

Abstract
Background:
The gut microbiota is altered in patients with chronic kidney disease (CKD), and cardiovascular risk increases with progressive CKD. This study examined the potential link between short chain fatty acids (SCFAs), which are produced by the gut microbiota, and cardiovascular outcomes in patients with CKD.
Methods:
SCFAs were measured using a targeted liquid chromatography-mass spectrometry platform in baseline plasma samples from 214 patients with CKD enrolled in the Clinical Phenotyping Resource and Biobank Core (CPROBE); 81 patients with coronary artery disease (CAD) and 133 without were randomly assigned to training and validation subsets. The primary outcome was a history of CAD and the secondary outcome was a composite history of cardiovascular disease (CVD) at enrollment.
Results:
We found a significantly higher levels of the SCFA valerate among patients with CAD as compared with patients without CAD in the training set (p<0.001). The Valerate concentrations were also significantly higher among subjects with composite outcomes of CVD compared to those without CVD (p=0.006). These results were subsequently replicated in the validation set. Logistic regression analysis revealed a strong independent association between plasma valerate levels and CVD in both training and validation sets. When Valerate was added to the base clinical model comprising of diabetes, hypertension, urinary protein-creatinine ratio and eGFR, it increased the c-statistics for predicting CVD from 0.68 to 0.79(p=0.02) in the training set, an observation which was confirmed in the validation set.
Conclusion:
This study provides evidence for alterations in gut-microbiota-derived SCFAs with advancing CKD, demonstrates the association of higher plasma valerate levels with pre-existing CVD, and reveals areas for future exploration of cardiovascular risk in patients with CKD.
Keywords: Short chain fatty acids, Valerate, Coronary artery disease, Cardiovascular outcomes, Chronic kidney disease
Introduction:
Chronic kidney disease (CKD) is a major healthcare problem, is currently estimated to affect ~30 million Americans, and is associated with a significantly higher risk of cardiovascular morbidity and mortality [1–4]. Although CKD and cardiovascular disease (CVD) share several major risk factors, traditional cardiovascular risk factors, such as diabetes and hypertension, do not completely account for the high risk of cardiovascular disease in patients with CKD [5]. Identifying additional risk factors is one focus of current research to understand the pathophysiology of atherosclerosis, including coronary artery disease (CAD), and its accelerated phenotype in patients with CKD. There is a graded increase in cardiovascular mortality with increasing renal dysfunction [5, 6], which may increase as renal excretory function declines either from accumulation of toxins or loss of a protective mechanism.
Research conducted over the last decade has established that gut microbiota has a significant influence on human health and disease. The relative abundance of different bacterial phyla and their biological impact are modified by a number of genetic, dietary and environmental factors [7, 8]. Gut microbiota produce a variety of products through saccharolytic and proteolytic breakdown of food material [9]. Some of these gut-microbiota-derived products have a significant regulatory impact on host metabolism, immunity, and nutrition. Additionally, the gut microbiota has been long recognized as the source of uremic toxins [10, 11], which can have a potentially deleterious impact on host health. Changes in gut microbiota have been documented in CKD [12, 13] due to changes in the gut biochemical milieu, because increasing quantities of urea, uric acid and oxalate are transported via the gut as kidney function declines. This dysbiosis is postulated to result in enhanced generation of toxins associated with kidney disease progression [14, 15] and increased cardiovascular risk [16–18].
In addition to the accumulation of toxins, kidney disease also alters the synthesis of beneficial metabolites as the composition of the microbiome changes. Recently, short chain fatty acids (SCFAs) have attracted considerable interest. The primary source of SCFAs in humans is as metabolic byproducts of complex starch fermentation in the gut by commensal bacteria yielding 1-6 carbon structures with variable acyl chain attached to the carboxylic group [19]. The relative concentrations of SCFAs are modified by the composition of the microbiota and the amount of indigestible starch consumed [20]. SCFA-synthesizing bacteria change with the development of CKD [21], which raises questions about the possible role of SCFAs in CKD progression and its associated cardiovascular complications. However, the impact of CKD on the plasma concentrations of SCFAs or their association with cardiovascular mortality in patients with CKD had not been studied.
We hypothesized that plasma SCFAs would be altered in patients with CAD or composite CVD. In this study, we compared the plasma concentrations of the SCFAs in CKD patients with and without CVD and assessed the odds of cardiovascular outcomes associated with alteration of any identified SCFA markers.
Methods:
Study Cohort:
This was a cross sectional study utilizing participant data from the Clinical Phenotyping Resource and Biobank Core (C-PROBE) with details published previously [22, 23] . In brief, C-PROBE is a multicenter cohort of 1235 adult CKD patients (recruitment as of December 2017) with high- quality biological samples and clinical data to conduct translational research.
From the entire cohort, a representative subcohort of CKD patients (stages 1-5) recruited between January 2009 to July 2012 was selected for the downstream analysis. The selected samples comprised of 81 patients with and 133 patients without a self-reported history of CAD. Patients with and without CAD were frequency-matched by sex, race, and CKD stage. Inclusion criteria were age >18 years and stage 1-5 CKD (according to the CKD-EPI formula). We showed previously that the selected patients were highly similar to the rest of the patients in CPROBE, suggesting that they are unbiased representatives of the entire CPROBE population [22]. We used the baseline clinical data, laboratory data, and plasma samples collected at the time of enrollment for downstream biomarker identification and analyses. We randomly divided the study participants into two equal subsets for training and validation (Figure 1).
Figure 1:
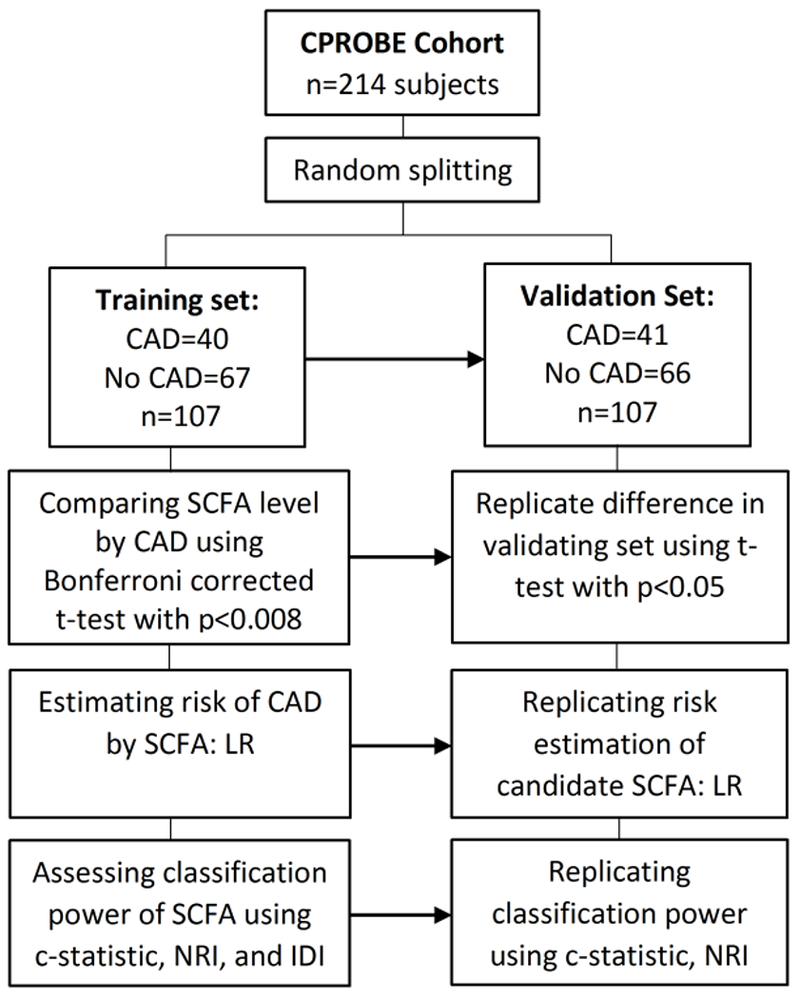
Flow diagram of analysis employed for identification and validation of SCFAs that independent predict CAD and cardiovascular outcomes. CAD, coronary artery disease; IDI, Integrated discrimination improvement; LR, Logistic regression; NRI, net reclassification index; SCFA, short chain fatty acid.
Outcomes:
The primary outcome was presence of CAD, defined as a history of myocardial infarction, angina, coronary artery bypass grafting, or angioplasty/stenting of a coronary artery. However, because patients without CAD may have other cardiovascular disorders, we defined a secondary cardiovascular outcome, composite CVD, as the composite of self-reported CAD, stroke, peripheral arterial disease, congestive heart failure or arrhythmia at the time of enrollment.
Reagents and materials:
Analytical-reagent-grade 3-nitrophenylhydrazine (3-NPH), N-(3-dimethylaminopropyl)-N1-ethylcarbodiimide (EDC), hydrochloric acid (HCl), liquid chromatography-mass spectrometry (LCMS)-grade acetonitrile, and high-performance liquid chromatography (HPLC)-grade pyridine were purchased from Sigma-Aldrich/ Millipore-Sigma. HPLC-grade water and HPLC-grade formic acid were purchased from Honeywell Fluka.
Sample processing:
A previously described derivatization strategy for measurement of SCFA in stool samples [24] was modified after considerable experimentation and optimization. A 200 mM 3-NPH.HCl solution in 50% acetonitrile and a 120 mM EDC.HCl solution in 6% pyridine were prepared for the derivatization reaction. The extraction solvent was 50% acetonitrile with deuterium-labelled D4 butyrate and D11 hexanoate as internal standards for analytical measurement. Each biological sample (25 μL of serum) was mixed with 175 μl of the extraction solvent for protein precipitation. After mixing thoroughly, the sample was incubated on ice for 10 minutes and subsequently centrifuged for 10 minutes at 12000 rpm. The supernatant (20 μl) was then mixed with 10 μl 200 mM 3-NPH.HCl and 10 μl 120 mM EDC.HCl. The samples were kept at 40°C for 30 minutes for the derivatization reaction prior to transfer into inserts and injection for liquid chromatography and mass spectrometry.
Mass Spectrometry:
An Agilent 6410 triple quadrupole mass spectrometer coupled to an Agilent 1290 infinity series chromatography system equipped with an electrospray ionization (ESI) source was used in negative-ion mode for measurement. An Acquity UPLC BEH C18 column (2.1×100 mm, 1.7 μm; Waters, Milford MA) was used for liquid chromatography and the mobile phase. Solvent A was water: formic acid (100:0.01%, v/v), and solvent B was acetonitrile: formic acid (100:0.01%, v/v). SCFAs were identified and quantitated using Agilent Mass Hunter Analyst software.
Statistical analysis:
Normal distribution of continuous variables was checked using the univariate Kolmogorov-Smirnov (KS) test. If a skewed distribution was identified, we used median and interquartile range for description and the KS test for independent samples to test the significance by study subgroup. To compare the mean of normally distributed continuous variables between 2 groups, we used a t-test. For more than 2 groups, we used analysis of variance (ANOVA) with the Dunnett correction for multiple comparisons. To test the association of categorical variables by presence of CAD or by CKD stage, we used chi-square analysis. To identify whether the concentration of an SCFA differed significantly by study outcomes, we used compound-by-compound t-tests with post-hoc Bonferroni correction. We applied a significance threshold of p<0.0083 (0.05/6) in the compound-by-compound comparisons, because six SCFAs were examined.
We applied unadjusted, estimated glomerular filtration rate (eGFR)-adjusted, and fully adjusted logistic regression models in the training subset to estimate the odds of study outcomes (CAD and composite CVD) by alteration of the SCFA valerate. The covariates for the fully adjusted model consisted of age, etiology of CKD, diabetes, hypertension, use of statins, and eGFR. We applied the coefficients of the logistic regression models that were developed in the training set to calculate the z-score (standardized probabilistic risk score) in the validation set to test the reproducibility of risk estimations seen in the training set. We applied a forward, stepwise, iterative inclusion algorithm in the logistic regression models.
We then calculated c-statistic, net reclassification index (NRI) and integrated discrimination improvement (IDI) to predict CAD in the training set when valerate values were added to a baseline clinical model consisting of diabetes, hypertension, eGFR, and urine protein-creatinine ratio (UPCR)[23]. We replicated the c-statistic, NRI, and IDI of the proposed model as compared with the base model in the validation set by calculating the corresponding probabilistic risk score using the coefficients of the logistic regression models developed in training set. We applied the coefficients of variation of acetate, propionate, butyrate, isovalerate, valerate, and caproate in the sample pools, which were 11.78%, 3.29%, 0.82%, 2.28%, 1.8% and 7.28%, respectively. The study had 88% power to detect a 1 μmol/L difference in the concentration of valerate at alpha=0.05 using a 2-sided t-test in the training set. The power to detect the valerate mean difference observed in case and control groups in the entire cohort at alpha=0.05 using a 2-sided t-test was 95%. SPSS version 24 (Armonk, NY) and STATA version 10 (College Station. TX) were applied for the analysis.
Results:
Baseline characteristics:
We examined the distribution of baseline demographic characteristics, medications, and comorbidities in the entire cohort (Table 1) and in the training and validation sets (Supplementary Table 1). Patients with CAD were older than those without CAD (p<0.001), and also had a significantly higher incidence of hypertension, diabetes mellitus, statin use, stroke, and peripheral vascular disease. There were no other significant differences in clinical characteristics. Because the selected patients were randomly split in to the training and validation sets, the overall characteristics of the two sets were similar, except a higher proportion of patients with CAD in the training set had diabetes. There were no differences in drugs such as fibrates, niacin, angiotensin converting enzyme inhibitor (ACEI), angiotensin receptor blocker (ARB), proton pump inhibitor (PPI), antibiotic/probiotics or erythropoiesis stimulating agent (ESA) use among those with and without CAD and CVD in both training and validation sets (Table 1 and Supplementary Table 1).
Table 1:
Baseline characteristics of study participants according to coronary artery disease. BMI, body mass index; CAD, coronary artery disease; CRP, C-reactive protein; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate; HDL, high density lipoprotein; LDL, low density lipoprotein; PVD, peripheral artery disease; ACEI, Angiotensin converting enzyme inhibitor; ARB, Angiotensin Receptor Blocker, PPI, proton pump inhibitor; ESA, erythropoietin stimulating agent; SBP, systolic blood pressure; UPCR, urine protein-creatinine ratio.
| N | With CAD | Without CAD | P value |
|---|---|---|---|
| Anthropometric parameters | |||
| Sample size | 81 | 133 | |
| Age (years) | 68 ± 10 | 55 ± 16 | <0.001 |
| Male Sex (%) | 45 (55.6) | 65 (48.9) | 0.40 |
| White Race (%) | 56 (69.1) | 94 (70.7) | 0.88 |
| SBP (mmHg) | 141 ± 23 | 132 ± 20 | 0.05 |
| DBP (mmHg) | 75 ± 11 | 75 ± 12 | 0.88 |
| Height (m) | 1.71 ± 0.11 | 1.69 ± 0.14 | 0.39 |
| Weight (kg) | 92 ±22 | 89 ± 21 | 0.46 |
| BMI (Kg/m2) | 31 ± 6 | 31 ± 7 | 0.47 |
| Smoking (%) | 9 (11.1) | 12 (9.0) | 0.64 |
| Etiology of CKD | |||
| Hypertension (%) | 21 (25.9) | 23 (17.3) | <0.001 |
| Diabetes (%) | 20 (24.7) | 27 (20.3) | |
| Glomerular (%) | 13 (16.0) | 60 (45.1) | |
| Tubulointerstitial (%) | 2 (2.5) | 5 (3.8) | |
| Others (%) | 25 (30.9) | 18 (13.5) | |
| Comorbidities | |||
| Hypertension (%) | 73 (90.1) | 103 (77.4) | 0.03 |
| Diabetes (%) | 44 (54.3) | 45 (33.8) | 0.004 |
| Stroke (%) | 17 (21.0) | 13 (9.8) | 0.03 |
| Heart failure (%) | 26 (32.1) | 5 (3.8) | <0.001 |
| PVD (%) | 21 (25.9) | 8 (6.0) | <0.001 |
| Arrhythmia (%) | 18 (22.2) | 15 (11.3) | 0.05 |
| Medications | |||
| Statins (%) | 53 (65.4) | 61 (45.9) | 0.007 |
| Fibrates (%) | 8 (9.9) | 10 (7.5) | 0.61 |
| Niacin (%) | 3 (3.7) | 4 (3.0) | 1.000 |
| ACEI/ARB (%) | 60 (74.1) | 90 (67.7) | 0.321 |
| PPI (%) | 27 (33.3) | 31 (23.3) | 0.164 |
| ESA (%) | 10 (12.3) | 9 (6.8) | 0.164 |
| Probiotics/Antibiotics (%) | 2 (2.5) | 3 (2.3) | 1.000 |
| Biochemical parameters | |||
| Albumin (g/dL) | 4.0 ± 0.5 | 4.1 ± 0.4 | 0.65 |
| Hemoglobin (g/dL) | 12.2 ± 1.7 | 11.8 ± 1.7 | 0.08 |
| CRP (mg/dL)† | 1.4 ± 0.4 | 2.6 ± 0.4 | 0.72 |
| Cholesterol (mg/dL) | 160 ± 53 | 174 ± 51 | 0.44 |
| LDL (mg/dL) | 81 ± 38 | 89 ± 41 | 0.23 |
| HDL (mg/dL) | 36 ± 16 | 40 ± 18 | 0.32 |
| Triglycerides (mg/dL) | 157 ± 114 | 167 ± 108 | 0.65 |
| UPCR, median [IQR] | 1.2 [0.4 – 1.9] | 1.0 [0.2 – 1.9] | 0.56 |
| eGFR, mL/min, median [IQR] | 33 [23 to 46] | 38 [26 to 55] | 0.06 |
| eGFR categories | |||
| >60 ml/min (%) | 8 (9.9) | 28 (21.0) | 0.11 |
| 30-59 ml/min(%) | 42 (51.8) | 57 (42.9) | |
| 15-29 ml/min(%) | 26 (32.1) | 35 (26.3) | |
| <15 ml/min(%) | 5 (6.2) | 13 (9.8) | |
Sample size for CRP is 12 for CAD and 45 for no CAD.
SCFAs and cardiovascular outcomes:
We detected 6 different SCFAs is the plasma of the CKD patients examined (Table 2). Valerate was the only SCFA that had a significantly higher plasma concentration in patients with CAD or CVD as compared to those without, in the training set. This finding was replicated in the validation set (Table 2, Figure 2). As such, the valerate level in the entire cohort (n=214) was also significantly higher in patients with the primary or the secondary outcomes of CAD or CVD as compared to those without CAD or CVD.
Table 2:
Comparing SCFA concentrations by primary and secondary study outcomes in the training set, validation set, and the entire cohort. Values are mean ± standard deviation. P values reflect comparison of CAD or CVD with no CAD or CVD with each set. Values are mean ± standard deviation. CAD, coronary artery disease; CVD, cardiovascular disease.
| Training set | Validation set | Entire cohort | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Primary outcome: | No CAD | CAD | p | No CAD | CAD | p | No CAD | CAD | p |
| n | 67 | 40 | 66 | 41 | 133 | 81 | |||
| Acetate (μm/L) | 214.7 ± 44.9 | 215.4 ± 35.0 | 0.93 | 210.6 ± 39.3 | 199.6 ± 39.2 | 0.16 | 212.7 ± 42.2 | 207.4 ± 37.8 | 0.36 |
| Propionate (μm/L) | 95.3 ± 3.9 | 96.3 ± 2.7 | 0.16 | 95.4 ± 3.1 | 96.2 ± 3.5 | 0.25 | 95.4 ± 3.5 | 96.3 ± 3.1 | 0.07 |
| Butyrate (μm/L) | 65.5 ± 0.6 | 65.6 ± 0.7 | 0.50 | 65.5 ± 0.7 | 65.5 ± 0.6 | 0.89 | 65.5 ± 0.6 | 65.5 ± 0.7 | 0.56 |
| Isovalerate (μm/L) | 79.9 ± 5.9 | 80.3 ± 4.5 | 0.76 | 78.6 ± 3.3 | 78.3 ± 1.8 | 0.61 | 79.3 ± 4.9 | 79.3 ± 3.5 | 0.98 |
| Valerate (μm/L) | 28.6 ± 1.1 | 29.5 ± 1.6 | <0.001 | 28.8 ± 1.3 | 29.4 ± 1.1 | 0.019 | 28.7 ± 1.2 | 29.4 ± 1.4 | <0.001 |
| Caproate (μm/L) | 26.2 ± 1.6 | 26.4 ± 2.2 | 0.64 | 25.6 ± 1.2 | 26.0 ± 1.2 | 0.35 | 26.0 ± 1.4 | 26.2 ± 1.8 | 0.39 |
| Secondary outcome: | No CVD | CVD | No CVD | CVD | No CVD | CVD | |||
| n | 50 | 57 | 50 | 57 | 100 | 114 | |||
| Acetate (μm/L) | 223.3 ± 43.5 | 207.7 ± 38.2 | 0.05 | 208.2 ± 40.2 | 204.7 ± 39.2 | 0.65 | 216.0 ± 42.4 | 206.2 ± 38.5 | 0.08 |
| Propionate (μm/L) | 95.4 ± 3.7 | 96.0 ± 3.3 | 0.39 | 95.1 ± 2.9 | 96.2 ± 3.4 | 0.09 | 95.3 ± 3.3 | 96.1 ± 3.4 | 0.07 |
| Butyrate (μm/L) | 65.6 ± 0.6 | 65.5 ± 0.6 | 0.47 | 65.4 ± 0.7 | 65.5 ± 0.6 | 0.74 | 65.5 ± 0.6 | 65.5 ± 0.6 | 0.87 |
| Isovalerate (μm/L) | 80.3 ± 5.8 | 79.8 ± 5.1 | 0.60 | 78.4 ± 3.5 | 78.5 ± 2.1 | 0.94 | 79.4 ± 4.5 | 79.1 ± 3.9 | 0.65 |
| Valerate (μm/L) | 28.6 ± 1.2 | 29.3 ± 1.5 | 0.006 | 28.7 ± 0.9 | 29.3 ± 1.5 | 0.017 | 28.6 ± 1.1 | 29.3 ± 1.5 | <0.001 |
| Caproate (μm/L) | 26.2 ± 1.6 | 26.4 ± 2.0 | 0.56 | 25.9 ± 1.3 | 25.8 ± 1.1 | 0.80 | 26.0 ± 1.5 | 26.1 ± 1.7 | 0.77 |
Figure 2:
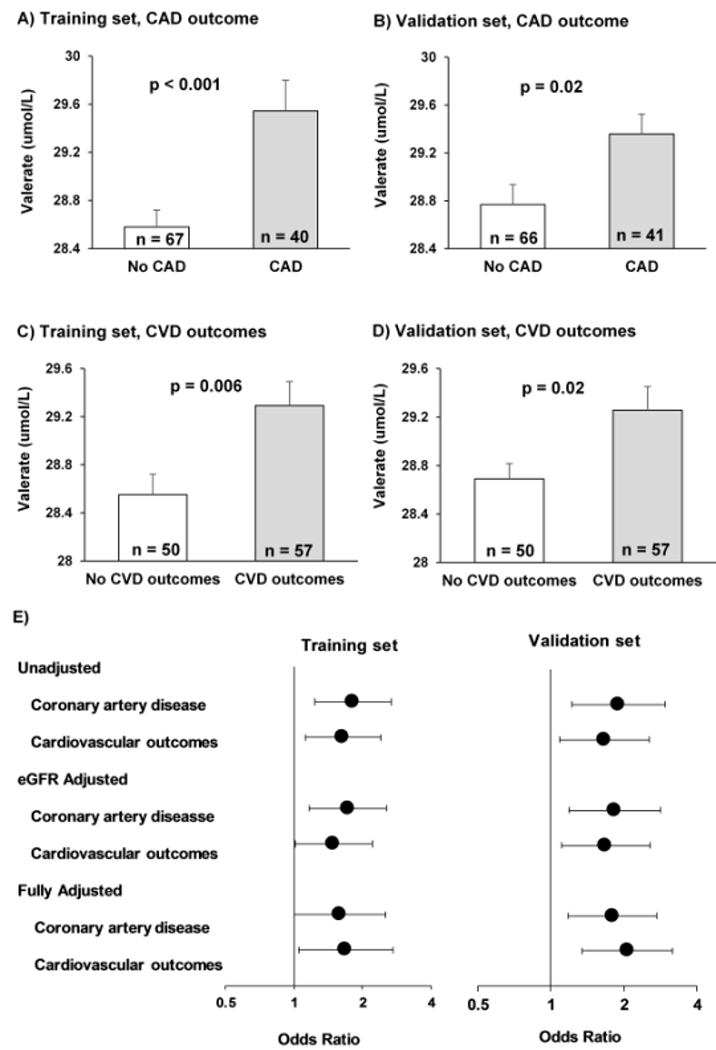
Valerate concentrations by coronary artery disease and cardiovascular outcomes in training and validation sets. A significantly higher valerate level is observed in patients with CAD in the training (A) and the validation subsets (B). Similarly, its significantly higher level is observed in patients with CVD in the training (C) and the validation subsets (D). Plots show mean and standard error. Panel (E) compares the odds ratio of study outcomes in unadjusted, eGFR-adjusted, and fully adjusted models. Odds ratios are compared by each 1 μmol/L alteration in plasma valerate in the training set, and by each 1 standard alteration in the standardized probabilistic risk score (developed from the coefficients of the logistic regression models of the training set) in the validation set. Covariates of fully adjusted models consist of age, diabetes, hypertension, use of statins, and eGFR. eGFR, estimated glomerular filtration rate; CAD, coronary artery disease; CVD, cardiovascular disease.
SCFA levels with CKD stage:
When we examined SCFA concentrations in patients with CKD in different stages, we observed a significant graded decrease in the concentration of acetate (228.6 ± 44.5 umol/L in CKD stage 1/2 vs. 195.2 ± 29.6 umol/L in CKD stage 5, P<0.01). We additionally found that the plasma valerate concentration increased with advancing CKD, from 28.4 ± 0.8 umol/L in CKD stage ½ to 29.5 ± 1.4 umol/L in CKD stage 5 (p=0.045). There were no significant differences in the concentrations of any other SCFAs between CKD stage 1/2 and CKD stage 5 as measured in this population (Supplementary Table 2). When comparing the results of the training with the validation subsets, the significant increase in valerate by CKD stage observed in the training subset was not replicated in the validation subset (Supplementary Table 3).
Risk estimation of study outcomes by levels of valerate:
Using logistic regression, we identified a significant increase in the odds of having CAD, as well as the odds of composite CVD, with increasing plasma valerate level in all models applied to the training subset (unadjusted, eGFR-adjusted, fully adjusted), an observation which was replicated in the validation set (Figure 2, Supplementary Table 4). In the unadjusted model, each 1 μmol/L increase in plasma valerate was associated with 1.81 fold higher odds of association with CAD (95% CI: 1.23 to 2.67, p=0.003) and 1.64 fold higher odds of association with composite CVD (95% CI: 1.12 to 2.41, p=0.01) in the training set. The odds of association were 1.59 fold higher for CAD (95% CI: 1.00 to 2.51, p=0.049) and 1.69 fold higher for CVD (95% CI: 1.05 to 2.71, p=0.03) in the fully adjusted model. The findings were similar in the validation set (Figure 2, Supplementary Table 4).
Classification power and test characteristics of valerate:
When valerate concentration was added to a base model, the classification power of the model to predict CAD improved in both the training and validation sets. In the training set, the c-statistic increased significantly from c=0.68 (95% CI: 0.58-0.79) to c=0.79 (95% CI: 0.69-0.88) with a p value of 0.019. Similarly, in the validation set, c-statistic increased from 0.59 (95% CI: 0.47-0.70) to 0.70 (95% CI: 0.60-0.80) with a p value of 0.011. Addition of valerate to the base model provided an event NRI of 0.29 (12/41) and a non-event NRI of 0.61 (40/66) altogether translated to a significant overall NRI of 0.90 (95% CI: 0.52 to 1.30, p<0.0001) in the training set. Similarly, in the validation set an event NRI of 0.32 (13/40) and a non-event NRI of 0.43 (29/67) contributed to a significant overall NRI of 0.75 (95% CI: 0.55 to 0.95, p<0.001), confirming the finding observed in the training set. The IDI showed a similar improvement after addition of valerate to the base model with event IDI of 0.14 (95%CI: 0.08 to 0.20, p<0.001) in training set and 0.09 (95% CI: 0.03 to 0.15, p=0.002) in the validation set.
Discussion:
In this study, we showed for the first time that a higher level of plasma valerate, a SCFA generated by the gut microbiota, was independently associated with pre-existing CVD in patients with CKD. This association was independent of imbalanced baseline characteristics such as age, diabetes, hypertension, and use of drugs such as statins. Furthermore, we showed that the addition of valerate to a baseline model, consisting of UPCR, eGFR, hypertension, and diabetes mellitus, significantly improved the classification power of the model in identifying preexisting CVD.
The accumulation of metabolites and uremic toxins derived from the commensal bacteria and the changes in gut mucosal permeability allowing translocation of bacterial products have been proposed as potential mechanisms underlying atherosclerosis and CVD [25–27]. Animal studies have also shown a reduction in the size of myocardial infarctions with manipulation of the gut microbiome [28, 29]. Changes in the microbiota alter a variety of physiological processes including the immune response, vascular physiology and metabolic programming, but the precise mechanisms responsible for these changes are not completely defined. In human studies, gut-microbiota-derived trimethylamine N-oxide (TMAO) has been shown to be associated with cardiovascular outcomes in high risk patients [27, 30], but its demonstration as a causative influence and role in the general chronic kidney disease population remains unproven.
The majority of microbiota studies in CKD patients have clearly outlined the changes in microbiota seen with advancing kidney disease [12, 31], but there has not been a concerted effort to define the resulting changes in microbiota-derived metabolites, especially SCFAs. Our study documents these alterations. We identified a progressively graded decrease in acetate concentration with CKD progression. This decline is postulated to be caused by changes in diet, medications, the gastrointestinal milieu, and host immunity. There was, however, no association between this change and cardiovascular outcomes in our population; its impact on other physiological processes remains unexplored. The concentrations of propionate, butyrate, and hexanoate did not change significantly, which was unexpected given recent literature identifying a significant decline in butyrate-producing bacteria with advancing kidney disease[32].
A graded increase in valerate concentration was strongly associated with CAD and our composite CVD secondary outcome, even after correcting for relevant covariates including eGFR. A model predicting CAD and CVD that incorporated valerate produced a c-statistic, beyond what was possible with the use of only diabetes mellitus, hypertension, UPCR and eGFR in the model, and highlights the potential of valerate as a non-invasive marker of CVD. This may be particularly useful given the presence of subclinical CVD in many CKD patients who would benefit from better risk stratification and interventions to reduce their likelihood of a clinically significant cardiovascular event. These findings also highlight potential avenues of future exploration of the pathophysiological basis of atherosclerosis and its complications.
Valerate is the salt of a straight-chain alkyl carboxylic acid with 5 carbon atoms, which is a naturally occurring compound in the plant kingdom and is also produced in significant amounts by the commensal gut bacteria. Stool concentrations of valerate are in the millimolar range (our unpublished work), so we believe the gut microbiota is the most likely source of the observed plasma valerate. Valerate, unfortunately, has not been studied as extensively as other SCFAs, and details of its physiological effects are sparse. Previous work has identified valerate as a ligand for the G-protein coupled receptors [33], which influence a variety of metabolic, immune, and vascular processes [34–36].
G-protein coupled receptors are the largest family of transmembrane signaling molecules and detect changes in the extracellular environment SCFAs act as ligands for at least 4 different types of G-protein coupled receptors in humans, namely GPR41, GPR43, GPR109a and OR51E2. GPR43 is present in high concentrations on immune cells, and SCFAs have been shown to mediate chemotaxis [37] and increase inflammatory mediator release [36, 38], thereby promoting a robust inflammatory response. GPR41 is widely present on adipocytes, sympathetic neurons, entero-endocrine cells and vascular smooth muscle, where it regulates adipocyte function, sympathetic discharge, glucose sensitivity, and vascular tone [39]. Both GPR41 and GPR43 respond to stimulation by valerate, and this may enhance the immune response [36], modulate adipocyte metabolism [40] and alter vascular physiology [34], leading to the resultant increase in CVD witnessed in our population. Whether valerate acting through G-protein coupled receptors is responsible for the increased cardiovascular risk requires further investigation, but the results of this study provide a testable biological explanation for the observed progressive increase in cardiovascular risk with worsening kidney function. The plasma concentration of all the SCFAs are, however, much lower than the EC50 reported for GPR41 and GPR43, which raises questions about the physiological role of GPR 41 receptors on vascular smooth muscles and in the kidney. GPR43 present on the immune cells may be activated as these cells move in the intestinal immune system and may modulate the immune response, but again the mechanisms are not clear and require further study.
This study has both strengths and limitations. The excellent phenotyping and quality samples at baseline contributed to minimizing noise and allowing identification of signal. We applied rigorous quality control measures including the use of pooled samples with sequential measures and continuous calibration of our instruments. There was no missing data requiring imputation. By design, we applied training and validation sets, which strengthens of our findings. All samples from patients with CKD stage 5 were obtained prior to dialysis or transplant, and therefore, the plasma SCFA levels were not impacted by renal replacement therapies. The persistence of statistically significant association of valerate level with study outcomes even after adjusting for imbalanced baseline covariates including age, diabetes, hypertension, and use of statins suggests that the observed associations were unlikely to have been explained by such covariates. A number of medications such as PPIs and antibiotics may impact the gut microbiota, but the distribution of these agents were not different by the study outcomes and therefore they are unlikely to be confounding factors for the association of valerate and cardiovascular outcomes. The limitations of our study include the use of biological samples and clinical data from a single clinical cohort, which raises questions about the generalizability of the results. Reproduction of our results in additional cohorts will greatly strengthen the case for valerate as a non-invasive marker of CVD in patients with CKD. While the changes in valerate levels are modest, the biological relevance of our findings need to be investigated in mechanistic studies which will be the subject of further and future research. The observational nature of our study prevents us from drawing causal inferences, and studies in animal models are needed to identify a causal relationship. The lack of dietary history, longitudinal clinical data and the retrospective nature of the outcomes are also limitations.
In conclusion, this study defines changes in plasma SCFA levels with advancing kidney disease and identifies a significant association of plasma valerate levels with cardiovascular outcomes. Improving stratification of cardiovascular risk in patients with CKD using a non-invasive test may help identify patients with subclinical disease in need of comprehensive evaluation and aggressive treatment of cardiovascular risk factors that would allow us to improve clinical outcomes through personalized medicine. These findings, however, need further exploration to clarify the role of valerate in atherosclerosis and cardiovascular risk, which may lead to the identification of novel therapeutic strategies for the prevention and treatment of CVD in patients with CKD and perhaps enhance our understanding of the pathophysiology of atherosclerosis to the benefit of all patients at risk for this major healthcare problem.
Supplementary Material
Acknowledgment
* The Michigan Kidney Translational Core Clinical Phenotyping Resource and Biobank Core Investigator Group includes Matthias Kretzler (University of Michigan), Keith Bellovich (St. Clair Nephrology Research, Detroit), Zeenat Bhat (Wayne State University), Susan Massengill (Levin Children’s Hospital), and Kalyani Perumal, JH Stroger Hospital.
Support: K08HL130944 (AM), K08DK106523 (FA), P30DK089503, DK082841, P30DK081943, P30DK020572, DK097153 (SP)
Footnotes
Conflict of Interest: Authors declare that they do not have any conflict of interest.
References:
- 1.Control, C.f.D., National Chronic Kidney Disease Fact Sheet. 2017; Available from: http://www.cdc.gov/ckd
- 2.Damsgaard EM, et al. , Microalbuminuria as predictor of Increased mortality In elderly people. BMJ, 1990. 300(6720): p. 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drey N, et al. , A population-based study of the Incidence and outcomes of diagnosed chronic kidney disease. Am J Kidney Dis, 2003. 42(4): p. 677–84. [DOI] [PubMed] [Google Scholar]
- 4.Culleton BF, et al. , Cardiovascular disease and mortality in a community-based cohort with mild renal insufficiency. Kidney Int, 1999. 56(6): p. 2214–9. [DOI] [PubMed] [Google Scholar]
- 5.Go AS, et al. , Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med, 2004. 351(13): p. 1296–305. [DOI] [PubMed] [Google Scholar]
- 6.Chronic Kidney Disease Prognosis, C., et al. , Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet, 2010. 375(9731): p. 2073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodrich JK, et al. , Human genetics shape the gut microbiome. Cell, 2014. 159(4): p. 789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conlon MA and Bird AR, The impact of diet and lifestyle on gut microbiota and human health. Nutrients, 2014. 7(1): p. 17–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sekirov I, et al. , Gut microbiota in health and disease. Physiol Rev, 2010. 90(3): p. 859–904. [DOI] [PubMed] [Google Scholar]
- 10.Einheber A and Carter D, The role of the microbial flora in uremia. I. Survival times of germfree, limited-flora, and conventionalized rats after bilateral nephrectomy and fasting. J Exp Med, 1966. 123(2): p. 239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wikoff WR, et al. , Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A, 2009. 106(10): p. 3698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaziri ND, et al. , Chronic kidney disease alters intestinal microbial flora. Kidney Int, 2013. 83(2): p. 308–15. [DOI] [PubMed] [Google Scholar]
- 13.Ramezani A, et al. , Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am J Kidney Dis, 2016. 67(3): p. 483–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Satoh M, et al. , Uremic toxins overload accelerates renal damage in a rat model of chronic renal failure. Nephron Exp Nephrol, 2003. 95(3): p. e111–8. [DOI] [PubMed] [Google Scholar]
- 15.Tang WH, et al. , Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res, 2015. 116(3): p. 448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CJ, et al. , Meta-Analysis of the Associations of p-Cresyl Sulfate (PCS) and Indoxyl Sulfate (IS) with Cardiovascular Events and All-Cause Mortality in Patients with Chronic Renal Failure. PLoS One, 2015. 10(7): p. e0132589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Organ CL, et al. , Choline Diet and Its Gut Microbe-Derived Metabolite, Trimethylamine N-Oxide, Exacerbate Pressure Overload-Induced Heart Failure. Circ Heart Fail, 2016. 9(1): p. e002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barreto FC, et al. , Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol, 2009. 4(10): p. 1551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoverstad T and Midtvedt T, Short-chain fatty acids in germfree mice and rats. J Nutr, 1986. 116(9): p. 1772–6. [DOI] [PubMed] [Google Scholar]
- 20.Trompette A, et al. , Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med, 2014. 20(2): p. 159–66. [DOI] [PubMed] [Google Scholar]
- 21.Wong J, et al. , Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am J Nephrol, 2014. 39(3): p. 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Afshinnia F, et al. , Impaired beta-Oxidation and Altered Complex Lipid Fatty Acid Partitioning with Advancing CKD. J Am Soc Nephrol, 2018. 29(1): p. 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Afshinnia F, et al. , Myeloperoxidase Levels and Its Product 3-Chlorotyrosine Predict Chronic Kidney Disease Severity and Associated Coronary Artery Disease. Am J Nephrol, 2017. 46(1): p. 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han J, et al. , An isotope-labeled chemical derivatization method for the quantitation of short-chain fatty acids in human feces by liquid chromatography-tandem mass spectrometry. Anal Chim Acta, 2015. 854: p. 86–94. [DOI] [PubMed] [Google Scholar]
- 25.Koren O, et al. , Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A, 2011. 108 Suppl 1: p. 4592–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karlsson FH, et al. , Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun, 2012. 3: p. 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang WH, et al. , Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med, 2013. 368(17): p. 1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gan XT, et al. , Probiotic administration attenuates myocardial hypertrophy and heart failure after myocardial infarction in the rat. Circ Heart Fail, 2014. 7(3): p. 491–9. [DOI] [PubMed] [Google Scholar]
- 29.Lam V, et al. , Intestinal microbiota determine severity of myocardial infarction in rats. FASEB J, 2012. 26(4): p. 1727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z, et al. , Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature, 2011. 472: p. 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramezani A and Raj DS, The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol, 2014. 25(4): p. 657–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang S, et al. , A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie Van Leeuwenhoek, 2016. 109(10): p. 1389–96. [DOI] [PubMed] [Google Scholar]
- 33.Le Poul E, et al. , Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem, 2003. 278(28): p. 25481–9. [DOI] [PubMed] [Google Scholar]
- 34.Pluznick JL, et al. , Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A, 2013. 110(11): p. 4410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Z, et al. , Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes, 2009. 58(7): p. 1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mirmonsef P, et al. , Short-chain fatty acids induce pro-inflammatory cytokine production alone and in combination with toll-like receptor ligands. Am J Reprod Immunol, 2012. 67(5): p. 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinolo MA, et al. , SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS One, 2011. 6(6): p. e21205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vieira AT, et al. , A Role for Gut Microbiota and the Metabolite-Sensing Receptor GPR43 in a Murine Model of Gout. Arthritis Rheumatol, 2015. 67(6): p. 1646–56. [DOI] [PubMed] [Google Scholar]
- 39.Bolognini D, et al. , The Pharmacology and Function of Receptors for Short-Chain Fatty Acids. Mol Pharmacol, 2016. 89(3): p. 388–98. [DOI] [PubMed] [Google Scholar]
- 40.Ge H, et al. , Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology, 2008. 149(9): p. 4519–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.