHH3 immunopositivity (Supplementary Figure S4B), contrasting with the result obtained with the flank tumor model with the same tumor cells (Figure 3B and Figure 3C). The lack of activity of these agents in the brain may be due to their poor blood brain barrier penetration in our models.
Figure 3. PLK1 inhibitor suppresses MMR-deficient, TMZ-resistant glioma tumor growth in vivo.
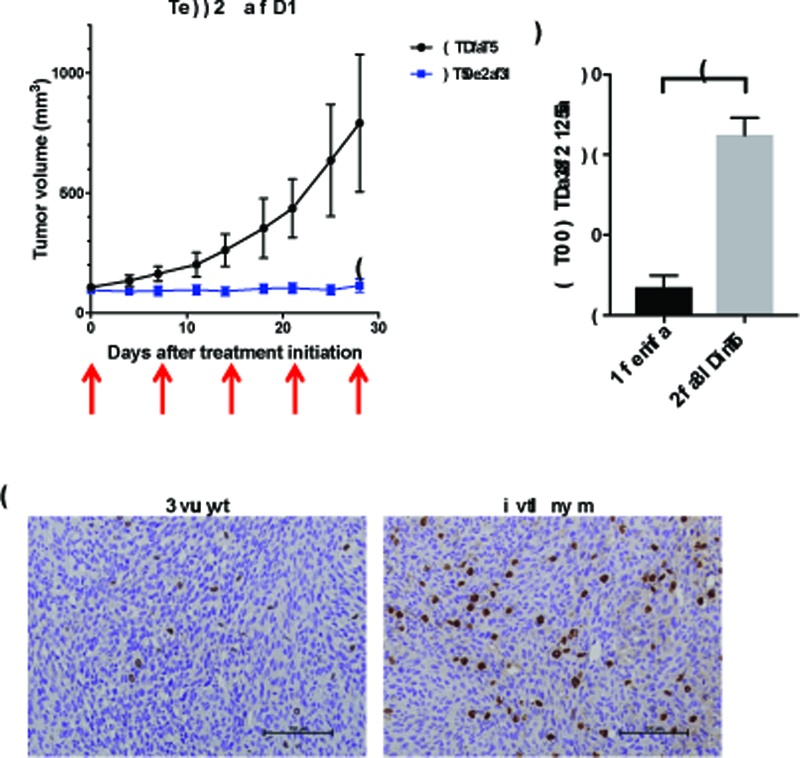
(A) Tumor growth curves in LN229shMSH6 xenograft model. Animals were treated with Volasertib (40 mg/kg i.v. once a week x 5 cycles)(N=5) or PBS (Control, N=6). Data are presented as mean tumor volume and SD in each group. Arrows indicate dates of treatment. *, P=0.017, PBS vs Volasertib on day 27 (Mann-Whitney test). (B, C) Nude mice bearing established flank LN229shMSH6 tumors were treated i.v. with a single dose of 40mg/kg Volasertib (N=3) or PBS (Control, N=3). Flank tumors were collected at 24 hours after dosing, and pHH3 immunohistochemistry was performed (brown) (B). Scale bars, 100 μm. (C) Quantification of B is shown. *, P=0.006, PBS (Control) vs Volasertib (student t-test).
Testing an extended cell line panel identifies Myc as a potential molecular marker of PLK1 inhibitor efficacy.
Both PLK1 inhibitors (Volasertib, GSK461364) demonstrated nanomolar potency to inhibit the growth of glioblastoma cell lines and patient-derived glioma sphere lines (Figure 1D and Supplementary Figure S2A). We confirmed that Volasertib and GSK461364 had only modest toxicity to proliferating normal human astrocytes (Supplementary Figure S5A). To begin seeking predictive molecular biomarkers for PLK1 inhibitor effects, we furthered testing of PLK1 inhibitors in extended panels of glioblastoma cell lines and patient-derived glioma sphere lines (Figure 4A and Supplementary Figure S5B). We did not find that PLK1 protein levels were associated with sensitivity (Figure 4A, Figure 4B, and Supplementary Figure S5B). Previously, it was shown that PLK1 stabilizes N-Myc via inhibition of FBW7, and in turn N-Myc activates PLK1 transcription (40). This report motivated us to probe Myc and N-Myc as a potential predictor of response to PLK1 inhibitor. We found a potential association of response with cellular Myc/N-Myc expression status. Patient-derived glioma sphere lines with strong Myc (e.g., MGG4, MGG18) or N-Myc (e.g., MGG8, MGG152) expression due to corresponding gene amplifications (19, 21, 22) were very sensitive to PLK1 inhibitors, while cell lines that do not have detectable Myc or N-Myc expression (MGG23, MGG75) were rather resistant (Figure 4A, Figure 4B, and Supplementary Figure S5B). In established cell lines, Gli36 that has a very high Myc level was the most sensitive to PLK1 inhibitor among the 7-line mini panel (Figure 4A, Figure 4B, and Supplementary Figure S5B). We therefore chose Gli36 and MGG4 as Myc dysregulated lines and U87 and MGG75 as non-Myc lines to further characterize the effect of Volasertib. Following Volasertib treatment, Gli36 cells exhibited multi-lobular, giant nuclear and/or highly fragmented nuclei, a characteristic nuclear morphology described as “mitotic catastrophe” (Figure 4C and Figure 4D). Immunofluorescence of pHH3 showed a dose response increase in the fraction of pHH3-positive cells (Figure 4C and Figure 4E), which was validated by western blot (Figure 4F). These Volasertib-induced phenotypic changes in Myc-high Gli36 cells were seen to a greater degree than in Myc-low U87 cells (Figure 4C, Figure 4D, Figure 4E, and Figure 4F). Similarly, much greater increases in mitotic catastrophe and pHH3 were observed in Myc-high MGG4 compared to non-Myc MGG75 after Volasertib treatment (Figure 4C, Figure 4D, Figure 4E, and Figure 4F). These mitotic abnormalities caused by PLK1 inhibition in sensitive, Myc-overexpressing Gli36 and MGG4 cells reflected selective accumulation of DNA damage (phosphor-H2Ax) and induction of apoptosis (cleaved caspase 3 and cleaved PARP), which was not obvious in low-Myc U87 and non-Myc MGG75 cells (Figure 4G). Active induction of cell death in Gli36, but not U87, was confirmed by a time course cellular assay (Supplementary Figure S5C). Thus, Myc status of glioblastoma cells was associated with PLK1 inhibitor-induced sensitivity, aberrant mitosis, DNA damage and apoptosis.
Figure 4. Testing PLK1 inhibitors in an extended panel identifies Myc as a candidate molecular marker associated with efficacy.
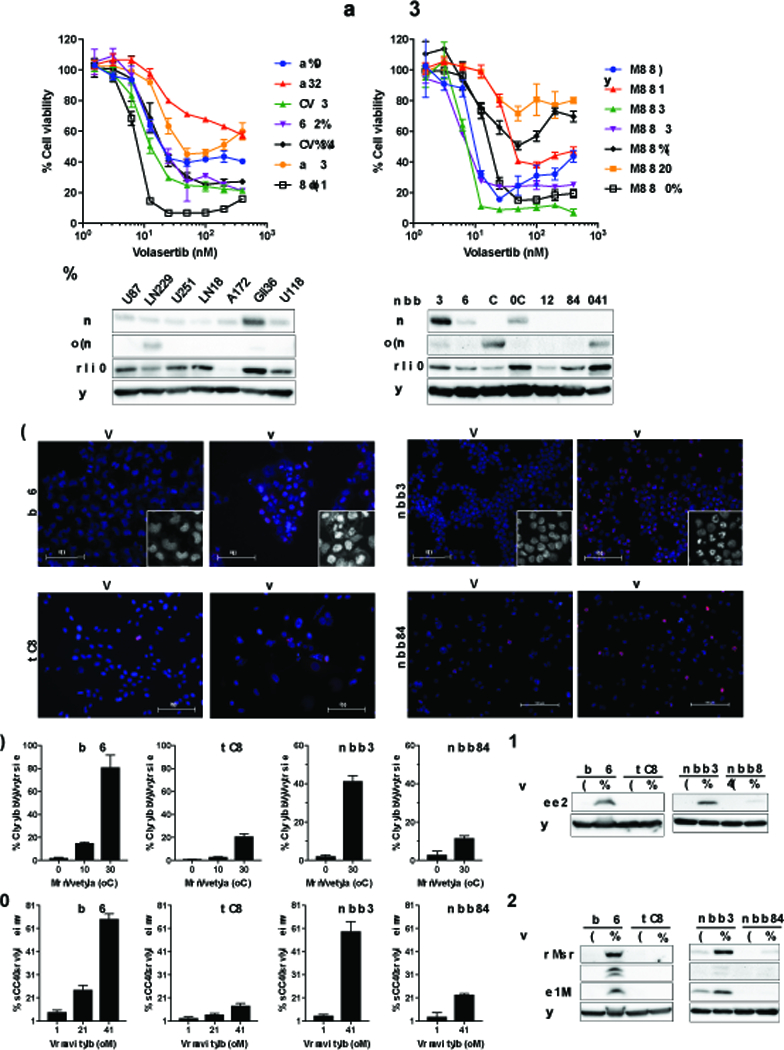
(A) A cohort of glioblastoma cell lines (left) and patient-derived glioma sphere lines (right) were treated with different concentrations of Volasertib. Cell viability was measured by CellTiter Glo at 72 hours. (B) Immunoblot of Myc, N-Myc and PLK1 in glioblastoma cell lines (left) and patient-derived glioma sphere lines (right). Actin was used as a loading control. (C-G) Myc dysregulated glioblastoma preferentially induces apoptosis, DNA damage and mitotic catastrophe in response to Volasertib treatment. (C-E) Myc expressers (Gli36, MGG4) and low/no Myc expressers (U87, MGG75) were treated with and without 30 nM Volasertib for 24 hours and immunostained with pHH3 (red) followed by counterstaining with DAPI (C). Insets are magnified images to better show nuclear morphology. Scale bars, 100 μm. % cells with abnormal mitosis (mitotic catastrophe) were quantified in at least 100 cells per experiment (D). % pHH3 positive cells were measured in at least 100 cells per experiment (E). (F) Immunoblot of pHH3 in Gli36, U87, MGG4, MGG75 cells treated with control or Volasertib (30 nM, 24 hours). (G) Immunoblot of cleaved PARP (c-PARP), cleaved caspase3 (c-caspase3) and p-H2Ax in Gli36, U87, MGG4, MGG75 cells treated with Volasertib (30 nM, 24 hours) compared to control treatment. In F and G, actin was used as a loading control.
Myc overexpression upregulates PLK1 expression and sensitizes glioblastoma cells to PLK1 inhibitors.
We next tested our hypothesis that hyperactive Myc mediated sensitivity to PLK1 inhibitors. We used lentivirus constructs carrying either GFP or Myc to transduce U87 and MGG75 which have low or no detectable Myc/N-Myc expression, respectively, at baseline. Myc overexpression significantly sensitized both cells to PLK1 inhibitors, with effects being more pronounced in altering the phenotype of MGG75 (Figure 5A and Supplementary Figure S6A). Consistent with prior reports (40, 41), Myc overexpression resulted in upregulation of PLK1 expression (Figure 5B). Compared with GFP control, enforced Myc expression increased the number of cells undergoing mitotic catastrophe after Volasertib treatment (Figure 5C and Figure 5D), which was accompanied by a large increase in the pHH3 mitosis marker in MGG75-Myc cells (Figure 5C and Figure 5E). Ectopic Myc also induced DNA damage (p-H2Ax) and apoptosis (cleaved caspase 3 and/or cleaved PARP and increased caspase 3/7 activity) in cells exposed to Volasertib (Figure 5F and Figure 5G). PLK1 inhibition has been shown to decrease phosphorylation of Myc (p-Myc) at Serine 62 and induce reduction of Myc protein levels (42). However, Myc levels were only slightly reduced in U87-Myc cells and were not altered in Gli36 cells by Volasertib, which was associated with an increase in p-Myc (S62) within 24 hours in both glioma cells (Supplementary Figure S6B). Time course cell viability assays showed that Myc overexpression accelerated cell proliferation of MGG75 cells, suggesting Myc-addicted status rendered cells vulnerable to PLK1 inhibition (Supplementary Figure S6C and Supplementary Figure S6D). These data show that Myc overexpression can induce sensitivity to PLK1 inhibitors in glioblastoma cells, generating a cellular phenotype that responds to PLK1 inhibition with mitotic and DNA damage effects.
Figure 5. Myc overexpression upregulates PLK1 expression and sensitizes glioblastoma cells to PLK1 inhibitors.
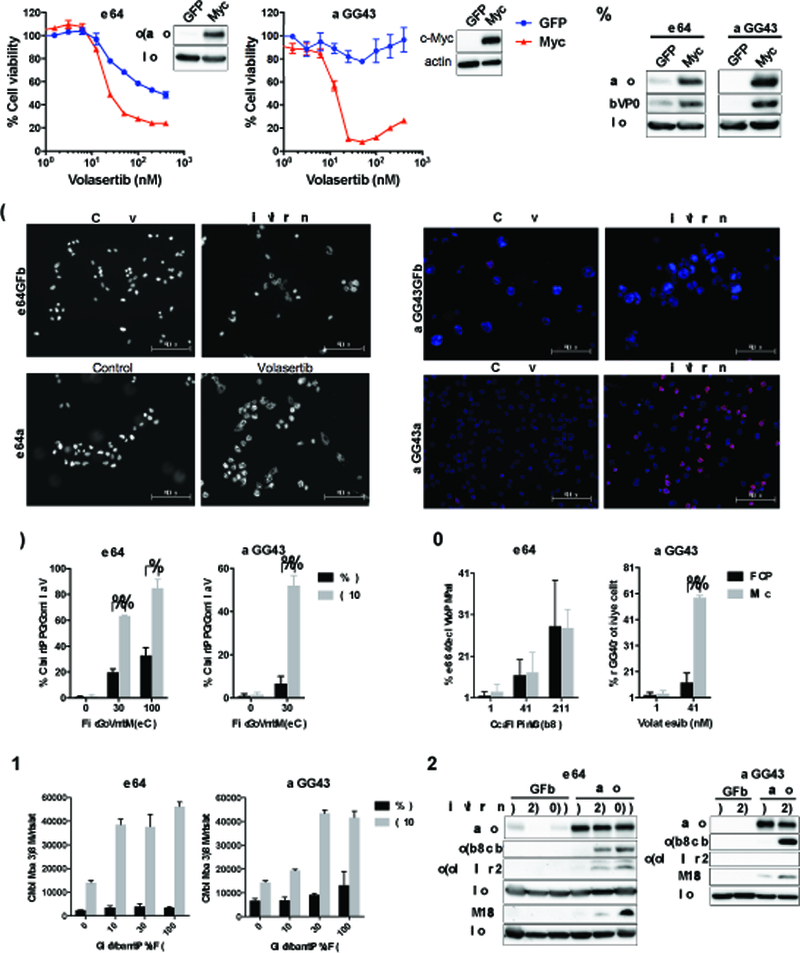
(A) U87 and MGG75 cells stably transduced with GFP or Myc cDNA were established. Immunoblot confirmed Myc expression with actin used as loading control. GFP (blue) and Myc (red) cells were treated with specified concentrations of Volasertib, and cell viability evaluated at 72 hours by CellTiter Glo. (B) Myc overexpression upregulates PLK1 expression. Immunoblot of Myc and PLK1 in GFP and Myc cells (U87, MGG75). Actin blot as loading control. (C-G) Myc overexpressing lines (U87, MGG75) preferentially induce mitotic catastrophe, DNA damage and apoptosis in response to Volasertib treatment. (C-E) GFP and Myc cells were treated with/without Volasertib at 30 nM for 24 hours and immunostained for pHH3 with DAPI nuclear staining. Black and white pictures are shown for U87GFP and U87Myc to allow better visualization of nuclear morphology. % cells with abnormal mitosis (mitotic catastrophe)(D) and % pHH3 positive cells (E) were measured in at least 100 cells per experiment. In D and E, *, P<0.005. **, P<0.0001 (student t-test). (F) GFP and Myc cells were treated with indicated concentrations of Volasertib for 24 hours. Caspase3/7 activity was evaluated by Caspase-Glo 3/7 assay. (G) Immunoblotting of Myc, cleaved PARP (c-PARP), cleaved caspase3 (c-caspase 3) and p-H2Ax expression in GFP and Myc cells after treatment with Volasertib (0, 30, 100 nM) for 24 hours. Actin was used as a loading control.
Myc downregulation desensitizes glioblastoma cells to PLK1 inhibitors.
To further elucidate the role of Myc in regulating response to PLK1 inhibition, we used tetracyclin-regulatable shRNA knockdown of Myc expression in Myc-high Gli36 cells (Figure 6A). ShRNA sequence 2 (shRNA2) was effective at silencing Myc, while sequence 1 (shRNA1) was not, making it additional negative control to non-silencing (NS) sequence (Figure 6A). We found Myc knockdown rendered Gli36 cells less sensitive to PLK1 inhibitors (Figure 6B). Myc knockdown in Gli36 decreased the number of cells displaying mitotic catastrophe and pHH3 positivity (Figure 6C, Figure 6D, and Figure 6E) and inhibited pHH3 induction (Figure 6F) after Volasertib treatment. Furthermore, Myc knockdown decreased caspase3/7 activity, and accumulations of cleaved PARP and p-H2Ax after Volasertib treatment (Figure 6G and Figure 6H). Time course cell viability assay showed that Gli36 is Myc-addicted as Myc silencing slowed cell proliferation (Supplementary Figure S6E). These results suggest that Myc down-regulation protected Gli36 cells from cytotoxic effects of Volasertib. Taken together, our data show that inappropriate Myc activation renders glioblastoma cells sensitive to PLK1 inhibitors.
Figure 6. Myc silencing desensitizes glioblastoma cells to PLK1 inhibitors.
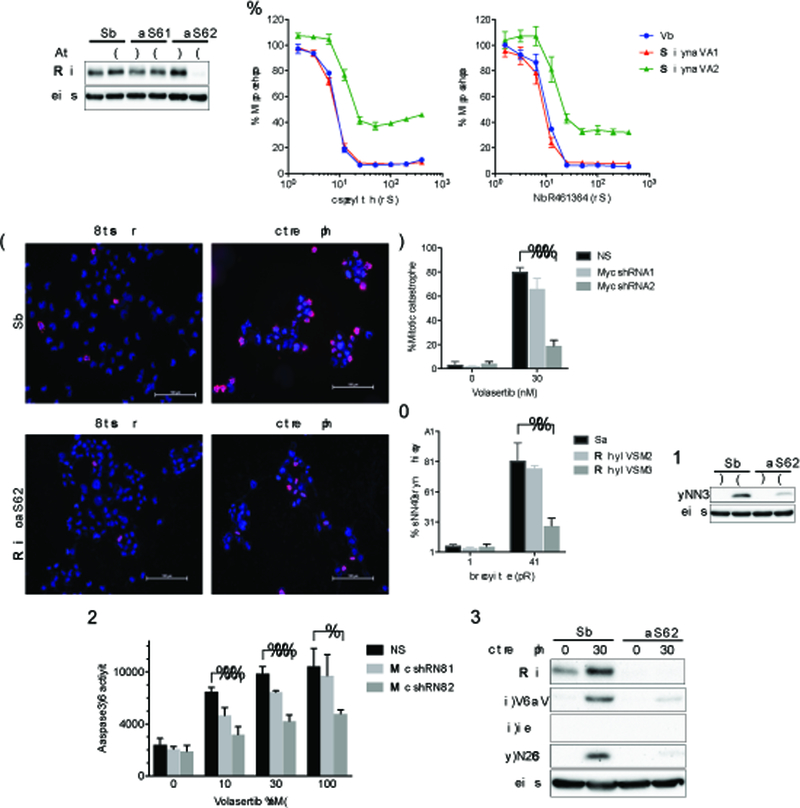
(A) Gli36 cells stably transduced with a non-targeting (shNS) or tetracycline-regulatable cMyc shRNA (shRNA1, shRNA2) vectors were established. Cells were lysed after exposed to doxycycline (Dox) for 72 hours. Immunoblotting confirmed a decrease in Myc by shRNA2. Actin was used as a loading control. (B) Gli36-shNS (NS, blue), Gli36-MycshRNA1 (Myc shRNA1, red) and Gli36c-MycshRNA2 (Myc shRNA2, green) cells were treated with different concentrations of Volasertib (left) and GSK461364 (right). Cell viability was evaluated by CellTiter Glo at 72 hours. (C-E) Gli36-NS (NS) and Gli36-MycshRNA2 (Myc shRNA2) cells were treated with/without Volasertib at 30 nM for 24 hours and immunostained for pHH3 (red) and counterstained with DAPI. Scale bars, 100 μm. % cells with abnormal mitosis (mitotic catastrophe) (D) and pHH3 positive cells (E) were evaluated in at least 100 cells per experiment. (F) Immunoblot of pHH3 expression in Gli36-NS and Gli36-MycshRNA2 cells with/without Volasertib treatment (30 nM, 24 hours). Actin was used as a loading control. (G) Gli36NS (NS), Gli36-MycshRNA1 (Myc shRNA1) and Gli36-MycshRNA2 (Myc shRNA2) cells were treated with indicated concentrations of Volasertib for 24 hours. Caspase3/7 activity was evaluated by Caspase-Glo 3/7 assay. (H) Immunoblot of Myc, cleaved PARP (c-PARP), cleaved caspase3 (c-caspase3) and p-H2Ax expression in Gli36-NS (NS) and Gli36-MycshRNA2 (shRNA2) cells after treatment with Volasertib (0, 30 nM) for 24 hours. Actin blot was used as a loading control. In D, E and G, *, P<0.01; **, P<0.005; ***, P<0.001 (student t-test).
Discussion
A major subset of recurrent glioblastoma and gliomas that have progressed after initial treatment are characterized by MMR deficiency due to mutations in MSH6 and other MMR relevant genes. Acquisition of MMR deficiency in gliomas is tightly associated with clinical history of TMZ chemotherapy and causes a characteristic hypermutant phenotype, which contributes to increased malignancy. MMR deficiency is a well-established mechanism of acquired resistance to TMZ, as intact MMR-mediated futile DNA repair is the major mechanism-of-action of TMZ. Effective therapy for MMR-defective, TMZ resistant recurrent glioma is lacking. Our screening of DNA damage modulators in isogenic MSH6-intact and knockdown TMZ-resistant glioma cells identified PLK1 inhibitors as a potent therapeutic that is independent of MMR status. PLK1 is a serine/threonine kinase which plays important roles throughout M phase of the cell cycle, including mitotic entry, centrosome maturation, bipolar spindle maturation, sister chromatid separation and cytokinesis, as well as regulating cell cycle and DNA damage response (34, 35, 43). PLK1 is highly expressed in a subgroup of glioblastoma compared with normal human astrocytes and expression is associated with patient prognosis (44, 45). These clinical data, as well as in vitro kinase screening assays using glioma cells, have previously identified PLK1 as a potential target in glioblastoma (37, 46). Here, we show that Volasertib, a small molecule inhibitor of PLK1, disrupted proliferation of glioblastoma cell lines and patient-derived glioma sphere lines with MSH6 knockdown and induced G2/M cell cycle arrest, mitotic catastrophe and DNA damage leading to apoptosis. Importantly, these Volasertib-mediated effects were completely equivalent in glioblastoma cells with proficient MSH6 and inactivated MSH6, whereas TMZ sensitivity was greatly impacted by MSH6 status. Previously, PLK1 inhibitor was shown to sensitize mutant IDH1-transduced astrocytes to TMZ (47), and to suppress glioblastoma cells resistant to TMZ via MGMT (48). Our current study showed that PLK1 inhibitor did not sensitize MSH6-knockdowned glioblastoma cells to TMZ. Therefore, our results indicate that cellular signaling pathways involved in response to PLK1 inhibitor and TMZ are distinct. Glioma cells are dependent on cell cycle and DNA damage signaling that is regulated by PLK1, and disruption of PLK1 signaling is lethal; deficiency in MMR that is typically acquired during the clinical course of glioma management does not alter the dependency of glioma cells on PLK1.
A second significant finding in the current work is the identification of Myc as a molecular marker of response to PLK1 inhibition. Being one of the Yamanaka pluripotency factors, Myc promotes the maintenance of glioma cancer stem cells capable of self-renewal and multi-lineage differentiation (14, 15). Previous studies have shown that PLK1 directly interacts with Myc and promotes Myc stabilization by phosphorylating Myc or protecting Myc from proteasome-mediated degradation (40–42). On the other hand, Myc directly activates PLK1 transcription, highlighting a PLK1-MYC axis that forms a positive feed-forward loop and reinforces Myc-regulated multiple oncogenic programs (40). Consistent with these studies, our study demonstrates that Myc overexpression upregulated PLK1 expression and sensitized glioma cells to PLK1 inhibition while Myc knockdown rendered glioma cells less responsive to PLK1 inhibition. Our data, however, showed that PLK1 inhibition did not induce substantive reduction of Myc protein levels, suggesting that Myc destabilization is not the primary mechanism of PLK1-induced cytotoxicity. Our genetic studies involving Myc overexpression and knockdown in glioma cells revealed that unrepaired DNA damage and mitotic catastrophe led to robust induction of apoptosis in Myc-deregulated cells, underscoring the role of PLK1 in maintaining the genomic integrity in those cells. In glioblastoma MYC or MYCN amplification has been found in 4.1% of the cases, less frequent than mutations of EGFR, PTEN, and PI3K-related genes (49). However, recent studies identified Myc amplification or mutations activating Myc pathway are one of the most frequent alterations that occur during malignant progression of lower grade gliomas (11). Myc appears to play a vital role in the evolution and progression of glioma by sustaining cancer stem cell subsets able to escape therapy and drive tumor relapse. We envision that recurrent glioma driven by dysregulated Myc may be particularly dependent on PLK1 to enable aberrant cell cycle progression and cell proliferation. PLK1 inhibition-mediated disruption of the PLK1-Myc malignant positive feedback could eliminate glioma stem cells that survived prior treatment and acquired resistance to chemotherapy, providing a promising targeted strategy for recurrent gliomas. It is possible that Myc and N-Myc are not functionally equivalent in terms of induction of sensitivity to PLK1 inhibition, and additional research is needed to elucidate this aspect.
We show proof-of-principal in vivo activity of PLK1 blockade in a subcutaneous model of MSH6-defective, TMZ-resistant glioma. Volasertib, however, does not have a favorable pharmacokinetics property to treat tumors in the brain (23), and our initial testing of systemic Volasertib as well as GSK461364 and BI2536 in an orthotopic model indicated poor activity. Efforts to improve and optimize pharmacological characteristics of newer generations of PLK1 inhibitors are essential in order to evaluate whether PLK1 inhibition represents a novel therapeutic option for patients with recurrent gliomas, including those MMR-deficient and TMZ-resistant.
Supplementary Material
Acknowledgements.
This work was supported by the Burroughs Wellcome Fund Career Award (D. Cahill) and NIH R01CA227821 (D. Cahill and H. Wakimoto). We thank Sakurako Endo for her assistance of animal experiments.
Footnotes
Conflicts of Interest:
All authors have no conflicts of interest to report with regard to this manuscript.
References
- 1.Wahl M, Phillips JJ, Molinaro AM, Lin Y, Perry A, Haas-Kogan DA, et al. Chemotherapy for adult low-grade gliomas: clinical outcomes by molecular subtype in a phase II study of adjuvant temozolomide. Neuro Oncol. 2017;19:242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Bent MJ, Baumert B, Erridge SC, Vogelbaum MA, Nowak AK, Sanson M, et al. Interim results from the CATNON trial (EORTC study 26053–22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: a phase 3, randomised, open-label intergroup study. Lancet. 2017;390:1645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology. 2009;10:459–66. [DOI] [PubMed] [Google Scholar]
- 5.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006;66:3987–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Thuijl HF, Mazor T, Johnson BE, Fouse SD, Aihara K, Hong C, et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta neuropathologica. 2015;129:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15:4622–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coskun S, Simon M, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nature reviews Cancer. 2004;4:562–8. [DOI] [PubMed] [Google Scholar]
- 13.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3:e3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tateishi K, Iafrate AJ, Ho Q, Curry WT, Batchelor TT, Flaherty KT, et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin Cancer Res. 2016;22:4452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wakimoto H, Kesari S, Farrell CJ, Curry WT, Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakimoto H, Mohapatra G, Kanai R, Curry WT, Yip S, Nitta M, et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012;14:132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res. 2014;20:2898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, et al. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15:3094–102. [DOI] [PubMed] [Google Scholar]
- 24.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. [DOI] [PubMed] [Google Scholar]
- 25.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–30. [DOI] [PubMed] [Google Scholar]
- 26.Sloane DA, Trikic MZ, Chu ML, Lamers MB, Mason CS, Mueller I, et al. Drug-resistant aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS Chem Biol. 2010;5:563–76. [DOI] [PubMed] [Google Scholar]
- 27.Mortlock AA, Foote KM, Heron NM, Jung FH, Pasquet G, Lohmann JJ, et al. Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J Med Chem. 2007;50:2213–24. [DOI] [PubMed] [Google Scholar]
- 28.Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol Cancer Ther. 2011;10:591–602. [DOI] [PubMed] [Google Scholar]
- 29.Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett. 2004;14:6083–7. [DOI] [PubMed] [Google Scholar]
- 30.Kuntz K, Salovich J, Mook R, Emmitte K, Chamberlain S, Rheault T, et al. Identification of GSK461364, a novel small molecule polo-like kinase 1 inhibitor for the treatment of cancer. 98th Annual Meeting of the American Association for Cancer Research; 2007. [Google Scholar]
- 31.Liu X Targeting Polo-Like Kinases: A Promising Therapeutic Approach for Cancer Treatment. Transl Oncol. 2015;8:185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48:8045–54. [DOI] [PubMed] [Google Scholar]
- 33.Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Current biology : CB. 2007;17:316–22. [DOI] [PubMed] [Google Scholar]
- 34.Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014;15:433–52. [DOI] [PubMed] [Google Scholar]
- 35.Weng Ng WT, Shin JS, Roberts TL, Wang B, Lee CS. Molecular interactions of polo-like kinase 1 in human cancers. J Clin Pathol. 2016;69:557–62. [DOI] [PubMed] [Google Scholar]
- 36.Pezuk JA, Brassesco MS, Morales AG, de Oliveira JC, de Paula Queiroz RG, Machado HR, et al. Polo-like kinase 1 inhibition causes decreased proliferation by cell cycle arrest, leading to cell death in glioblastoma. Cancer gene therapy. 2013;20:499–506. [DOI] [PubMed] [Google Scholar]
- 37.Tandle AT, Kramp T, Kil WJ, Halthore A, Gehlhaus K, Shankavaram U, et al. Inhibition of polo-like kinase 1 in glioblastoma multiforme induces mitotic catastrophe and enhances radiosensitisation. Eur J Cancer. 2013;49:3020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–60. [DOI] [PubMed] [Google Scholar]
- 39.Gilmartin AG, Bleam MR, Richter MC, Erskine SG, Kruger RG, Madden L, et al. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res. 2009;69:6969–77. [DOI] [PubMed] [Google Scholar]
- 40.Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, et al. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol Cell. 2016;64:493–506. [DOI] [PubMed] [Google Scholar]
- 41.Padmanabhan A, Li X, Bieberich CJ. Protein kinase A regulates MYC protein through transcriptional and post-translational mechanisms in a catalytic subunit isoform-specific manner. J Biol Chem. 2013;288:14158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan J, Li Z, Lee PL, Guan P, Aau MY, Lee ST, et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013;3:1156–71. [DOI] [PubMed] [Google Scholar]
- 43.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nature reviews Cancer. 2006;6:321–30. [DOI] [PubMed] [Google Scholar]
- 44.Dietzmann K, Kirches E, von B, Jachau K, Mawrin C. Increased human polo-like kinase-1 expression in gliomas. Journal of neuro-oncology. 2001;53:1–11. [DOI] [PubMed] [Google Scholar]
- 45.Lee C, Fotovati A, Triscott J, Chen J, Venugopal C, Singhal A, et al. Polo-like kinase 1 inhibition kills glioblastoma multiforme brain tumor cells in part through loss of SOX2 and delays tumor progression in mice. Stem Cells. 2012;30:1064–75. [DOI] [PubMed] [Google Scholar]
- 46.Danovi D, Folarin A, Gogolok S, Ender C, Elbatsh AM, Engstrom PG, et al. A high-content small molecule screen identifies sensitivity of glioblastoma stem cells to inhibition of polo-like kinase 1. PLoS One. 2013;8:e77053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koncar RF, Chu Z, Romick-Rosendale LE, Wells SI, Chan TA, Qi X, et al. PLK1 inhibition enhances temozolomide efficacy in IDH1 mutant gliomas. Oncotarget. 2017;8:15827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Triscott J, Lee C, Hu K, Fotovati A, Berns R, Pambid M, et al. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget. 2012;3:1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.