

Abstract
Unresectable hepatic metastases of colon cancer respond poorly to existing therapies and are a major cause of colon cancer lethality. In this study, we evaluated the therapeutic viability of targeting the Mediator kinase CDK8, an early clinical stage drug target, as a means to suppress metastasis of colon cancer. CDK8 was amplified or overexpressed in many colon cancers and CDK8 expression correlated with shorter patient survival. Knockdown or inhibition of CDK8 had little effect on colon cancer cell growth but suppressed metastatic growth of mouse and human colon cancer cells in the liver. This effect was due in part to inhibition of already established hepatic metastases, indicating therapeutic potential of CDK8 inhibitors in the metastatic setting. In contrast, knockdown or inhibition of CDK8 had no significant effect on the growth of tumors implanted subcutaneously, intrasplenically, or orthotopically in the cecum. CDK8 mediated colon cancer growth in the liver through downregulation of matrix metalloproteinase (MMP) inhibitor TIMP3 via TGFβ/SMAD-driven expression of a TIMP3-targeting microRNA, miR-181b, along with induction of Mmp3 in murine or MMP9 in human colon cancer cells via Wnt/beta-catenin-driven transcription. These findings reveal a new mechanism for negative regulation of gene expression by CDK8 and a site-specific role for CDK8 in colon cancer hepatic metastasis. Our results indicate the utility of CDK8 inhibitors for the treatment of colon cancer metastases in the liver and suggest that CDK8 inhibitors may be considered in other therapeutic settings involving TGFβ/SMAD or Wnt/β-catenin pathway activation.
Keywords: CDK8, colon cancer, hepatic metastasis, TIMP3, TGFβ/BMP
Introduction
Metastatic growth in the liver is a leading cause of death from colon cancer (1). Patients with unresectable hepatic metastases receive chemotherapy in combination with EGFR- or VEGFR-targeting antibodies, but these treatments have limited efficacy and novel therapeutic approaches are urgently needed.
Cyclin-dependent kinase 8 (CDK8) and its closely related paralog CDK19 are alternative subunits of the regulatory CDK module of the transcriptional Mediator complex (2). CDK8/19, unlike better-known members of the CDK family, are not required for cell cycle progression (2,3). Instead, CDK8 is a co-regulator of several transcription factors, such as Wnt/β-catenin (4), serum response network (5), TGFβ/Smad (6,7), HIF1A (8), ERα (9), STAT1 (10) and NFκB (11). CDK8 has been identified as an oncogene implicated in colon (4), breast (3,9,12,13), pancreatic (14) and prostate cancers (15), melanoma (16) and leukemia (17,18). CDK8 inhibition also stimulates natural killer cells (19) and increases innate immunity (20). Our work has identified CDK8 as a mediator of damage-induced gene expression associated with tumor-promoting paracrine activities (3). CDK8 provides an attractive anticancer drug target (2), and the first selective CDK8/19 inhibitor, Senexin B (21) has recently entered clinical trials.
CDK8 is an oncogene frequently amplified or overexpressed in colon cancer (4), where it was reported to be a positive regulator of HIF1A (8) and glycolysis (22); CDK8 expression is predictive of colon cancer-specific mortality (23). CDK8 knockdown was reported to inhibit colon cancer cell growth (4,24) but various CDK8/19 kinase inhibitors showed no significant effect on colon cancer cells (3,17,25). Here we show that the tumor-suppressive effects of CDK8 knockdown or inhibition in colon cancer are site-specific, affecting tumor growth in the liver but not in the cecum, the spleen or subcutaneously (s.c.) The effect of CDK8 on liver growth of colon cancer is exerted primarily through downregulation of matrix metalloproteinase (MMP) inhibitor TIMP3 via TGFβ/SMAD-driven expression of a TIMP3-targeting microRNA, miR-181b. CDK8 also stimulates expression of MMP3 in murine or MMP9 in human colon cancer cells via Wnt/β-catenin-driven transcription. Our results indicate potential utility of CDK8 inhibitors for treatment of colon cancer hepatic metastases and potentially in other settings involving TGFβ/SMAD or Wnt/β-catenin pathways.
Materials and Methods.
Cell lines and vectors.
Sources of cell lines, plasmids, antibodies and other materials and software are listed in Table S1. All cell lines were confirmed mycoplasma-free (MycoAlert PLUS kit, Lonza) and authenticated by STR profiling (University of Arizona). Plasmid pLenti6-TIMP3 was constructed by cloning full-length human TIMP3 cDNA from pRK5M-TIMP3 into the XbaI site of lentiviral vector pLenti6-TR. The resulting construct was named VTIMP3; GFP cDNA sequence cloned into the same vector was used as a control named VGFP. Most of constitutively expressing shRNA lentiviral plasmids were in pLKO.1 lentiviral vector; pLKO.1 insert-free vector and vector with scrambled shRNA (sh-scramble) were used as controls. shRNA targeting the 3’ UTR of human CDK8 was designed for highest knockdown efficacy and target selectivity score using the Broad Institute website and cloned into Tet-pLKO-puro lentiviral vector. Lentiviral production was carried out by the Functional Genomics Core (FGC) of the Center for Targeted Therapeutics at the University of South Carolina (USC) as described (9). shRNA was induced in puromycin selected cells using 1 μg/mL doxycycline for indicated time. For microRNA studies, cells were transfected with miRIDIAN microRNA Mimics and Inhibitors (2.5 μg per well of 6-well plate) using Lipofectamine™ 3000 per manufacturer’s protocol.
Gene expression measurements.
The concentration of Senexin B used in all the in vitro assays was 1 μM unless indicated otherwise. Microarray analyses were performed on Illumina microarray expression platform by Proteogenomics Facility at Medical University of South Carolina and on Affymetrix Mouse gene 2.0 X ST arrays by the FGC. Data were analyzed using Expression Console and deposited: GEO accession number GSE104963. Immunoblotting and qPCR mRNA measurements were carried out as previously described (9). qPCR primers are listed in Table S2. For miRNA analysis, RNA was isolated with QIAzol Reagent and polyadenylated with E. coli Poly(A) Polymerase; cDNA was prepared by Tetro Reverse Transcriptase kit using Universal RT primer (URT). miRNA expression was quantified by qPCR using Universal qPCR primer (UPCR) and the miRNA specific primer. qPCR assays were performed in triplicates unless indicated otherwise.
Mouse Studies.
All mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) of USC. Female BALB/cJ mice at 7–8 weeks for CT26 model or female nude mice at 7–8 weeks for HCT116 model were obtained from The Jackson Laboratory and housed at the Animal Research Facility at USC. CDK8/19 inhibitor Senexin B was administered to mice by one of three regimens, as indicated in figure legends: (i) daily intraperitoneal (i.p.) injection of 8 mg/mL Senexin B-dichloride in 10mM citrate buffer, 150 mM NaCl, pH6.0, to achieve 35mg/kg dose (i.p. daily); (ii) twice daily by oral gavage with 10 mg/mL Senexin B-dimaleate in 1% Dextrose, 6.25% 2-Hydroxypropyl-β-Cyclodextrin, to achieve 33 mg/kg dose (oral b.i.d.); (iii) by feeding with medicated diet (350 mg Senexin B-dimaleate per kg control diet, Research Diets, D12450B), supplemented with daily oral gavage at 33 mg/kg (diet + oral).
For splenic injection studies, mice were anesthetized with 2–5% isoflurane in oxygen delivered by an EZ anesthesia vaporizer. Buprenorphine was administered i.p. (0.05–0.1 mg/kg) as preemptive analgesia prior to surgery. A small abdominal incision was made about 1 cm above the spleen and spleen was exteriorized. 2 × 105 CT26 cells in 20 μl DMEM(HG) media (or 1×106 HCT116 cells in 100 μl of media) were injected into spleen. For standard metastasis studies on CT26 model, the site of injection was sealed with Tissue Adhesive (3M Vetbond™) and for survival studies on CT26 model and standard metastasis studies on HCT116 model, spleen was removed 1 min after injection. The abdominal incision was sutured; mice were given 500 μl of saline i.p. for hydration and kept in cages on a heating pad (37°C) for recovery from anesthesia. Buprenorphine was administered as a post-operative analgesic (0.05 mg/kg, s.c.) as needed. For standard metastasis studies, mice were euthanized 14–16 days (CT26 model) or 6 weeks after tumor inoculation (HCT116 model). For survival studies, mice were monitored daily and mice with limiting clinical signs such as continuous weight loss, anemia, impaired respiration, trembling convulsions, or permanent recumbency were euthanized.
For cecal injection studies, experiments were carried out as described (26). Briefly, 2 ×106 cells in 20 μL media were implanted subserosally under a stereo zoom microscope using the same procedures for anesthesia, sterilization and post-operative care as above. Mice were euthanized 30 days after injection.
For s.c. injection studies, cells (1×106 cells in 100 μL media) were injected s.c. into the right flanks of anesthetized mice. Tumor volumes were measured twice a week by measuring perpendicular tumor diameters (length and width, L>W); tumor volumes (V) were calculated using the formula V = L x W2/ 2. Mice received treatment when the tumor volume reached ~200 mm3. At the end of the study, mice were euthanized and tumor weights recorded.
Microscopic analysis.
The formalin-fixed tissues were processed, paraffin-embedded, sectioned at 5 μm and H&E stained. Quantification of tumor area was performed on H&E sections using ImageJ software. For immunofluorescence (IF) staining, sections were deparaffinized, rehydrated and blocked in PBS containing 5% goat serum and 0.3% Triton-X-100 for 1 h at room temperature and incubated with primary antibodies in TBST with 1% goat serum overnight at 4°C followed by goat secondary antibodies conjugated with fluorescent dyes for 1 hour at room temperature. After counterstaining with 300 nM DAPI in PBS for 1 min, sections were mounted with 80% glycerol in PBS. The H&E staining images were acquired with Leica DM1000 LED microscope with LEICA ICC50 HD camera, with 4 x objective lens (0.10 N.A.), using acquisition software Leica Application Suite EZ. IF images were taken with LEICA DMIRE2 microscope with photometric coolSNAP HQ camera, with 10 x objective lens (0.25 N.A.), using acquisition software Micro-Manager 1.4.22.
Statistical Analysis.
Most data were presented as Mean ± SEM of biological replicates. Meta-analysis of clinical data was conducted using SurvExpress. Survival data were analyzed by Kaplan Meier survival curves and the comparisons were performed through LogRank test using GraphPad Prism 5 software. Statistical significance was tested using unpaired Student’s t-test (two-tailed) and P values of less than 0.05 were considered significant.
Results
Inhibition of CDK8 does not block colon cancer cell proliferation in culture.
CDK8 is frequently amplified in colon cancers (4), and elevated CDK8 protein was associated with increased colon cancer specific mortality (23). Since CDK8 or its paralog CDK19 function in complex with cyclin C (CCNC), MED12 and MED13 (2), we used SurvExpress RNASeq database to investigate whether CDK8, CDK19, CCNC, MED12, MED13 and MED13L (a paralog of MED13) are differentially expressed between equal-size high-risk and low-risk populations of 797 colon cancer patients. Most of these genes, except CCNC and MED12, were expressed at much higher levels in the high-risk group, with CDK8 showing the greatest differential (Fig 1A). Kaplan-Meyer analysis showed shorter disease-specific survival of patients with highest CDK8 RNA expression (Fig. 1B), in agreement with previous protein studies (23).
Fig. 1. CDK8 expression correlates with colon cancer survival but does not affect colon cancer cell proliferation.
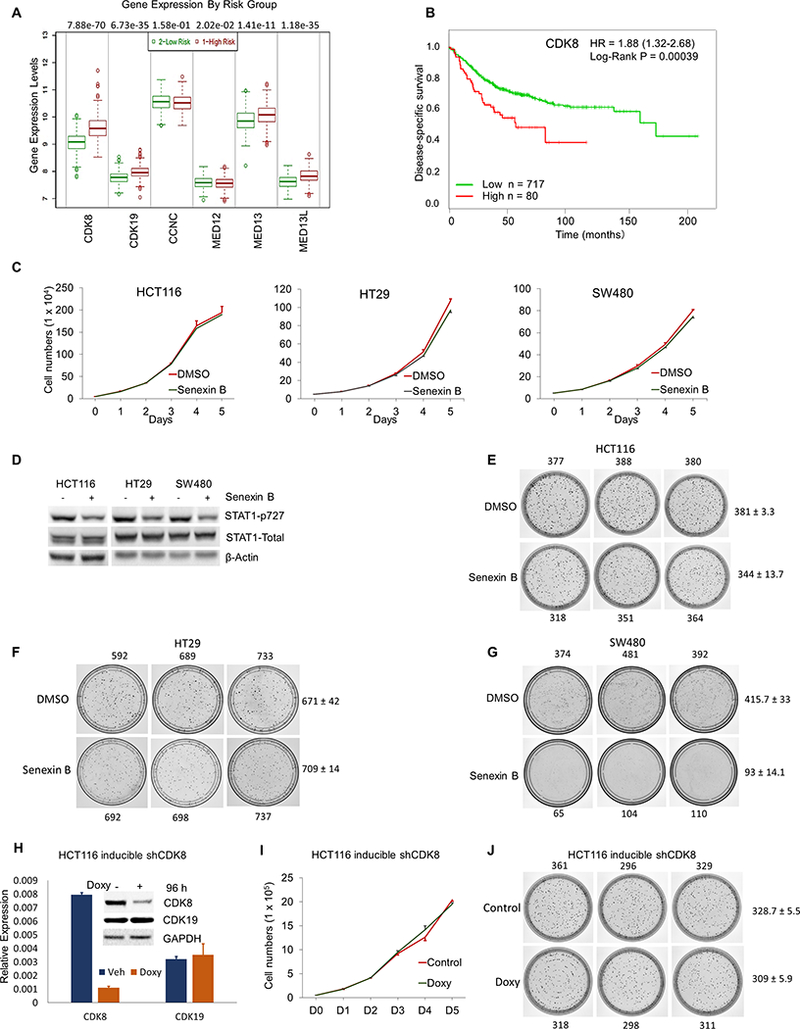
(A) Expression of Mediator-associated CDK module components in equal-sized high-risk and low-risk groups of 797 colon cancer patients. (B) Association of CDK8 expression with disease specific patient survival in the same dataset. (C) Effect of 1 μM Senexin B on the growth of human colon cancer cell lines (in triplicates). (D) Effects of Senexin B (3 hrs, 1 μM) on STAT1S727 phosphorylation and expression of STAT1 and CDK8 in human colon cancer cell lines. (E-G) Effects of 1 μM Senexin B on colony formation by HCT116 (E), HT29 (F) and SW480 (G) cells (14 days), colony numbers are indicated. (H) Effects of inducible CDK8 shRNA expression (96 hrs) on the expression of CDK8 and CDK19 RNA (qPCR) and proteins (inset) in HCT116 cells. Effects of inducible CDK8 knockdown on (I) HCT116 cell growth (in triplicates) and (J) colony formation by HCT116 cells.
We analyzed the effects of CDK8/19 inhibition on in vitro growth of three CDK8-overexpressing human colon cancer cell lines: HT29 and SW480 where CDK8 gene is amplified and HCT116 where CDK8 is overexpressed without amplification (4). CDK8 knockdown in these cells has been associated with decreased proliferation (4) but several CDK8/19 kinase inhibitors failed to suppress their growth (3,17,25). We treated these cell lines with a highly selective CDK8/19 kinase inhibitor Senexin B (21), which inhibits the growth of leukemias (18) and breast cancers (9). 5-day treatment with 1 μM Senexin B (corresponding to ~20 times IC50 in cell-based CDK8 activity assays) did not inhibit the growth of any cell line (Fig. 1C). In contrast, STAT1 phosphorylation at S727, which is exerted in part by CDK8 (10), was decreased by Senexin B (Fig. 1D). We have also carried out long-term (14-day) colony formation assays. 1 μM Senexin B did not decrease the colony number in HCT116 and HT29 compared to vehicle control (Fig. 1E,F) but image analysis showed that average colony size was moderately reduced (1.39-fold in HCT116, 1.85-fold in HT29). In contrast, SW480 (Fig. 1G) showed a decrease in both colony number (4.47-fold) and size (2.78-fold) upon Senexin B treatment. Hence, CDK8 kinase inhibition in CDK8-overexpressing colon cancer cells does not affect cell proliferation in the short term but moderately inhibits long-term cell growth in some cell lines.
Stable CDK8 knockdown in HCT116 and HT29 reduced cell growth in prior studies (4). However, establishment of stable CDK8 knockdown derivatives could involve additional changes; indeed, HCT116 cells with CDK8 overexpressed CDK19 (8). To avoid changes associated with cell selection, we introduced CDK8-targeting shRNA into HCT116 cells using a doxycycline-inducible vector. Doxycycline treatment decreased CDK8 expression in these cells, with no effect on CDK19 (Fig. 1H), but did not inhibit cell growth in a 5-day proliferation assay (Fig. 1I). In 14-day colony formation assays (Fig. 1J), doxycycline-induced CDK8 shRNA expression had minor effects on the colony number (1.06-fold) and average colony size (1.22-fold).
CDK8 inhibition selectively suppresses colon cancer growth in the liver.
As the primary model for in vivo studies, we used murine CT26 colon cancer cells, which form syngeneic tumors in Balb/c mice. CT26 carry a G12D KRAS mutation, homozygous deletion of CDKN2A and are wild-type for APC and TP53 (27) but Wnt/β-catenin responsive (28). Treatment with 1 μM Senexin B did not inhibit CT26 growth in short-term culture (Fig. 2A), despite inhibition of STAT1S727 phosphorylation (Fig. 2B) (CT26 do not form colonies). We introduced two different CDK8 shRNAs (to control for off-target effects) into CT26 cells, which robustly express CDK8 but not its isoform CDK19 (Fig. 2C); stable knockdown of CDK8 alone had no effect on the low CDK19 expression (Fig. 2C). Both CDK8 shRNAs gave only modest (statistically insignificant) growth inhibition in vitro relative to control cells expressing either insert-free vector or scrambled shRNA (Fig. 2D).
Fig. 2. Effects of CDK8 knockdown or inhibition on CT26 murine colon cancer cell growth in vitro and in vivo.
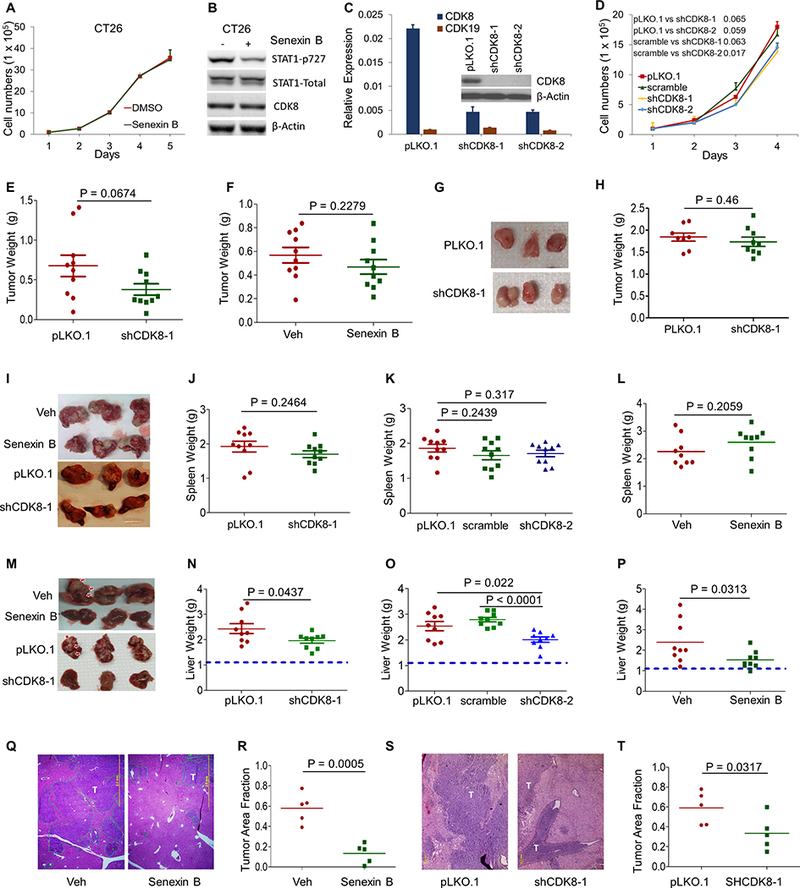
(A) Effect of Senexin B on the growth of CT26 cells (in duplicates). (B) Effects of Senexin B (3 hrs, 1 μM) on STAT1S727 phosphorylation and expression of STAT1 and CDK8 in CT26 cells. (C) Effects of CDK8 knockdown on the expression of CDK8 and CDK19. Knockdown was confirmed by qPCR and immunoblotting (inset; CDK19 was undetectable by immunoblotting). (D) Cell growth of control CT26 and CT26 with CDK8 knockdown (in duplicates). (E) Effect of CDK8 knockdown on the growth of s.c. tumors relative to control (CT26-pLKO.1) (n=10). (F) Effects of treatment with Senexin B (oral b.i.d.) on the growth of s.c. CT26 tumors relative to vehicle control (n=10). (G,H) Effect of CDK8 knockdown on orthotopic tumor growth (cecal injection) relative to pLKO.1 (n=10). Representative tumor images (G); final tumor weights (H). (I-T) Effects of CDK8 knockdown or inhibition on CT26 tumor growth in spleens and livers after splenic injection. (I, M) Representative images of tumor-bearing spleens (I) or livers (M). (J,K,N,O) Effect of CDK8 knockdown (shCDK8–1 and shCDK8–2) on tumor growth in spleens (J,K) or livers (N,O). Weights of tumor-bearing spleens and livers are shown. Dotted lines indicate average weight of corresponding normal livers. (L, P) Effects of Senexin B on tumor growth in spleens (L) or livers (P). Senexin B (i.p. daily) or vehicle control (n=9). (Q,S) Representative H&E staining of hepatic metastases from experiments in P and N, respectively. Scale bars: 2 mm. T: tumor. (R,T) Quantification of relative tumor area in the liver from experiments in P and N, respectively (n = 5).
We tested the effects of CDK8 knockdown and Senexin B treatment on in vivo growth of cells implanted subcutaneously (s.c.) in BALB/c mice and found only weak effects on tumor growth (Fig. 2E,F; Fig. S1A,B). CDK8 knockdown also had no effect on tumor growth in an orthotopic model (cecal injection) (Fig. 2G,H).
To evaluate the effects on metastatic tumor growth in the liver, we used a splenic injection model, where tumor cells are injected in the spleen, from where they rapidly migrate to the liver via the portal vein (29). 16 days after splenic injection, mice were euthanized and weights of tumor-bearing spleens and livers determined. Representative spleen images are in Fig. 2I. Neither CDK8 knockdown (two shRNAs, Fig. 2J,K) nor Senexin B treatment (Fig. 2L) had a significant effect on splenic tumor growth. In contrast, both CDK8 knockdown and Senexin B strongly decreased tumor growth in the liver (representative images in Fig. 2M), as indicated by liver weight (Fig. 2N-P) and histological measurements of tumor areas in liver sections (Fig. 2Q-T).
To determine the effect of CDK8 inhibition on mouse survival of hepatic metastasis, spleens were removed 1 min after tumor inoculation. Mice, euthanized upon morbidity, showed extensive liver metastasis (Fig. S1C). Kaplan-Meier plots show that both CDK8 knockdown in tumor cells and systemic Senexin B treatment significantly affect survival, with the combination of CDK8 knockdown and Senexin B further increasing this effect (Fig. 3A). Figs. 3B,C show independent experiments confirming the effects of Senexin B treatment on the survival of mice injected intrasplenically with parental (Fig. 3B) or CDK8 knockdown CT26 (Fig. 3C). The effect of Senexin B was much stronger on parental than CDK8 knockdown cells.
Fig. 3. Effects of CDK8 inhibition at different stages of liver metastatic growth.
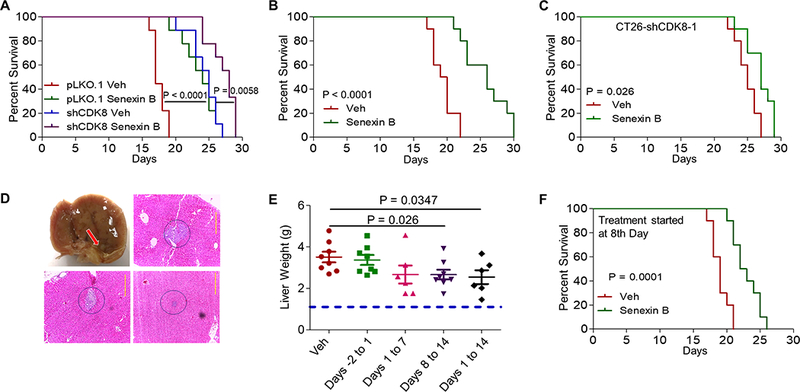
(A-C) Survival studies, Kaplan-Meier plots (n = 10 in all experiments). (A) pLKO.1 control and shCDK8–1 cell lines, treated with vehicle or Senexin B (oral b.i.d). (B) Parental CT26 cells treated with vehicle or Senexin B (diet + oral). (C) CT26-shCDK8–1 cells treated with vehicle or Senexin B as in (B). (D) Metastatic tumors in the livers of mice euthanized 7 days after splenic inoculation of CT26 cells, detectable macroscopically (upper left, tumor marked with arrow) and microscopically. Representative examples are shown. Scale bars: 0.2 mm. (E) Effects of Senexin B treatment at different stages of liver metastasis on tumor growth in liver. Mice (n=10) were treated with Senexin B (oral b.i.d.) over two days prior to splenic inoculation, during the first week after inoculation, during the second week after inoculation, or over two weeks after inoculation of CT26 cells. (F) Mouse survival study conducted as in (B) except drug treatment was initiated on day 8 after inoculation.
To determine if the effects of CDK8 inhibition on liver metastases were due to prevention of metastatic foci establishment or suppression of already established metastases, we asked whether CDK8/19 inhibition can affect tumor growth in the liver when the inhibitor is administered after establishment of metastases. Consistent with previously described time to liver metastasis in this model (30), we found macroscopically and microscopically detectable metastases in 3/3 livers of mice 7 days after splenic inoculation (Fig. 3D), indicating that any effects after this point would involve suppression of hepatic tumor growth. We compared the effects of Senexin B administered (i) for two days prior to splenic inoculation, (ii) on days 1–7 (day 1 is the inoculation day), (iii) on days 8–14, or (iv) on days 1–14. The weights of tumor-bearing livers are in Fig. 3E. Senexin B administration prior to inoculation had no effect on liver metastasis. Drug administration on days 1–7 decreased liver weights although this effect did not reach statistical significance. On the other hand, treatments administered on days 1–14 or 8–14 were efficacious in suppressing hepatic tumor growth, indicating that CDK8 inhibition suppresses already-established liver metastases (Fig. 3E). This conclusion was confirmed by a survival study, where Senexin B administration initiated on day 8 after splenic inoculation significantly extended mouse survival (Fig. 3F).
Effects of CDK8 are mediated via TIMP3 and MMP3 expression.
To understand the molecular mechanism of the effects of CDK8, a transcriptional regulator, on liver metastatic growth, we carried out transcriptomic analysis of the effects of CDK8 knockdown or Senexin B treatment in CT26 cells. Microarray hybridization revealed strong effects on several members of the matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinases (TIMP) families, specifically TIMP3 that was upregulated and MMP3, MMP10 and MMP13 that were downregulated by CDK8 knockdown or inhibition (Fig. 4A-C), as validated by qPCR (Fig. 4D). Senexin B mimicked the effects of CDK8 shRNA in control cells but not in cells with CDK8 knockdown (Fig. 4D). The effects of Senexin B on gene expression were concentration and time dependent (Fig. S2A, S2B). Immunoblotting (Fig. 4E) confirmed concentration-dependent TIMP3 induction and MMP3 inhibition by Senexin B. CT26 tumors growing in the livers of mice continuously treated with Senexin B also showed increased TIMP3 and decreased MMP3, MMP10 and MMP13 (Fig. S2C).
Fig. 4. CDK8 regulates the expression of TIMP3 and MMPs.
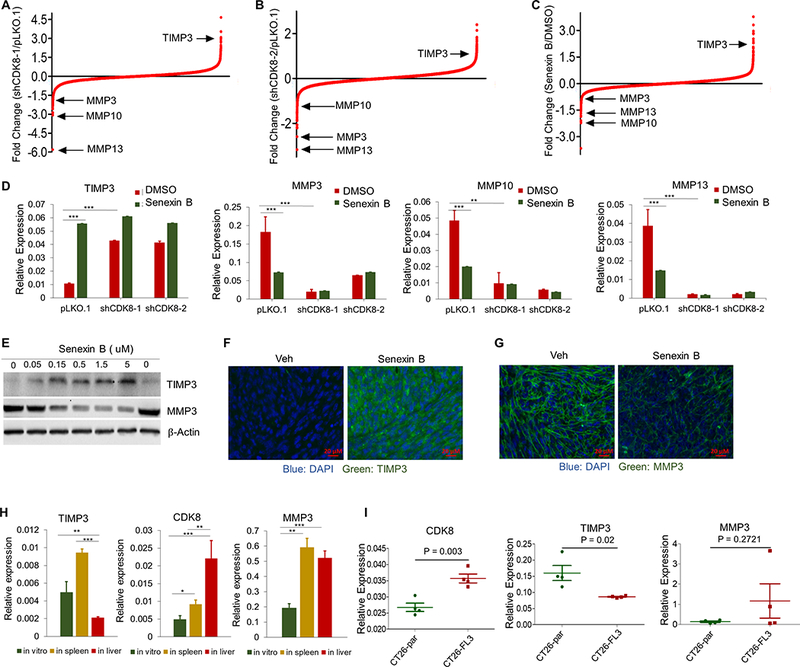
(A-C) Microarray analyses identify TIMP3, MMP3, MMP10 and MMP13 as CDK8 targets in CT26 cells. (A) Illumina microarray comparing CT26-shCDK8–1 vs. CT26-pLKO.1. (B) Affymetrix microarray comparing CT26-shCDK8–2 vs. CT26-pLKO.1. (C) Affymetrix microarray comparing parental CT26 treated with DMSO or Senexin B. (D) qPCR analysis of the effects of CDK8 knockdown on TIMP3, MMP3, MMP10 and MMP13 in the absence or presence of Senexin B (24 h treatment). (E) Effects of different Senexin B concentrations on TIMP3 and MMP3 protein expression. (F,G) Immunofluorescence analysis of TIMP3 (F) and MMP3 (G) proteins in hepatic metastases treated with Senexin B (diet + oral) on days 11–14 after splenic inoculation. Additional representative images are shown in Fig. S4. Scale bars: 20 μm. (H) Expression of CDK8, TIMP3 and MMP3 in CT26 cells growing in vitro or as tumors in spleen or in liver. Tumors were isolated from vehicle-treated mice (n= 3) 16 days after splenic inoculation. (I) Expression of CDK8, TIMP3 and MMP3 in tumors derived from parental CT26 cells and their highly metastatic CT26-FL3 derivatives (data from GEO: GSE67675). * denotes P<0.05, ** denotes P<0.01 and *** denotes P<0.001.
We compared RNA levels of TIMP3, MMP3, MMP10 and MMP13 in the normal intestine and CT26 s.c. tumors and found significant decrease in TIMP3 expression in the tumor relative to the normal intestine (Fig S2D). All three MMPs were elevated in tumors relative to the intestine but MMP3 was expressed at 1–2 orders of magnitude higher levels than MMP13 or MMP10 (Fig. S2E). Hence, subsequent testing was concentrated on MMP3, as well as TIMP3, a known inhibitor of CT26 liver metastasis (31). To determine if TIMP3 induction or MMP inhibition could be pharmacodynamic markers of CDK8 inhibition in colon cancer hepatic foci, we treated mice carrying established liver metastases with Senexin B or vehicle control, on days 11–14 after splenic injection. Immunofluorescence analysis showed strong TIMP3 induction in hepatic tumors (Fig. 4F, Fig. S3A) but not in normal liver tissues (Fig. S3B). Different effects of CDK8 in tumor and liver cells are not surprising since CDK8 functions are cell context specific (11). MMP3 inhibition in Senexin B-treated tumors was also readily apparent in tumor cells (Fig. 4G, Fig. S3C; MMP3 was undetectable in liver cells).
To determine if hepatic tumor growth is associated with altered CDK8, TIMP3 and MMP3 expression, we compared RNA expression of these genes between CT26 grown in culture or as tumors in spleen or liver. CDK8 was moderately elevated in splenic tumors relative to cells in culture but greatly elevated in hepatic tumors (Fig. 4H). TIMP3 was moderately increased in splenic tumors relative to cultured cells but decreased in hepatic tumors. In contrast, MMP3 was increased to a similar degree in both splenic and hepatic tumors relative to cells in culture (Fig. 4H). We also analyzed transcriptomic data from tumors formed by the parental CT26 and their derivative CT26-FL3 selected in vivo for a high rate of hepatic metastasis following cecal implantation (26). Remarkably, CDK8 was significantly elevated and TIMP3 downregulated in highly metastatic relative to parental cells, whereas MMP3 elevation was not statistically significant (Fig. 4I).
To test the effect of TIMP3 on CT26 growth in the liver, we overexpressed human TIMP3 in CT26 cells (Fig. 5A, Fig. S3D). This overexpression had no effect on endogenous murine TIMP3 (Fig. S3D), CDK8 or MMP3 (Fig. S3E). TIMP3 overexpression did not affect cell growth in vitro (Fig. S3F) or in vivo, when injected s.c. (Fig. 5B, Fig. S3G) or in the cecum (Fig. 5C, Fig. S3H). However, TIMP3 expression significantly increased mouse survival of liver metastasis in the splenic injection model (Fig. 5D). The effect of Senexin B treatment on mouse survival of tumors with TIMP3 overexpression (Fig. 5E) was much weaker than its effect on parental CT26 tumors (Fig. 3A,B) and similar to its effect on tumors with CDK8 knockdown (Fig. 3A,C).
Fig. 5. Effects of TIMP3 and MMP3 on CT26 tumor growth.
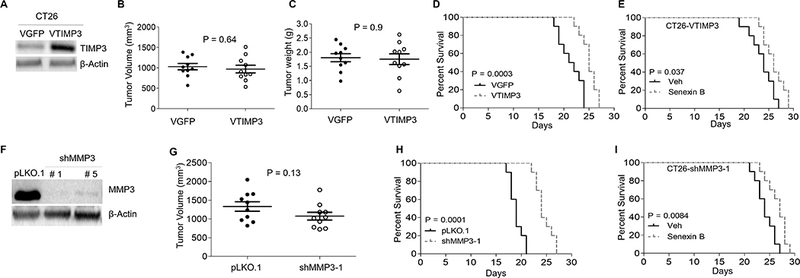
(A) Expression of TIMP3 protein in CT26-VTIMP3 and CT26-VGFP determined by immunoblotting. (B) Final tumor volumes of VTIMP3 and VGFP derivatives injected s.c. (n=10). (C) Weights of orthotopic VTIMP3 and VGFP tumors (n=10). (D) Kaplan-Meier survival plots of mice following splenic injection of VTIMP3 and VGFP cells. (E). Kaplan-Meier survival plots of mice following splenic injection with VTIMP3 cells and treatment with vehicle or Senexin B (diet + oral). (F) Immunoblotting analysis of MMP3 protein knockdown with different shRNAs. (G) Final tumor volumes of control and MMP3 knockdown derivatives injected s.c. (n=10. (H) Kaplan-Meier survival plots of mice following splenic injection of control and MMP3 knockdown cells. (E). Kaplan-Meier survival plots of mice following splenic injection with MMP3 knockdown cells treated with vehicle or Senexin B (diet + oral).
MMP3 knockdown (Fig. 5F, Fig. S3I) had no significant effect on CDK8 or TIMP3 expression (Fig. S3J), cell growth in vitro (Fig. S3K) or tumor growth s.c. (Fig. 5G, Fig. S3L) but strongly increased mouse survival in the splenic injection model (Fig. 5H) and lessened the survival effect of Senexin B (Fig. 5I). These results identify TIMP3 upregulation and MMP3 downregulation as events mediating the suppression of hepatic metastasis by CDK8 inhibition.
CDK8 regulates TIMP3 and MMP3 expression and hepatic metastasis through effects on TGFβ/SMAD-regulated miR-181b expression and the Wnt/β-catenin pathway.
TIMP3 can be regulated by TGFβ/SMAD both positively (32) and negatively, via SMAD-dependent microRNA expression (33,34). Importantly, SMAD activity is potentiated by CDK8 (6,7). To examine the role of TGFβ/SMAD in TIMP3 regulation by CDK8, we knocked down SMAD4, the common mediator of R-SMAD transcription factors (35) (Fig. 6A,B). SMAD4 knockdown had no significant effect on cell growth in vitro (Fig. S4A). While CDK8 inhibition had no effect on SMAD4 expression (Fig. 6A), SMAD4 knockdown, like CDK8 inhibition, induced TIMP3 both at RNA (Fig. 6A) and protein levels (Fig. 6B). The relative effect of Senexin B on TIMP3 was greatly diminished by SMAD4 knockdown and the relative effect of SMAD4 knockdown was similarly reduced by Senexin B (Fig. 6A). On the other hand, SMAD4 knockdown, in contrast to CDK8 inhibition, appeared to induce rather than inhibit MMP3 expression and did not interfere with MMP3 inhibition by Senexin B (Fig. S4B). These results indicate that the effect of CDK8 on TGFβ/SMAD mediates to a large extent the effect of CDK8 on TIMP3 (but not MMP3) transcription.
Fig. 6. Regulatory mechanisms of CDK8-controlled TIMP3 and MMP3 expression.
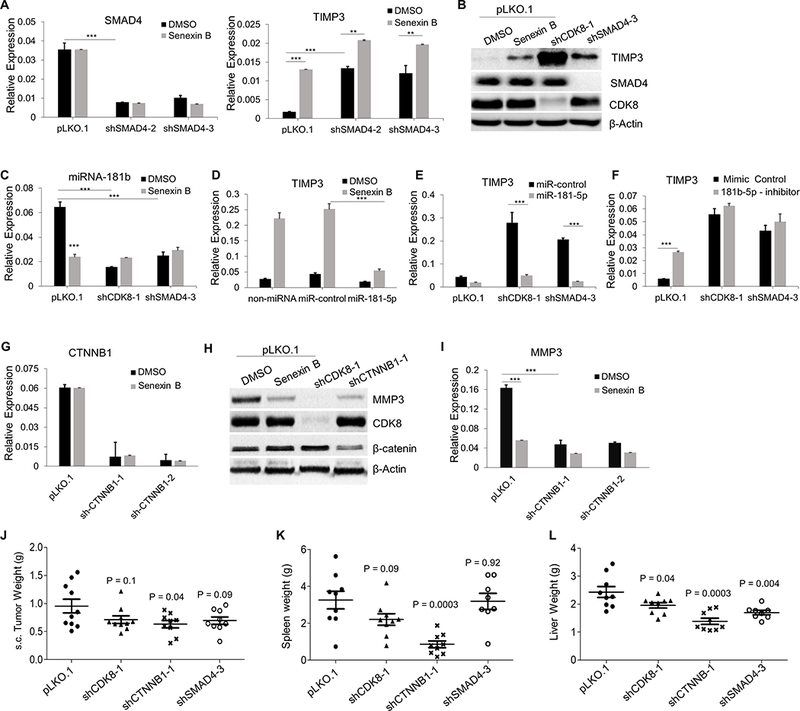
(A,B) Effects of SMAD4 knockdown on the expression of SMAD4 and TIMP3 in CT26 cells treated with Senexin B (24 hr): (A) RNA (qPCR, in duplicates), (B) protein (immunoblotting). (C) Effects of CDK8 and SMAD4 knockdown on the expression of miR-181b (qPCR, in duplicates) in CT26 cells treated with Senexin B (24 hr). (D) RNA expression of TIMP3 in CT26 cells transfected with control miRNA mimic or miR-181b mimic and treated with Senexin B (18 h); Senexin B treatment started 8 h after transfection (qPCR, in duplicates). (E) Expression of TIMP3 in CT26-pLKO.1, CT26-shCDK8 and CT26-shSMAD4 cells transfected with control miRNA mimic or miR-181b mimic (qPCR, in duplicates). (F) Expression of TIMP3 in CT26-pLKO.1, CT26-shCDK8 and CT26-shSMAD4 cells transfected with control miRNA mimic or miR-181b inhibitor (qPCR, in duplicates). (G-I) Effects of CTNNB1 knockdown on the expression of MMP3 in CT26 cells treated with Senexin B or vehicle control (24 hr) (G,I qPCR, in duplicates; H, immunoblotting). (J) Effects of CDK8, CTNNB1 and SMAD4 knockdown on the growth of CT26 s.c. tumors. (K, L) Effects of CDK8, CTNNB1 and SMAD4 knockdown on CT26 growth in spleen (K) and liver (L) after splenic injection. * denotes P<0.05, ** denotes P<0.01 and *** denotes P<0.001.
Since TIMP3 was reported to be inhibited by several microRNAs including TGFβ/SMAD-regulated miR-181b, as well as miR-21a, miR-21b, miR-125b1, miR-125b2 and miR-712 (36–38), we measured the effects of Senexin B and CDK8 or SMAD4 knockdown on the expression of these microRNAs. While most microRNAs were unaffected by CDK8 inhibition, Senexin B and CDK8 or SMAD4 knockdown decreased miR-181b levels (Fig. S4C). The relative effect of Senexin B was greatly decreased in CDK8 or SMAD4 knockdown cells (Fig. 6C). A mimic of miR-181b transfected into parental CT26 cells prevented TIMP3 induction upon treatment with Senexin B or by CDK8 or SMAD4 knockdown compared to control cells (Fig. 6D,E). Conversely, transfection of miR-181b antagonist into parental CT26 strongly induced TIMP3 expression, with a much weaker effect in cells with CDK8 or SMAD4 knockdown (Fig. 6F). These results indicate that CDK8 inhibition induces TIMP3 via SMAD-regulated expression of miR-181b. Notably, while CDK8 regulated miR-181b (Fig. 6C), miR-181b or its antagonist had no effect on CDK8 expression (Fig. S4D).
Several MMPs, including MMP3, are known transcriptional targets of wnt/β-catenin (39), which is regulated by CDK8 (4). To determine if the effect of CDK8 on MMP3 could be mediated through wnt/β-catenin, we generated β-catenin (CTNNB1) knockdown derivatives of CT26 (Fig. 6G,H). CTNNB1 knockdown had no significant effect on cell growth in vitro (Fig. S4A). Senexin B did not affect CTNNB1 expression (Fig. 6G) but CTNNB1 knockdown mimicked CDK8 inhibition by downregulating MMP3 at both protein (Fig. 6H) and RNA levels (Fig. 6I). The relative effect of Senexin B on MMP3 was greatly diminished upon CTNNB1 knockdown and the relative effect of CTNNB1 knockdown was similarly reduced by Senexin B (Fig. 6I). In addition to the strong effect on MMP3, CTNNB1 knockdown moderately induced TIMP3 and showed a modest effect on TIMP3 induction by Senexin B (Fig. S4E). These results suggest that the Wnt/β-catenin pathway mediates to a large extent the effect of CDK8 on MMP3 and to a lesser extent on TIMP3.
We have also tested if STAT1, another pleiotropic transcription factor known to be co-regulated by CDK8 (10), is involved in TIMP3 and MMP3 regulation. While Senexin B inhibited STAT1 phosphorylation at S727 (Fig. 2B), STAT1 knockdown (Fig. S4F) had no effect on TIMP3 or MMP3 expression (Fig. S4G).
We compared the effects of the knockdowns of CDK8, SMAD4 and CTNNB1 on tumor growth in vivo, using s.c. and splenic injection models. CTNNB1 knockdown significantly inhibited tumor growth s.c. (Fig. 6J) and strongly inhibited tumor growth in the spleen (Fig. 6K), whereas neither CDK8 nor SMAD4 knockdown had significant effects at these sites (Fig. 6J,K). In contrast, hepatic tumor growth was significantly suppressed by CDK8, CTNNB1 or SMAD4 knockdown (Fig. 6L). Hence, both TGFβ/SMAD and Wnt/β-catenin pathways regulate the growth of hepatic metastases in this model, but only TGFβ/SMAD inhibition, like CDK8 inhibition, selectively affects liver metastasis.
CDK8 inhibition induces TIMP3, inhibits MMP9 and suppresses hepatic metastasis in human colon cancer cells.
The effects of Senexin B on TIMP3 and different MMPs were analyzed in a panel of 10 human colon cancer cell lines. Senexin B induced TIMP3 expression, with statistically significant induction in 7 of 10 lines (Fig. 7A). Among the MMPs, MMP9 was inhibited by Senexin B in most cell lines (Fig. 7B), whereas MMP3, MMP10, MMP13, MMP2 and MMP7 were not significantly affected by the inhibitor (Fig. S5A). Inducible expression of CDK8 shRNA in HCT116 cells also induced TIMP3 and inhibited MMP9, suppressing the effects of Senexin B on these genes (Fig. 7C-D). We analyzed the effect of Senexin B on liver metastatic growth of human HCT116 cells in nude mice (Fig. 7E-H). Following splenic injection and spleen resection, mice were treated with Senexin B or vehicle control. 7 weeks after tumor inoculation, mice were euthanized and livers, all of which contained tumor metastases (Fig. 7E), were weighed. Analysis of liver weights (Fig. 7F) and tumor areas in the liver (Fig. 7G,H) showed strong inhibition of hepatic tumor growth by Senexin B. Notably, even the long (7-week) Senexin B treatment showed no apparent toxicity (mouse body weights over the course of the study increased by 9.4±1.3% in the control group and by 7.1±1.4% in the Senexin B-treated group). In contrast to its effect on tumor growth in the liver, Senexin B did not inhibit the growth of HCT116 tumors s.c. (Fig. S5B).
Fig. 7. Effects of CDK8 inhibition in human colon cancer models.
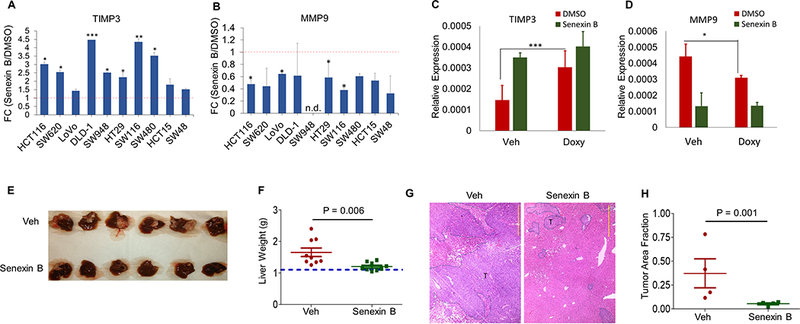
(A,B) qPCR analysis of the effects of Senexin B (24 hr) on the expression of (A) TIMP3 and (B) MMP9 in human colon cancer cell lines. (B) qPCR analysis of the effects of Senexin B (24 hr) on the expression of MMP9 in the indicated human colon cancer cell lines. (C-D): qPCR analysis of the effects of inducible CDK8 shRNA on TIMP3 and MMP9 expression in HCT116 cells. Cells were treated with doxycycline or vehicle control for 72 hrs, Senexin B was added for the last 24 h of treatment (qPCR in duplicates). (E-H) Effect of Senexin B on HCT116 hepatic metastasis. Vehicle or Senexin B (oral b.i.d., 5 days a week) (n=9) were administered for 6 weeks. E: Representative images of HCT116 tumor-bearing livers in mice treated with Senexin B or vehicle control. F: Weights of tumor-bearing livers. G: Representative H&E staining of hepatic metastases. T: tumor. Scale bars: 1 mm. H: quantitation of relative tumor areas in the liver (n = 4). * denotes P<0.05, ** denotes P<0.01 and *** denotes P<0.001.
Discussion
CDK8 is amplified in a subset of human colon cancers (4), where its expression is a negative prognostic marker (23). Our analysis confirms this association and shows that not only CDK8 but also CDK19 and their interactive proteins MED13 and MED13L are expressed at a higher level in high-risk colon cancers (Fig. 1A-B). CDK8 knockdown was reported to inhibit proliferation of CDK8-overexpressing colon cancer cells in vitro (4) and in vivo (24,40) but various CDK8/19 kinase inhibitors failed to suppress colon cancer growth (3,17,25). (Moderate effects on some cell lines were observed with CDK8/19 inhibitors that also showed pronounced toxicity (41) and transcriptomic effects suggesting possible off-target activities (18)). Here CDK8 kinase inhibition or inducible knockdown did not affect cell proliferation in three colon cancer cell lines with CDK8 amplification or overexpression in a 5-day assay and inhibited long-term colony formation in only one cell line.
Using colon cancer cells that do not respond to CDK8 inhibition in vitro, we investigated the in vivo effects on tumors implanted at different sites and on liver metastases arising after splenic injection. CDK8 inhibition or knockdown had no significant effect on tumor growth s.c., in the spleen or in the cecum. However, both CDK8 knockdown and kinase inhibition strongly suppressed metastatic growth in the liver, in both mouse CT26 and human HCT116 models. Importantly, the effects of the CDK8 inhibitor were associated with no apparent toxicity and no loss in mouse body weights. Furthermore, CDK8 expression was elevated in CT26 tumors in the liver and in a subline of CT26 cells selected for increased propensity for liver metastasis. Remarkably, a recent study identified CDK8 as one of the few genes that showed selective gene amplification in clinical liver metastases relative to matched primary tumors (42).
CDK8 inhibitor treatment initiated after the establishment of liver metastases suppressed metastatic growth, indicating that this effect is exerted at least in part through the suppression of already established metastases. Many anticancer drug candidates are efficacious in primary but not metastatic tumor models and subsequently fail in clinical trials of metastatic cancer (43). Our results reveal CDK8/19 inhibitors as a remarkable exception to this rule, active against the metastatic disease even when inefficient against the primary tumors.
Remarkably, one of the genes most strongly induced by CDK8 inhibition was TIMP3 and three of the most downregulated genes were MMPs. The network of MMPs and TIMPs (the natural MMP inhibitors) plays a major role in tumor progression in cancer. MMPs play key roles in a variety of biological processes aside from matrix degradation, including growth factor receptor signaling, angiogenesis, cell adhesion and apoptosis (44). The principal affected MMP in murine cells was MMP3, whereas MMP9 was inhibited in a panel of human cell lines; remarkably, MMP3 is a proteolytic activator of MMP9 (45). Interestingly, MMP9 gene, like CDK8, was amplified in liver metastases relative to matched primary colon cancers (46). CDK8 was found to regulate MMP3 expression primarily through wnt/β-catenin, previously shown to be co-regulated by CDK8 (4). To the best of our knowledge, this is the first example of an in vivo effect of CDK8/19 kinase inhibitors on the wnt/β-catenin pathway.
The induction of TIMP3, a suppressor of invasion, metastasis and angiogenesis in different cancers, including colorectal (31), was the most consistent pertinent transcriptional effect of CDK8 inhibition in all the colon cancer cell lines. Furthermore, TIMP3 was induced even by the lowest tested concentrations of Senexin B that had no effect on MMPs. Both liver-derived tumors and a CT26 derivative with increased propensity for liver metastasis (26) showed concurrent upregulation of CDK8 and downregulation of TIMP3. Both TIMP3 induction and MMP3 downregulation were observed upon CDK8 inhibition at primary and metastatic sites, as well as in cell culture. In contrast, TIMP3 overexpression or MMP3 knockdown suppressed the growth of liver metastases but not of primary tumors, indicating that the site selectivity of CDK8 action likely reflects environment-specific roles of TIMP3 and MMPs. Notably, the later stages of liver metastatic growth require angiogenesis (which is suppressed by TIMP3) and involve deposition of fibrosis-like matrix (susceptible to proteinases) that restrains metastases (47).
TIMP3 inhibition by CDK8 was mediated via induction of miR-181b that directly targets TIMP3 (33,34). While there are reports of microRNAs that regulate CDK8 expression, this is the first example of a microRNA regulated by CDK8 and mediating its biological activity. Many mechanisms of positive regulation of transcription by CDK8 have been elucidated (5,8,9,11), but there are fewer known mechanisms of negative regulation by CDK8 in mammalian cells (48). Our findings raise the possibility that positive regulation of microRNAs could be a general mechanism for negative regulation of gene expression by CDK8.
miR-181b induction by CDK8 was found to be mediated via TGFβ/SMAD, previously shown to be regulated by CDK8 through R-SMAD linker phosphorylation (6). BMP-4, a member of the TGFβ/BMP family, is universally upregulated in colon cancer cells and tissues (49) and TGFβ/SMAD have been implicated in colon cancer metastasis (50). We found that SMAD4 knockdown selectively inhibits hepatic metastasis but not colon cancer cell growth in vitro, s.c. or in the spleen. The remarkably similar site-specificity of the effects of SMAD4 knockdown and CDK8 inhibition identifies CDK8 as a druggable mediator of in vivo tumor-promoting effects of TGFβ/SMAD.
Given the major contribution of hepatic metastasis to colon cancer mortality, the observed effects of CDK8 inhibition on metastatic growth in the liver warrants exploring the use of CDK8-targeting drugs for the treatment of hepatic metastasis of colon cancer. Furthermore, the results of the present study suggest that CDK8 inhibitors may potentially be useful in other therapeutic settings involving TGFβ/SMAD and wnt/β-catenin pathways.
Supplementary Material
Significance: Findings demonstrate that inhibition of the transcription-regulating kinase CDK8 exerts a site-specific tumor-suppressive effect on colon cancer growth in the liver, representing a unique therapeutic opportunity for the treatment of advanced colon cancer.
Acknowledgments.
We thank Diego Altomare and Functional Genomics Core of the USC Center for Targeted Therapeutics and Proteogenomics Facility at Medical University of South Carolina for microarray analysis.
Financial support: NIH P30GM103336 (I.B. Roninson, M.M. Pena, E.V. Broude), NIH P20RR016461 (I.B. Roninson, J. Liang), NIH P20GM109091 (E.V. Broude, K. Mythreye, I.B. Roninson), NIH R01CA154731 (M.M. Pena) and SPARC Graduate Student Research grant from the University of South Carolina (J. Liang).
Footnotes
Conflict of Interest statement: I.B.R. is the Founder and J.L., M.C. and E.V.B. are consultants of Senex Biotechnology, Inc.
References
- 1.Abdalla EK, Adam R, Bilchik AJ, Jaeck D, Vauthey JN, Mahvi D. Improving resectability of hepatic colorectal metastases: expert consensus statement. Ann Surg Oncol 2006;13:1271–80 [DOI] [PubMed] [Google Scholar]
- 2.Philip S, Kumarasiri M, Teo T, Yu M, Wang S. Cyclin-Dependent Kinase 8: A New Hope in Targeted Cancer Therapy? J Med Chem 2018;61:5073–92 [DOI] [PubMed] [Google Scholar]
- 3.Porter DC, Farmaki E, Altilia S, Schools GP, West DK, Chen M, et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc Natl Acad Sci U S A 2012;109:13799–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008;455:547–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donner AJ, Ebmeier CC, Taatjes DJ, Espinosa JM. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol 2010;17:194–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009;139:757–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serrao A, Jenkins LM, Chumanevich AA, Horst B, Liang J, Gatza ML, et al. Mediator kinase CDK8/CDK19 drives YAP1-dependent BMP4-induced EMT in cancer. Oncogene 2018. doi: 10.1038/s41388-018-0316-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013;153:1327–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDermott MS, Chumanevich AA, Lim CU, Liang J, Chen M, Altilia S, et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget 2017;8:12558–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013;38:250–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, Liang J, Ji H, Yang Z, Altilia S, Hu B, et al. CDK8/19 Mediator kinases potentiate induction of transcription by NFkappaB. Proc Natl Acad Sci U S A 2017;114:10208–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Broude EV, Gyorffy B, Chumanevich AA, Chen M, McDermott MS, Shtutman M, et al. Expression of CDK8 and CDK8-interacting Genes as Potential Biomarkers in Breast Cancer. Curr Cancer Drug Targets 2015;15:739–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu D, Li CF, Zhang X, Gong Z, Chan CH, Lee SW, et al. Skp2-macroH2A1-CDK8 axis orchestrates G2/M transition and tumorigenesis. Nat Commun 2015;6:6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu W, Wang Z, Zhang W, Qian K, Li H, Kong D, et al. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/beta-catenin signaling pathway. Cancer Lett 2015;356:613–27 [DOI] [PubMed] [Google Scholar]
- 15.Bragelmann J, Klumper N, Offermann A, von Massenhausen A, Bohm D, Deng M, et al. Pan-Cancer Analysis of the Mediator Complex Transcriptome Identifies CDK19 and CDK8 as Therapeutic Targets in Advanced Prostate Cancer. Clin Cancer Res 2017;23:1829–40 [DOI] [PubMed] [Google Scholar]
- 16.Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010;468:1105–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015;526:273–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rzymski T, Mikula M, Zylkiewicz E, Dreas A, Wiklik K, Golas A, et al. SEL120–34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget 2017;8:33779–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Putz EM, Gotthardt D, Hoermann G, Csiszar A, Wirth S, Berger A, et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep 2013;4:437–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johannessen L, Sundberg TB, O’Connell DJ, Kolde R, Berstler J, Billings KJ, et al. Small-molecule studies identify CDK8 as a regulator of IL-10 in myeloid cells. Nat Chem Biol 2017;13:1102–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roninson IB, Porter DC, Wentland MP CDK8-CDK19 selective inhibitors and their use in anti-metastatic and chemopreventative methods for cancer. U.S. Patent 9,321,737, filed 2013, issued 2016.
- 22.Galbraith MD, Andrysik Z, Pandey A, Hoh M, Bonner EA, Hill AA, et al. CDK8 Kinase Activity Promotes Glycolysis. Cell Rep 2017;21:1495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Firestein R, Shima K, Nosho K, Irahara N, Baba Y, Bojarski E, et al. CDK8 expression in 470 colorectal cancers in relation to beta-catenin activation, other molecular alterations and patient survival. Int J Cancer 2010;126:2863–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adler AS, McCleland ML, Truong T, Lau S, Modrusan Z, Soukup TM, et al. CDK8 maintains tumor dedifferentiation and embryonic stem cell pluripotency. Cancer Res 2012;72:2129–39 [DOI] [PubMed] [Google Scholar]
- 25.Koehler MF, Bergeron P, Blackwood EM, Bowman K, Clark KR, Firestein R, et al. Development of a Potent, Specific CDK8 Kinase Inhibitor Which Phenocopies CDK8/19 Knockout Cells. ACS Med Chem Lett 2016;7:223–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Davis C, Ryan J, Janney C, Pena MM. Development and characterization of a reliable mouse model of colorectal cancer metastasis to the liver. Clin Exp Metastasis 2013;30:903–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castle JC, Loewer M, Boegel S, de GJ, Bender C, Tadmor AD, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics 2014;15:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong HJ, Jang GB, Lee HY, Park SR, Kim JY, Nam JS, et al. The Wnt/beta-catenin signaling/Id2 cascade mediates the effects of hypoxia on the hierarchy of colorectal-cancer stem cells. Sci Rep 2016;6:22966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lafreniere R, Rosenberg SA. A novel approach to the generation and identification of experimental hepatic metastases in a murine model. J Natl Cancer Inst 1986;76:309–22 [PubMed] [Google Scholar]
- 30.Vidal-Vanaclocha F The prometastatic microenvironment of the liver. Cancer Microenviron 2008;1:113–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin H, Zhang Y, Wang H, Xu D, Meng X, Shao Y, et al. Tissue inhibitor of metalloproteinases-3 transfer suppresses malignant behaviors of colorectal cancer cells. Cancer Gene Ther 2012;19:845–51 [DOI] [PubMed] [Google Scholar]
- 32.Qureshi HY, Ricci G, Zafarullah M. Smad signaling pathway is a pivotal component of tissue inhibitor of metalloproteinases-3 regulation by transforming growth factor beta in human chondrocytes. Biochim Biophys Acta 2008;1783:1605–12 [DOI] [PubMed] [Google Scholar]
- 33.Wang B, Hsu SH, Majumder S, Kutay H, Huang W, Jacob ST, et al. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene 2010;29:1787–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Q, Zheng X, Chen L, Xu B, Yang X, Jiang J, et al. Smad2/3/4 Pathway Contributes to TGF-beta-Induced MiRNA-181b Expression to Promote Gastric Cancer Metastasis by Targeting Timp3. Cell Physiol Biochem 2016;39:453–66 [DOI] [PubMed] [Google Scholar]
- 35.Massague J TGFbeta signalling in context. Nat Rev Mol Cell Biol 2012;13:616–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu J, Ni S, Cao Y, Zhang T, Wu T, Yin X, et al. The Angiogenic Effect of microRNA-21 Targeting TIMP3 through the Regulation of MMP2 and MMP9. PLoS One 2016;11:e0149537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi L, Wan Y, Sun G, Gu X, Qian C, Yan W, et al. Functional differences of miR-125b on the invasion of primary glioblastoma CD133-negative cells and CD133-positive cells. Neuromolecular Med 2012;14:303–16 [DOI] [PubMed] [Google Scholar]
- 38.Son DJ, Kumar S, Takabe W, Kim CW, Ni CW, Alberts-Grill N, et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat Commun 2013;4:3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamamura Y, Otani T, Kanatani N, Koyama E, Kitagaki J, Komori T, et al. Developmental regulation of Wnt/beta-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem 2005;280:19185–95 [DOI] [PubMed] [Google Scholar]
- 40.Cai WS, Shen F, Feng Z, Chen JW, Liu QC, Li EM, et al. Downregulation of CDK-8 inhibits colon cancer hepatic metastasis by regulating Wnt/beta-catenin pathway. Biomed Pharmacother 2015;74:153–7 [DOI] [PubMed] [Google Scholar]
- 41.Clarke PA, Ortiz-Ruiz MJ, TePoele R, Adeniji-Popoola O, Box G, Court W, et al. Assessing the mechanism and therapeutic potential of modulators of the human Mediator complex-associated protein kinases. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawamata F, Patch AM, Nones K, Bond C, McKeone D, Pearson SA, et al. Copy number profiles of paired primary and metastatic colorectal cancers. Oncotarget 2018;9:3394–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerbel RS, Shaked Y. The potential clinical promise of ‘multimodality’ metronomic chemotherapy revealed by preclinical studies of metastatic disease. Cancer Lett 2017;400:293–304 [DOI] [PubMed] [Google Scholar]
- 44.Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol 2015;44–46:147–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogata Y, Enghild JJ, Nagase H. Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J Biol Chem 1992;267:3581–4 [PubMed] [Google Scholar]
- 46.Mamlouk S, Childs LH, Aust D, Heim D, Melching F, Oliveira C, et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat Commun 2017;8:14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van den Eynden GG, Majeed AW, Illemann M, Vermeulen PB, Bird NC, Hoyer-Hansen G, et al. The multifaceted role of the microenvironment in liver metastasis: biology and clinical implications. Cancer Res 2013;73:2031–43 [DOI] [PubMed] [Google Scholar]
- 48.Zhao X, Feng D, Wang Q, Abdulla A, Xie XJ, Zhou J, et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J Clin Invest 2012;122:2417–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yokoyama Y, Watanabe T, Tamura Y, Hashizume Y, Miyazono K, Ehata S. Autocrine BMP-4 Signaling Is a Therapeutic Target in Colorectal Cancer. Cancer Res 2017;77:4026–38 [DOI] [PubMed] [Google Scholar]
- 50.Villalba M, Evans SR, Vidal-Vanaclocha F, Calvo A. Role of TGF-beta in metastatic colon cancer: it is finally time for targeted therapy. Cell Tissue Res 2017;370:29–39 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.