

Introduction
Idiopathic pulmonary hypertension (IPAH) is a rare progressive vasculopathy with an unknown etiology, and it is fatal if not diagnosed and treated in time (1). Moyamoya disease (MD) is characterized by spontaneous occlusion of the circle of Willis, associated with an exaggerated development of collateral vascular network at the base of the skull (2). MD associated with pulmonary hypertension is an unusual presentation. Herein, we reported a child who presented with exercise-associated syncope and was diagnosed with MD with pulmonary hypertension.
Case Report
A 15-year old boy presented with recurrent syncope while climbing stairs. He had three syncope attacks in 2 weeks, all of which were associated with exercise. On admission, his general appearance was stable. The vital signs were as follows: heart rate, 98/min; respiratory rate, 16/min; body temperature, 36.5°C; and blood pressure, 116/72 mm Hg. The pulse oximeter reading was 94% at room air. There was a narrowly split second heart sound with a loud pulmonic component. No respiratory distress was encountered. Mild mental retardation was apparent, and neurological examination was otherwise normal. Family history was unremarkable.
Regarding medical history, he had focal seizure twice at the age of 3 and 4 years. Carbamazepine was started after the second seizure. Electroencephalogram (EEG) was normal and cranial magnetic resonance imaging (MRI) revealed no major parenchymal abnormality. There was no seizure attack after the initiation of carbamazepine, and the drug was stopped after 5 years. The patient was asymptomatic, and EEG revealed no abnormality. However, he had mild mental retardation and his performance in school was not good.
Electrocardiography showed right ventricular hypertrophy criteria. Laboratory findings were normal except for pro b-type natriuretic peptide (pro-BNP), which was mildly elevated (110 pg/mL). The right atrium, right ventricle, and pulmonary artery were enlarged in echocardiogram. The right ventricular pressure was estimated to be 110 mm Hg via tricuspid regurgitation. There was neither heart defect nor any congenital abnormality in echocardiography.
For other etiologies of pulmonary hypertension, evaluations including thorax high-resolution computed tomography, pulmonary function test, serologic tests for connective tissue disorders and coagulation studies, thyroid functions, and Doppler ultrasound for liver were performed, which were all normal. The patient underwent cardiac catheterization for hemodynamic study and vazoreactivity test. Angiogram revealed no ventricular septal defect, patent ductus arteriosus, and aortopulmonary septal defect. The results of hemodynamic study and vazoreactivity test are shown on Table 1. The functional capacity of the patient was NYHA class 1.
Table 1.
Hemodynamic study of the patient on admission
| Before vazoreactivity test | After vazoreactivity test | |
|---|---|---|
| MAPpressure | 80 mm Hg | 78 mm Hg |
| Aorta systolicpressure | 100 mm Hg | 99 mm Hg |
| Rp/Rs | 0.66 | 0.67 |
MAP - mean pulmonary artery; Rp/Rs - pulmonary/systemic vascular resistance ratio
According to our initial findings, the patient was diagnosed with IPAH, and bosentan and aspirin therapy were started.
Cranial MRI performed 12 years ago in another center was re-evaluated, and small collateral vessels striking in both hemispheres were observed. Therefore, cranial MR angiography was performed, and it showed that the cavernous segments of both internal carotid arteries and the supraclinoid segment of right internal carotid artery were not demonstrated. Moreover, the calibration of supraclinoid segment of the left internal carotid artery was minute, and significant collateral vessels were seen in MR angiography. Conventional cerebral angiography confirmed the diagnosis of MD (Fig. 1).
Figure 1.
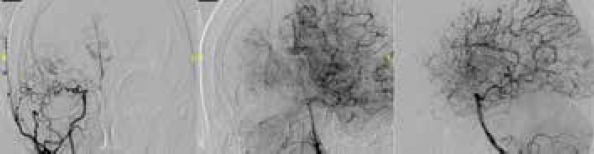
Bilateral internal carotid artery supraclinoid segment and right posterior cerebral artery P1 segment occlusion. “Puff of smoke” appearance indicating the collateralization of cerebrum
Discussion
MD is a chronic progressive, occlusive cerebrovascular disorder, and compensatory enlargement of the cerebral arteries leads to the development of collaterals that appear as a “puff of smoke”, thus, the Japanese term “Moyamoya” (3). Children with MD may present with various symptoms such as aphasia, dysarthria, hemiparesis, and seizures. Less common presentations such as syncope, visual changes, and chorea can occur (4). Although the first sign was seizure in our patient, MD was not diagnosed because of the nonspecific findings on cranial MRI. After experiencing syncope twice while exercising, he was diagnosed with pulmonary hypertension based on echocardiography findings, and the secondary causes were excluded. Since there was co-occurrence of pulmonary hypertension and seizure, the cranial MRI was re-evaluated, and MD was suspected due to collateral vessels seen in both hemispheres. This is a very rare presentation of MD in a child. Therefore, it was emphasized that MD must be kept in mind in all patients diagnosed with IPAH, particularly those who have had concomitant seizures.
The co-occurrence of pulmonary hypertension and MD has been previously reported in very few cases (5-7). Among them, RNF213 homozygosity was found in two patients. Therefore, this gene may cause a novel entity involving the brain and lung together. We also plan to investigate this gene mutation in our patient.
There is no consensus about therapy in children with MD and severe pulmonary hypertension because of the rarity of this entity. In one study, two cases with MD and pulmonary hypertension were reported, and both died after vasoreconstructive surgery for MD (8). Thus, surgery in patients with MD may have poor prognosis, particularly in those with severe pulmonary hypertension, and it should be discussed very carefully with the parents. The risk of surgery was expected to be high due to pulmonary hypertension in our patient, and the parents refused the surgery.
There was one report that showed bosentan therapy improved the blood flow in the cerebral hemispheres in a child with MD (9). Although the role of endothelin pathways in the pathogenesis of MD has not been studied, this case suggested the use of endothelin receptor antagonists to improve the cerebral circulation. However, bosentan and tadalafil combination therapy did not improve the pulmonary artery pressures in our patient; we do not know about the blood flow to both hemispheres yet. Additional studies are needed to determine whether the patients may benefit with bosentan or other specific pulmonary artery hypertension treatments to improve the circulation of cerebral hemispheres and pulmonary artery pressures.
Conclusion
In conclusion, MD should be considered in children with seizures accompanied with pulmonary hypertension, and the risk of shunt surgery is very high in these patients. It is not known whether bosentan or other specific pulmonary artery hypertension treatment influence the mortality or morbidity in this rare entity.
References
- 1.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France:results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 2.Natori Y, Ikezaki K, Matsushima T, Fukui M. “Angiographic moyamoya” its definition, classification, and therapy. Clin Neurol Neurosurg. 1997;99(Suppl 2):S168–72. doi: 10.1016/s0303-8467(97)00052-8. [DOI] [PubMed] [Google Scholar]
- 3.Fukui M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (‘moyamoya’disease). Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare, Japan. Clin Neurol Neurosurg. 1997;99(Suppl 2):S238–40. [PubMed] [Google Scholar]
- 4.Amlie-Lefond C, Ellenbogen RG. Factors Associated with the Presentation of Moyamoya in Childhood. J Stroke Cerebrovasc Dis. 2015;24:1204–10. doi: 10.1016/j.jstrokecerebrovasdis.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Fukushima H, Takenouchi T, Kosaki K. Homozygosity for moyamoya disease risk allele leads to moyamoya disease with extracranial systemic and pulmonary vasculopathy. Am J Med Genet A. 2016;170:2453–6. doi: 10.1002/ajmg.a.37829. [DOI] [PubMed] [Google Scholar]
- 6.Kapusta L, Daniëls O, Renier WO. Moya-Moya syndrome and primary pulmonary hypertension in childhood. Neuropediatrics. 1990;21:162–3. doi: 10.1055/s-2008-1071486. [DOI] [PubMed] [Google Scholar]
- 7.Ou P, Dupont P, Bonnet D. Fibromuscular dysplasia as the substrate for systemic and pulmonary hypertension in the setting of Moya-Moya disease. Cardiol Young. 2006;16:495–7. doi: 10.1017/S104795110600045X. [DOI] [PubMed] [Google Scholar]
- 8.Tokunaga K, Hishikawa T, Sugiu K, Date I. Fatal outcomes of pediatric patients with moyamoya disease associated with pulmonary arterial hypertension. Report of two cases. Clin Neurol Neurosurg. 2013;115:335–8. doi: 10.1016/j.clineuro.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Day RW, Brockmeyer DL, Feola GP. Safe treatment of pulmonary hypertension with bosentan in a patient with moyamoya disease and cerebral ischemia. J Child Neurol. 2010;25:504–7. doi: 10.1177/0883073809339360. [DOI] [PubMed] [Google Scholar]