

Abstract
While DNA sequencing is increasingly being used as a diagnostic tool, targeted assays, culture and other isolation techniques remain a critical component in effective diagnostic workflows.

Subject Categories: Microbiology, Virology & Host Pathogen Interaction; S&S: Health & Disease; S&S: Technology
Diagnostic tests for microbial pathogens have been used in clinical practice long before DNA sequencing technologies were developed. Many of us have personal experience with throat swabs to diagnose strep throat caused by the bacterium Streptococcus pyogenes or nose swabs to diagnose flu caused by the influenza virus. Public health officials are also interested in reliable diagnostic tests to protect us from emerging epidemics such as the recent Ebola and Zika outbreaks, or from food‐borne illnesses that all too frequently make the headlines. Doctors, public health officials, biodefense experts, and many others critically rely on pathogen detection and characterization technologies to diagnose, prevent, treat, and investigate infectious disease.
Are DNA sequencing technologies fast and accurate enough to transform the field of pathogen diagnostics?
Many of the existing tests rely on culturing on media that is known to enrich the targeted pathogen. Incubation times for a positive diagnosis can be as short as 1 or 2 days, but may require up to 5 days for many pathogens, and even weeks in certain cases, such as Nocardia and Actinomyces spp. High‐throughput DNA sequencing technologies have enormous potential to change this state of affairs. The hope is that these technologies will enable culture‐free pathogen diagnostic approaches that are fast, sensitive, and have broad diagnostic capabilities. One can easily imagine a situation where a doctor would place a sample into a microfluidic device that extracts its DNA and feeds it into a rapid sequencing device. The resulting data would be analyzed computationally to determine the identity of the pathogen or pathogens contained in the sample, to propose a strain‐specific treatment regime, and to provide the full sequence of the pathogen(s) for further analysis. The entire process could take just a few hours or perhaps minutes.
There is no doubt that DNA sequencing technologies have already transformed biomedical research. The increasing availability of full genome sequences of many organisms is a valuable tool for molecular biology experiments. Sequencing itself is being used as an assay, for example, to measure gene expression or interrogate the spatial organization of chromosome. Full exome or full genome sequencing is also now part of the standard of care in oncology or to diagnose rare diseases. These and many other applications are enabled by recent technological advances that have resulted in sequencing instruments with much higher throughput and lower costs than available just a few years ago. Yet, their use as a general diagnostics tool raises various questions. Are DNA sequencing technologies fast and accurate enough to transform the field of pathogen diagnostics? What further advances are necessary to enable the vision we outlined above?
Pathogen detection
At their core, all approaches to detect pathogens are similar: They try to identify unique features that distinguish specific pathogens from the many other organisms found in clinical or environmental samples. Culturing techniques are most commonly used, employing selective media to preferentially enrich the pathogens of interest over other organisms. Further analysis of the morphology of colonies within a Petri dish, or other characteristics, such as serotype, can provide more information for distinguishing between pathogens and similar but non‐pathogenic microbes.
In certain applications, such as epidemiology or forensics, it is also useful to finely type an organism in order to trace its origin and path of transmission…
A number of culture‐free diagnostic approaches have already been developed that provide faster response times and/or more accurate diagnoses than culture. Immune antigen‐based tests are generally the fastest as they do not require any additional processing of the samples because they target unique features on the surface of a pathogen's cells. Molecular approaches use one or more specific locations within the genome of an organism of interest for characterization and therefore require an additional step to isolate DNA. Some tests focus solely on characterizing the size of genomic segments, such as tandem repeats with variable copy number (VNTRs) or the size of genomic segments created through the use of a restriction enzyme. Other tests use DNA sequencing to interrogate a number of predetermined locations within the genome of an organism, an approach referred to as multi‐locus sequence typing (MLST), which is routinely used to identify and characterize Salmonella infections.
While molecular diagnostic approaches that are carefully designed for specific pathogens are effective in most clinical settings, they are powerless when faced with emerging threats.
Both culture‐based and molecular diagnostic approaches can also provide an estimate of the amount of pathogen found in a sample. Techniques such as quantitative culture or quantitative PCR are critical in a surveillance context where it is important to distinguish between a harmful pathogen load and a harmless level of environmental contamination.
Gaps in current diagnostic capabilities
Pathogen detection is not a simple “yes/no” question. In deciding on treatment or further action, it is important to know not just the identity of a pathogen, but also whether it is resistant to certain antibiotics, or whether it has evolved or has been intentionally modified. In certain applications, such as epidemiology or forensics, it is also useful to finely type an organism in order to trace its origin and path of transmission—information that can only be obtained by characterizing its genome. Such questions cannot be easily answered by targeted molecular diagnostic techniques; thus, culture remains a critical tool in the characterization of pathogens, as the cultured cells provide a substrate for further in‐depth assays, such as assessing drug resistance or genome sequencing.
In diagnosing food poisoning, for example, a clinician needs to know not only that the likely pathogen is Escherichia coli, but also whether this organism contains specific genes or toxins. While in some cases E. coli infections can be treated with antibiotics, in others, such as those caused by enterohemorrhagic E. coli (EHEC), antibiotic treatment can lead to hemolytic uremic syndrome, a potentially fatal complication when the dying bacteria release Shiga toxin into the gut lumen.
While molecular diagnostic approaches that are carefully designed for specific pathogens are effective in most clinical settings, they are powerless when faced with emerging threats. Prior to the recent Zika outbreak that prompted the development and use of Zika‐specific diagnostics, the infection was frequently mis‐diagnosed as the more commonly occurring dengue or chikungunya virus. Untargeted sequencing‐based assays can accurately diagnose such rare diseases or flag the presence of unexpected organisms even if their identity or pathogenic potential is not known.
Sequencing in outbreak investigations
The anthrax mailings of 2001 prompted the first use of whole‐genome sequencing as part of the outbreak investigation 1. While the diagnostics of Bacillus anthracis, the causative agent of anthrax, was well established at that time, existing diagnostic markers did not provide sufficient resolution to distinguish between the many different B. anthracis isolates that were characterized during the investigation—dubbed by the FBI “the Amerithrax investigation”. A number of isolates were subjected to whole‐genome shotgun sequencing—a technology that was still in its infancy—in hopes that small genomic changes that accumulated during culturing would help to pinpoint the origin of the strain used in the attack. This hope was borne out: Sequencing revealed a number of mutations, including a previously unknown sequence duplication within the genome of some of the B. anthracis isolates. The latter feature helped to link the attack strain to the specific isolate from which it likely originated.
Just a few years after the anthrax attacks, DNA sequencing was used in an investigation of a tuberculosis outbreak that occurred between August and September 2006 in British Columbia 2. And another few years later, in 2011, sequencing was used to characterize the source of a food‐borne E. coli O104:H4 outbreak in Germany and France, helping to identify the lateral transfer of genomic factors that rendered this strain particularly virulent 3.
These, and many other studies that followed, have cemented the role of sequencing in outbreak investigations. In the United States, pathogens identified during surveillance or outbreak investigations are now routinely sequenced and made publicly available through the GenomeTrakr database 4.
What does it take to be a pathogen?
The examples described above used DNA sequencing to finely type pathogens that had already been detected and isolated through culture; however, sequencing can also be used to fully eliminate the need for culturing. DNA extracted from a sample can be sequenced and computationally analyzed to determine the presence of pathogens and even specific genes of interest such as toxin or drug resistance genes. In its most basic form, the detection algorithm can simply search each DNA sequence against a database of known pathogens or genes. More efficiently, one could focus on just those segments that distinguish pathogens from non‐pathogenic close relatives.
… common logic is often a better diagnostic feature thatcannot be easily encoded in a traditional database.
A powerful demonstration of the limitations of such approaches is a 2015 study of the microbes found in the New York City subway 5. The authors used high‐throughput genome sequencing to characterize the microbial composition of more than 1,400 samples obtained from across the city's subway system. By searching public databases, they were able to create a “pathogen map” of the subway. They highlighted, in particular, the discovery of several significant human pathogens, such as B. anthracis (causative agent of anthrax) and Yersinia pestis (causative agent of plague). These findings turned out to be incorrect, particularly because of the way in which the sequencing data were interpreted 6. The genomic feature that led to the discovery of plague in the subway was a plasmid found not just in Y. pestis but also in several other organisms that do not cause plague. It just happened to only be found in Y. pestis within the database used by the researchers. Similarly, the sequence that positively identified B. anthracis belonged to a virus that happens to infect these organisms, but also non‐pathogenic relatives which were not included in the database at that time.
Databases matter
This example highlights the paramount importance of the database used to classify sequences. It is not sufficient that a database contains the pathogens of interest, but also their many non‐pathogenic relatives that could be found in the specific environment from where a sample is taken. Little is known, however, about the normal microflora inhabiting our world. Sequencing efforts have understandably focused on our immediate threats: the relatively few organisms that can harm or kill humans, animals, and crops. Funding for scientific research remains scarce and broader sequencing efforts are difficult to justify, especially given the tremendous genomic diversity of the world's microbiota.
It is certain, however, that sequencing‐based analytics require non‐trivial investments in computational infrastructures.
In the absence of a comprehensive database of non‐pathogenic organisms, incorrect diagnoses can be avoided through a careful selection of the diagnostic marker used to distinguish between a pathogen and its harmless relatives. In some cases, the distinguishing feature may be just a single nucleotide. For example, all known pathogenic strains of B. anthracis contain a single nucleotide mutation that inactivates the PlcR gene. This feature was used to determine that a bacterium found during routine monitoring of the International Space Station (ISS) was not B. anthracis, as suggested by its morphological features, but instead a previously uncharacterized strain of Bacillus cereus, a usually non‐pathogenic relative of B. anthracis that is commonly found in soil and even on food 7.
Other useful diagnostic markers include toxins and other genomic factors that directly influence an organisms' pathogenicity. The virulence of B. anthracis is largely due to two plasmids which carry toxin and capsule genes. These plasmids are perhaps an even better target for a diagnostic test—B. anthracis cells lacking these plasmids are unlikely to be harmful, but B. cereus strains that have acquired the plasmids would represent a health threat despite the usually non‐pathogenic nature of B. cereus.
Constructing databases that are an effective component of sequencing‐based diagnostic assays is a non‐trivial endeavor. The sheer size of the data that need to be stored, representing all known pathogens and a good representation of their non‐pathogenic relatives, is only a small part of this challenge. We currently lack a good understanding of the function of most genes, even for well‐studied organisms, yet it is exactly this understanding that is needed to identify those genomic features that are effective targets of diagnostic assays: the genes and other genomic segments that make a pathogen a pathogen. Continued and sustained funding of functional characterization studies is a critical prerequisite for developing this knowledge.
Further advances are also needed in the semantic structure of databases. Existing frameworks are effective at capturing what exists and is known: Does a sequence match a known pathogen or a known virulence gene? In some cases, however, the absence of a gene may be the distinguishing feature—for instance, some bacteria develop resistance to antibiotics by disrupting porin genes. Conclusively proving that a particular genomic region is absent from a sequencing data set is currently an unsolved problem. Furthermore, common logic is often a better diagnostic feature that cannot be easily encoded in a traditional database. In the case of the suspected B. anthracis contamination of the space station, months had passed between the time the sample had been isolated and the time it was being processed. If this had truly been a health threat, the astronauts would have already developed symptoms. Similarly, the detection of pathogen‐like organisms within artisanal cheese and meat products is counter‐balanced by the fact that these products have been safely consumed for centuries. Such logic is commonly used by physicians within their diagnostic process, yet cannot be easily encoded in the databases and software used to analyze DNA sequences.
Can sequencing fully replace culture?
Even if the databases and algorithms used to detect pathogens from sequencing data were perfect, the use of sequencing alone as a pathogen detection assay is unlikely to be effective in most cases. An example is the diagnosis of Listeria monocytogenes within ice cream samples associated with an outbreak of listeriosis 8. The authors used a sequencing‐based assay to interrogate samples extracted at regular intervals from an enrichment culture of the contaminated ice cream. While a small amount of L. monocytogenes could be detected through sequencing in the initial sample, the pathogen was dwarfed by a complex background comprising many other bacterial species. This background microbiota dominated the culture media for the first 24 h of culture, with L. monocytogenes falling below the limit of detection for the first 4–8 h. Only after 24 h, L. monocytogenes was detected at a high enough abundance to allow further characterization of the strains contaminating the samples.
… the question is not whether sequencing‐based or traditional diagnostic approaches are better, but how can we combine multiple approaches to leverage their complementary strengths.
Such situations are common in food safety and biodefense applications. Many bacteria, including pathogens, can be found at low levels of abundance throughout our environment. Whether or not they pose a risk to human health depends on whether one could be exposed to an infectious dose of the pathogen. The answer depends on the absolute abundance of the pathogen of interest, while sequencing‐based assays generally measure the relative abundance of organisms. These issues are compounded in clinical applications where pathogens occur within a background of commensal organisms and host DNA. Accurate detection and quantitation of pathogens are impossible from sequencing data alone.
Quantitation aside, the sequencing of complex mixtures of organisms does not afford the same analytic powers as the sequencing of isolated colonies. Many of the advantages of sequencing we have discussed so far derive from the ability to characterize the full genome of potential pathogens, yet the reconstruction of complete genomes from complex mixtures is currently an unsolved challenge.
In short, the sensitivity of sequencing‐based assays is currently low. To be an effective diagnostic tool, sequencing must be combined with culture or other enrichment approaches specifically targeted at the pathogens of interest.
You can't sequence your way out of the problem
A counter‐point to the arguments made above is that sequencing costs are rapidly dropping making it feasible to sequence a particular sample as deeply as necessary to achieve high sensitivity. This argument is partly what fuels the current enthusiasm for the promise of sequencing in pathogen diagnostics. It does, however, ignore the significant computational costs required to analyze the resulting data. As sequencing costs drop, the computational costs increase, not just in terms of CPU time, but also in terms of the storage space needed for the sequencing data (Fig 1).
Figure 1. The cost of sequencing versus the cost of computing.
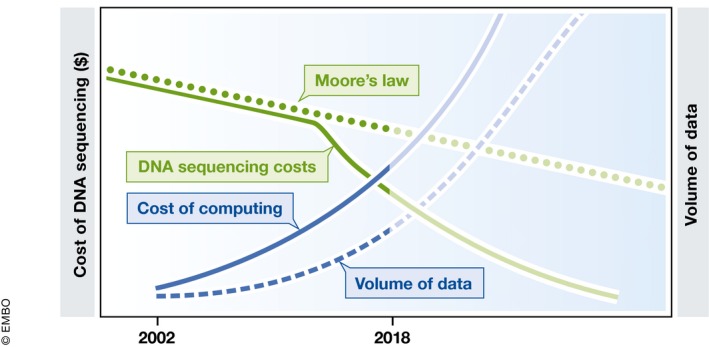
The costs of DNA sequencing (solid green line) have dropped significantly in recent years, leading to a rapid increase in the volumes of data being generated (dashed blue line). The reduction in sequencing costs, however, is outpacing the rate at which the cost of computing drops (Moore's law, dotted green line). As a result, the cost of computing (solid blue line) needed to analyze the resulting data is also rapidly increasing.
As an example, consider a typical data set generated by sequencing a human stool sample. The compressed data comprise more than 5 gigabytes or roughly 20–50 times more than a typical computed tomography (CT) scan. Aligning the sequences to a small set of just 50 complete bacterial genomes can take upwards of 3 h on a typical computer.
It is not yet clear whether the combined cost of sequencing and analysis is decreasing, staying stable or even possibly increasing. It is certain, however, that sequencing‐based analytics require non‐trivial investments in computational infrastructures. In many cases, even a single sample cannot be effectively analyzed on a typical modern laptop. Cloud computing can mitigate the costs of setting up and maintaining a complex computational infrastructure, but the increased use of such shared resources is raising significant security and privacy concerns. Data encryption and other approaches used to ensure the security of data stored in the cloud add to the already significant computational costs of analyses.
Independent of the volume of data being generated, the size of the database used to identify pathogens and to distinguish them from near neighbors is also rapidly increasing. As an example, as of the middle of 2018, the US FDAs GenomeTrakr database contained more than 200,000 bacterial genomes, with over 5,000 new genomes being deposited every month. This database comprises only bacterial pathogens, thus representing just part of the tremendous volume of data becoming available.
In the case of bacterial pathogens, database size is partly mitigated by the fact that detection schemes can rely on just a small fraction of the genomes, focusing on genes broadly conserved across bacteria. This is not the case for viral or eukaryotic pathogens. In viruses, rapid genome evolution, and in eukaryotes, large variation in gene structure at the DNA level even for a same class of proteins, makes it impossible to identify a small set of “marker” genes that can be effectively identified through DNA sequencing. As such, viral and eukaryotic pathogen databases must contain full genomic sequences, further compounding the computational costs of the diagnostic pipeline.
An outlook to the future
Our description so far has discussed sequencing‐based pathogen detection as a replacement for traditional culture‐based or targeted molecular diagnostic approaches. This is the most commonly held mindset—if only we could get cheaper and faster sequencing, we could do away with the awkward technologies of the past. As we have shown, the reality is a lot more nuanced. The sensitivity of sequencing‐based assays is relatively low, and the reduced costs and increased speed of sequencing are largely offset by the increased computational cost of analysis (Fig 1). At the same time, sequencing does provide analytical capabilities not previously available, particularly in the context of emerging diseases and in forensic applications. As such, the question is not whether sequencing‐based or traditional diagnostic approaches are better, but how can we combine multiple approaches to leverage their complementary strengths (Fig 2).
Figure 2. The value of pathogen culture.

When sequencing complex samples, the pathogen of interest may only represent a small fraction of the data generated (right side of figure). Differential culture helps amplify the amount of pathogen, thereby increasing the sensitivity of pathogen detection and identification (left side of the figure).
Nanopore sequencing—an exciting recent development—does open the possibility for effective sequencing‐based pathogen diagnostics. These devices directly read the DNA as it passes through a pore, generating data in real time. They can selectively reject individual DNA molecules based on the partially read segment of the molecule, effectively creating a real‐time microfluidic enrichment device 9. Furthermore, nanopore devices can detect not just the nucleotide composition of DNA fragment, but also epigenetic signals such as methylation 10. Such signals could distinguish, for example, between host and pathogen DNA. Coupled with selective sequencing, this functionality could provide high detection sensitivity even in the context of a complex background.
These technological innovations must be paralleled by improved algorithmic approaches. While nanopore devices generate data in real time, the analysis of the resulting data can be considerably slower. This is true even for the algorithms used to convert the electrical signal into base calls, let alone the more complex analytics to positively identify a pathogen. Effective real‐time filtering of DNA as it is being sequenced will require the development of new software able to process the data at the rate at which they are being generated.
Further advances are also needed to create and manage effective databases that can scale in both size and performance with the rapid increase in genomic sequences becoming available. While hundreds of thousands of genomes of pathogens and related organisms are currently available, no approaches exist today that can efficiently search them.
Sequencing technologies have certainly transformed pathogen diagnostics; however, the goal of creating rapid, sensitive, and broad sequencing‐based diagnostic approaches remains out of reach. Further technological advances in both sequencing and computational analytics techniques are necessary to achieve the vision we outlined at the beginning of this article.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
M.P. and T.J.T were supported by the FunGCAT program from the Office of the Director of National Intelligence (ODNI), Intelligence Advanced Research Projects Activity (IARPA), via the Army Research Office (ARO) under Federal Award No. W911NF‐17‐2‐0089. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of the ODNI, IARPA, ARO, or the US Government.
References
- 1. Rasko DA, Worsham PL, Abshire TG, Stanley ST, Bannan JD, Wilson MR, Langham RJ, Decker RS, Jiang L, Read TD et al (2011) Bacillus anthracis comparative genome analysis in support of the Amerithrax investigation. Proc Natl Acad Sci USA 108: 5027–5032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R et al (2011) Whole‐genome sequencing and social‐network analysis of a tuberculosis outbreak. N Engl J Med 364: 730–739 [DOI] [PubMed] [Google Scholar]
- 3. Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D et al (2011) Origins of the E. coli strain causing an outbreak of hemolytic‐uremic syndrome in Germany. N Engl J Med 365: 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allard MW, Strain E, Melka D, Bunning K, Musser SM, Brown EW, Timme R (2016) Practical value of food pathogen traceability through building a whole‐genome sequencing network and database. J Clin Microbiol 54: 1975–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Afshinnekoo E, Meydan C, Chowdhury S, Jaroudi D, Boyer C, Bernstein N, Maritz JM, Reeves D, Gandara J, Chhangawala S et al (2015) Geospatial resolution of human and bacterial diversity with city‐scale metagenomics. Cell Syst 1: 72–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ackelsberg J, Rakeman J, Hughes S, Petersen J, Mead P, Schriefer M, Kingry L, Hoffmaster A, Gee JE (2015) Lack of evidence for plague or anthrax on the New York City Subway. Cell Syst 1: 4–5 [DOI] [PubMed] [Google Scholar]
- 7. Venkateswaran K, Checinska Sielaff A, Ratnayake S, Pope RK, Blank TE, Stepanov VG, Fox GE, van Tongeren SP, Torres C, Allen J et al (2017) Draft genome sequences from a novel clade of Bacillus cereus Sensu Lato strains, isolated from the international space station. Genome Announc 5: e00680–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ottesen A, Ramachandran P, Reed E, White JR, Hasan N, Subramanian P, Ryan G, Jarvis K, Grim C, Daquiqan N et al (2016) Enrichment dynamics of Listeria monocytogenes and the associated microbiome from naturally contaminated ice cream linked to a listeriosis outbreak. BMC Microbiol 16: 275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loose M, Malla S, Stout M (2016) Real‐time selective sequencing using nanopore technology. Nat Methods 13: 751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W (2017) Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods 14: 407 [DOI] [PubMed] [Google Scholar]