

Abstract
During an infection, the detection of pathogens is mediated through the interactions between pathogen‐associated molecular patterns (PAMPs) and pathogen recognition receptors. β‐Heptose 1,7‐bisphosphate (βHBP), an intermediate of the lipopolysaccharide (LPS) biosynthesis pathway, was recently identified as a bacterial PAMP. It was reported that βHBP sensing leads to oligomerization of TIFA proteins, a mechanism controlling NF‐κB activation and pro‐inflammatory gene expression. Here, we compare the ability of chemically synthesized βHBP and Shigella flexneri lysate to induce TIFA oligomerization in epithelial cells. We find that, unlike bacterial lysate, βHBP fails to initiate rapid TIFA oligomerization. It only induces delayed signaling, suggesting that βHBP must be processed intracellularly to trigger inflammation. Gene deletion and complementation analysis of the LPS biosynthesis pathway revealed that ADP‐heptose is the bacterial metabolite responsible for rapid TIFA oligomerization. ADP‐heptose sensing occurs down to 10−10 M. During S. flexneri infection, it results in cytokine production, a process dependent on the kinase ALPK1. Altogether, our results rule out a major role of βHBP in S. flexneri infection and identify ADP‐heptose as a new bacterial PAMP.
Keywords: ADP‐heptose, ALPK1, HBP, pathogen recognition, TIFA
Subject Categories: Microbiology, Virology & Host Pathogen Interaction
Introduction
During an infection, an immune response is triggered by the detection of specific pathogen‐related antigens. This is mainly achieved by cells of the innate immune system including macrophages and neutrophils. Epithelial and endothelial cells are also involved in this process. They act as sentinels of the immune system and contribute, via the secretion of inflammatory mediators, to elicit an integrated immune response. At the molecular level, pathogen recognition is mediated by the interactions between pathogen‐associated molecular patterns (PAMPs) and pathogen recognition receptors. Well‐characterized bacterial PAMPs include among others lipopolysaccharide (LPS), flagellin, peptidoglycan, lipoteichoic acid, and DNA. An intermediate of the LPS biosynthesis pathway, β‐heptose 1,7‐bisphosphate (βHBP), was recently identified as a PAMP of Neisseria meningitidis 1. In N. meningitidis, it is produced by the d‐β‐d‐heptose 7‐phosphate kinase activity of the HldA protein that phosphorylates d‐glycero‐β‐d‐manno‐heptose 7‐phosphate into βHBP 2. Gaudet et al 1 showed that it can be secreted by N. meningitidis and sensed in the cytosol of eukaryotic cells after being internalized by endocytosis. Our laboratory reported that βHBP sensing is also involved in the inflammatory response of epithelial cells to Shigella flexneri and Salmonella typhimurium infection 3. This finding was based on the analysis of the hlde and gmhb deletion mutants of the LPS biosynthesis pathway. In these bacterial species closely related to Escherichia coli, hlde encodes a bifunctional protein that harbors both d‐β‐d‐heptose 7‐phosphate kinase and d‐β‐d‐heptose 1‐phosphate adenylyltransferase activities, responsible for the production of βHBP and ADP‐d‐glycero‐β‐d‐manno‐heptose (ADP‐d‐β‐d‐heptose), respectively 4. The dephosphorylation of βHBP into d‐glycero‐β‐d‐manno heptose 1‐phosphate is provided by the phosphatase activity of GmhB 5. We showed that infection with an hlde deletion (ΔhldE) mutant failed to induce the production of the inflammatory cytokine interleukin‐8 (IL‐8), whereas this latter was restored upon infection with a mutant deleted for gmhb (ΔgmhB) 3. S. flexneri infection leads to oligomerization of the proteins, TRAF‐interacting protein with FHA domain‐containing protein A (TIFA) and TNF receptor‐associated factor 6 (TRAF6), and activation of the transcription factor NF‐κB. In line with a previous study reporting that uninfected bystander epithelial cells constitute the main source of IL‐8 during S. flexneri infection 6, oligomerization of TIFA and TRAF6 as well as activation of NF‐κB were found in both infected and uninfected bystander cells 3. TIFA is a 20‐kDa protein that was first identified as a TRAF6‐interacting protein in a yeast two‐hybrid screen 7. It contains a forkhead‐associated (FHA) domain, known to bind phosphothreonines and phosphoserines 8, and a consensus TRAF6‐binding motif. TIFA oligomerization is dependent on the phosphorylation of a threonine at position 9 and on the FHA domain 9. Unphosphorylated TIFA is thought to exist as an intrinsic dimer constitutively linked to TRAF6. When T9 is phosphorylated, this is recognized by the FHA domain of other TIFA dimers leading to its oligomerization and thus the oligomerization of TRAF6. This enhances the E3 ubiquitin ligase activity of TRAF6 and leads to the activation of the NF‐κB pathway 10. We discovered that ALPK1, an atypical kinase of the α‐kinase family, regulates the oligomerization of TIFA in response to S. flexneri infection 3. More recently, the role of βHBP sensing and the contribution of the ALPK1/TIFA axis were also observed in Helicobacter pylori infection 11, 12, 13, reinforcing the broad implication of this newly emerging pathway in innate immunity. In addition to the use of LPS biosynthesis mutants, the pro‐inflammatory activity of βHBP was directly confirmed. It was first synthesized in vitro from the sedoheptulose 7‐phosphate precursor by the sequential activities of purified GmhA and HldE 1. βHBP was also chemically synthesized 14, 15, 16, 17. Its pro‐inflammatory activity was validated on the basis that it induced NF‐κB activation after several hours of treatment 15, 16. In this report, we compare the ability of chemically synthesized βHBP and S. flexneri lysate to induce TIFA oligomerization and IL‐8 production in human epithelial cells. In contrast to bacterial lysate, we find that βHBP is unable to induce rapid oligomerization of TIFA. Oligomerization is only significant 2 h post‐treatment, indicating that cellular processing of βHBP is necessary to elicit inflammatory signaling and that another PAMP was therefore responsible for the early mechanism of TIFA oligomerization that occurs during infection 3. By combining gene deletion mutants and functional complementation, we identify ADP‐heptose as a new PAMP involved in S. flexneri infection. Furthermore, we show that ADP‐heptose sensing leads to the production of cytokines via an ALPK1‐dependent mechanism.
Results and Discussion
βHBP fails to induce rapid oligomerization of TIFA
In order to characterize the role of βHBP in innate immunity, this bacterial metabolite was chemically synthesized. A synthetic route was implemented, which was inspired, for the most part, from the work of Vincent and coworkers 18 (Fig 1 and Materials and Methods). Briefly, starting from thiophenyl α‐d‐mannoside (2) 19, easily synthesized in three steps from d‐mannose (1), a sequence of protection/deprotection steps afforded the production of 2,3,4‐tri‐O‐benzylated derivative 3 on a large scale (55 mmol) and in good yield. Homologation of this thiomannoside intermediate by means of a Parikh–Doering oxidation/Wittig sequence gave the terminal olefin‐containing heptoside 4 20. The latter was subsequently reacted with catalytic osmium tetroxide to obtain a 7:3 diastereomeric mixture of diols, from which the major isomer was isolated by chromatography. A subsequent four‐step protection/deprotection procedure was applied to give hemiacetal 5. The hemiacetal was submitted to a Mitsunobu‐mediated bis‐phosphorylation at OH‐1 and OH‐7 by use of a combination of dibenzyl phosphate, DIAD, P(ClPh)3, and triethylamine in tetrahydrofuran 14. Chromatography of the obtained α/β mixture of the perbenzylated diphosphorylated monosaccharide gave the α‐anomer and β‐anomer in 30 and 41% yield, respectively. The purified β‐heptose was subjected to a palladium‐catalyzed hydrogenation followed by H+/Na+ exchange to give the expected βHBP (Fig EV1) 14. The pro‐inflammatory activity of chemically synthesized βHBP was first tested by measuring the production of IL‐8 after transient permeabilization of cell membranes with digitonin. In this assay, the cellular process of βHBP internalization is bypassed and the mechanisms of PAMP recognition and inflammatory signaling can thus be directly assessed. HeLa cells were treated for 30 min with digitonin and synthetic βHBP at indicated concentrations (Fig 2A and B). As a control, cells were treated with digitonin alone or digitonin and wild‐type (wt) S. flexneri bacterial lysate. Cells were then washed and treated with monensin to trap IL‐8 intracellularly, and IL‐8 was monitored in single cells after 6 h with automated microscopy. Data showed that synthetic βHBP induced the production of IL‐8 in a dose‐dependent manner (Fig 2A and B). Interestingly, the βHBP concentration required to induce IL‐8 synthesis to the level of S. flexneri lysate was unexpectedly high (Fig 2A and B). Since a millimolar‐range concentration of βHBP was likely not reached in bacterial lysates 21, this observation raised the possibility that additional bacterial factors contribute to elicit the IL‐8 response. βHBP‐induced IL‐8 production was previously shown to depend on the oligomerization of TIFA proteins, a process controlling NF‐κB activation 1, 3. We therefore analyzed the effect of synthetic βHBP on TIFA oligomerization. HeLa cells expressing GFP‐tagged TIFA protein (TIFA‐GFP) were treated as described above. TIFA‐GFP oligomerization was monitored after 6 h and quantified in single cells by automated image analysis. In line with IL‐8 data, βHBP induced the formation of large TIFA‐GFP oligomers in a dose‐dependent manner (Fig 2C and D). This result was in agreement with previous reports showing that synthetic βHBP induces NF‐κB activation after several hours of treatment 15, 16. Given that TIFA oligomerization is observed within minutes of S. flexneri, S. typhimurium, or H. pylori infections 3, 11, we also monitored TIFA oligomerization after 30 min of treatment. Strikingly, whereas S. flexneri lysate induced massive oligomerization of TIFA‐GFP within this time period, synthetic βHBP failed to do so (Fig 2E). At least 2 h was required to observe significant oligomerization of TIFA‐GFP in cells treated with 10−3 M synthetic βHBP (Fig 2F). Such delay in the response to synthetic βHBP suggests that this bacterial metabolite has to be processed intracellularly to trigger inflammatory signaling and that sensing of another bacterial PAMP is likely responsible for the rapid mechanism of TIFA oligomerization observed upon treatment with S. flexneri lysate.
Figure 1. Chemical synthesis of βHBP.
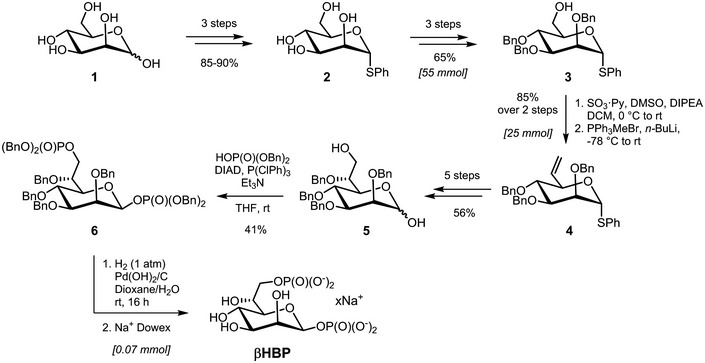
Chemical synthesis of βHBP from d‐mannose (1): overview.
Figure EV1. 1 H‐NMR spectrum of synthetic β‐heptose 1,7‐bisphosphate in its sodium salt form (400 MHz, D2O).
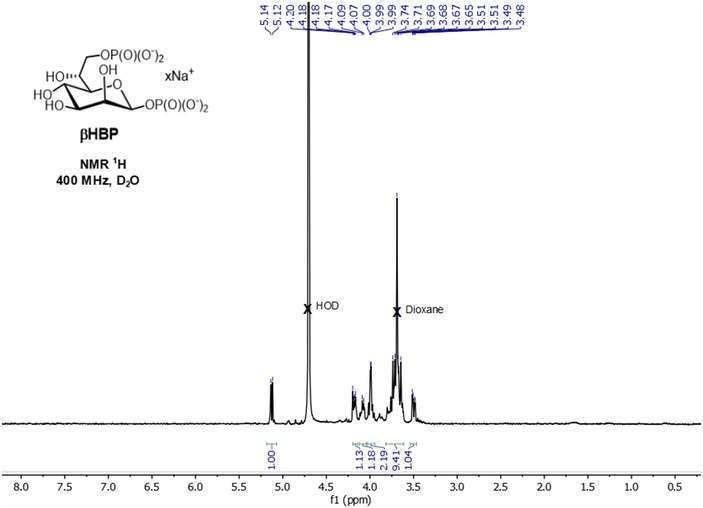
Figure 2. βHBP fails to induce rapid oligomerization of TIFA.
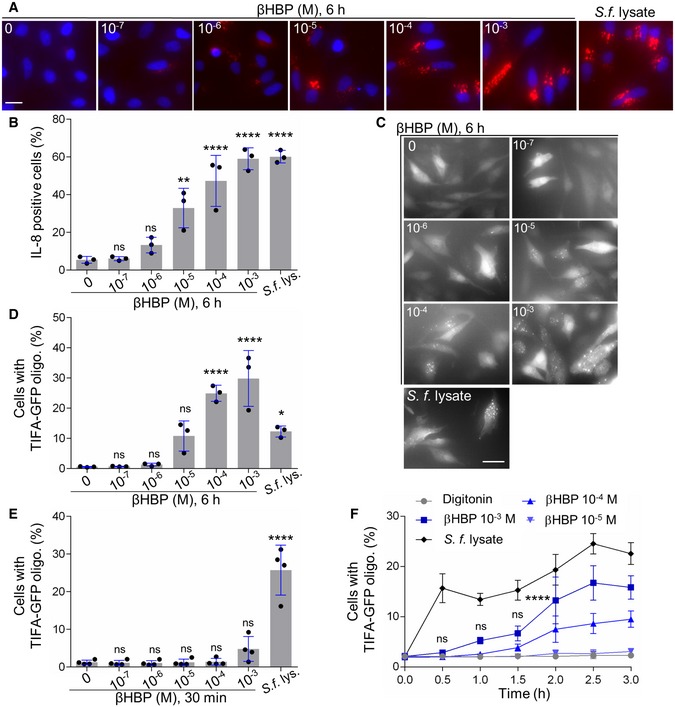
- IL‐8 production after 6 h of treatment with digitonin and βHBP at indicated concentrations or digitonin and wt Shigella flexneri lysate in HeLa cells (Hoechst in blue, IL‐8 in red).
- Quantification of IL‐8 as shown in (A). Data correspond to the mean ± SD of three independent experiments.
- TIFA oligomerization at 6 h in TIFA‐GFP‐expressing HeLa cells treated as in (A).
- Quantification of TIFA oligomerization as shown in (C). Data correspond to the mean ± SD of three independent experiments.
- TIFA oligomerization at 30 min in cells treated as in (A). Data correspond to the mean ± SD of four independent experiments.
- Kinetics of TIFA‐GFP oligomerization upon treatment with digitonin alone or in combination with βHBP or Shigella flexneri lysate. Data correspond to the mean ± SEM of four independent experiments.
ADP‐heptose is a newly identified bacterial PAMP
Our results showing that βHBP was unable to trigger rapid oligomerization of TIFA‐GFP (Fig 2E and F) opened a new avenue for the identification of new bacterial PAMPs inducing inflammatory signaling upon S. flexneri infection. Given that we previously reported that ΔhldE S. flexneri bacteria completely failed to trigger TIFA oligomerization and inflammatory gene expression 3, we further analyzed the role of the bifunctional enzyme HldE. More specifically, we analyzed the contribution of its d‐β‐d‐heptose 7‐phosphate kinase and d‐β‐d‐heptose 1‐phosphate adenylyltransferase activities that synthesize βHBP and ADP‐d‐β‐d‐heptose, respectively (Fig 3A). For this, we took advantage of the fact that in N. meningitidis, these enzymatic activities are harbored by two distinct proteins encoded by the hlda and hldc genes 2, and examined the ability of lysates from ΔhldE S. flexneri complemented with hlde, hlda, hldc, or hlda and hldc to induce TIFA‐GFP oligomerization in digitonin‐permeabilized cells. Whereas treatment with wt S. flexneri lysate induced oligomerization of TIFA‐GFP at 30 min, lysate from ΔhldE bacteria failed to do so (Fig 3B). As expected, complementation of ΔhldE bacteria with a plasmid encoding HldE (pHldE) fully restored fast oligomerization of TIFA‐GFP, confirming the critical role of HldE in this process. Interestingly, lysate from ΔhldE bacteria complemented with plasmid‐encoded HldA (pHldA) had no effect at 30 min (Fig 3B). This result was in line with synthetic βHBP data showing that this molecule was indeed unable to trigger rapid oligomerization of TIFA‐GFP (Fig 2E and F). By showing that bacterial d‐β‐d‐heptose 1‐phosphate adenylyltransferase activity was required, this new finding indicated that the PAMP eliciting rapid oligomerization was produced downstream of the adenylyltransferase enzymatic reaction within the LPS biosynthesis pathway (Fig 3A). Complementation of ΔhldE S. flexneri with plasmid‐encoded HldC (pHldC) also failed to trigger TIFA oligomerization at 30 min, showing that bacterial synthesis of βHBP, although not sufficient, was required to trigger early signaling. As expected, this process was restored with ΔhldE bacteria complemented with both pHldA and pHldC (Fig 3B). The observation that double complementation only led to partial restoration may be explained by a low expression of the HldA and HldC proteins encoded from two different plasmids, or by a difference in enzymatic efficacy between HldE on the one hand and HldA and HldC on the other hand. Together, these results showed that d‐β‐d‐heptose 7‐phosphate kinase and d‐β‐d‐heptose 1‐phosphate adenylyltransferase activities are both required to stimulate rapid pro‐inflammatory signaling, indicating that bacterial synthesis of βHBP and ADP‐d‐β‐d‐heptose is necessary. In order to further characterize the PAMP involved in rapid inflammatory signaling, we analyzed the hldD and waaC deletion mutants of the LPS biosynthesis pathway, named ΔhldD and ΔwaaC, respectively. HldD harbors an epimerase activity catalyzing the interconversion between ADP‐d‐β‐d‐heptose and ADP‐L‐glycero‐β‐d‐manno‐heptose (ADP‐L‐β‐d‐heptose; Fig 3A) 22. Directly downstream of HldD, WaaC, which has an ADP‐heptose‐lipopolysaccharide heptosyltransferase activity, allows heptose transfer to the LPS core 22. Data showed that ΔhldD and ΔwaaC lysates were both effective at triggering TIFA‐GFP oligomerization at 30 min, indicating that the PAMP controlling this process was produced upstream of HldD or WaaC, respectively. Altogether, by showing that the PAMP of interest was dependent on βHBP, produced downstream of the d‐β‐d‐heptose 1‐phosphate adenylyltransferase enzymatic reaction, and upstream of HldD or WaaC (Fig 3B), our results point to the role of ADP‐d‐β‐d‐heptose and ADP‐L‐β‐d‐heptose (named ADP‐heptose for simplification reason) in triggering rapid oligomerization of TIFA upon treatment with S. flexneri lysate. TIFA‐GFP oligomerization and IL‐8 production were also analyzed at 6 h. Based on synthetic βHBP data (Fig 2A–D and F), we hypothesized that, at this time point, both βHBP and ADP‐heptose contribute to inflammatory signaling. Data confirmed a critical role for bacterial expression of HldE in lysate‐induced TIFA‐GFP oligomerization (Fig 3C) and IL‐8 production (Fig 3D) at 6 h. In line with synthetic βHBP data (Fig 2A–D), pHldA‐mediated complementation partially restored TIFA‐GFP oligomerization (Fig 3C) and IL‐8 production (Fig 3D), showing that bacterial synthesis of βHBP is sufficient to trigger delayed inflammatory responses. Complementation of ΔhldE S. flexneri lysate with pHldC failed to trigger TIFA‐GFP oligomerization (Fig 3C) as well as IL‐8 production (Fig 3D), showing that bacterial synthesis of βHBP is strictly required for both processes. Finally, complementation with pHldA and pHldC partially restored both oligomerization and IL‐8 production (Fig 3C and D). The observation that expressing HldA or both HldA and HldC in ΔhldE bacteria had a rather similar effect may be explained by a higher expression of HldA in single complementation experiments and enhanced βHBP concentration in bacteria lacking d‐β‐d‐heptose 1‐phosphate adenylyltransferase activity. As expected, lysates from ΔhldD and ΔwaaC mutants induced TIFA‐GFP oligomerization and IL‐8 production (Fig 3C and D), confirming the role of ADP‐heptose in inflammation. The pro‐inflammatory activity of this bacterial metabolite was directly addressed by measuring TIFA‐GFP oligomerization 30 min after treatment with digitonin and chemically synthesized ADP‐L‐β‐d‐heptose (Fig EV2 and Materials and Methods). Unlike βHBP (Fig 2E and F), ADP‐L‐β‐d‐heptose induced rapid oligomerization of TIFA‐GFP in a dose‐dependent manner (Fig 3E). Interestingly, oligomerization was still significant at 10−10 M, showing that the cellular mechanism of ADP‐heptose sensing occurs down to this concentration limit. In line with these results, ADP‐L‐β‐d‐heptose induced IL‐8 production in a similar range of concentrations (Fig 3F), confirming its very potent pro‐inflammatory activity. Overall, our data confirm that βHBP can only elicit delayed inflammatory responses, and reveal that ADP‐heptose is a new potent bacterial PAMP that significantly contributes to the inflammatory response observed during treatment with S. flexneri lysate.
Figure 3. ADP‐heptose is a newly identified bacterial PAMP.
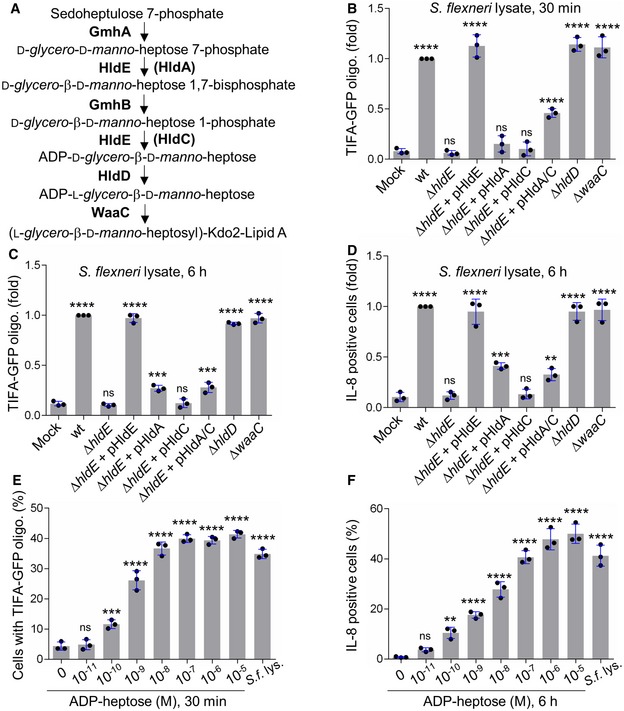
- LPS biosynthesis pathway. HldA and HldC from Neisseria meningitidis are in brackets.
- TIFA‐GFP oligomerization at 30 min in HeLa cells treated with digitonin (mock) or digitonin and a lysate from indicated Shigella flexneri strains (pHldA/C means plasmid‐encoded HldA and HldC).
- TIFA‐GFP oligomerization at 6 h in cells treated as in (B).
- IL‐8 production at 6 h in cells was treated as in (C).
- TIFA‐GFP oligomerization in cells treated for 30 min with digitonin alone, digitonin and wild‐type S. flexneri lysate, or digitonin and ADP‐L‐β‐d‐heptose at indicated concentrations.
- IL‐8 production after 6 h in cells treated as in (E).
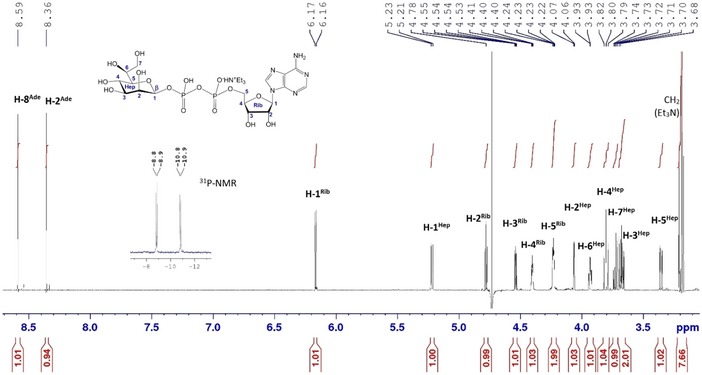
Figure EV2. 1 H‐NMR (600 MHz, D2O) and 31P‐NMR (243 MHz) spectra of synthetic ADP‐ l ‐ glycero ‐β‐ d ‐ manno ‐heptose.
ADP‐heptose sensing induces inflammation in Shigella flexneri infection
The use of digitonin‐permeabilized cells is a powerful method to directly address PAMP recognition and inflammatory signaling. However, it does not provide information on the actual involvement of a given PAMP during infection. The respective contribution of βHBP and ADP‐heptose was therefore tested in S. flexneri infection experiments. For this purpose, HeLa cells were infected with wt S. flexneri or with the mutants of the LPS biosynthesis pathway described above, and TIFA‐GFP oligomerization was visualized 30 min postinfection. Representative images are shown in Fig 4A. As previously reported 3, infection with wt bacteria induced rapid oligomerization of TIFA‐GFP in both infected cells and uninfected bystander cells. In contrast, the ΔhldE mutant failed to do so, whereas pHldE complementation fully restored oligomerization. Together, these results confirmed the critical implication of HldE in the early host response to S. flexneri infection. In line with data obtained with bacterial lysates (Fig 3B), complementing the ΔhldE mutant with pHldA or pHldC failed to trigger TIFA‐GFP oligomerization at 30 min (Fig 4A), showing that bacterial d‐β‐d‐heptose 7‐phosphate kinase and d‐β‐d‐heptose 1‐phosphate adenylyltransferase activities are both necessary to trigger rapid oligomerization of TIFA‐GFP. This mechanism is indeed restored when cells were infected with pHldA and pHldC‐complemented ΔhldE bacteria (Fig 4A). Finally, oligomerization was observed in cells infected with ΔhldD and ΔwaaC mutants, showing that the PAMP responsible for this process was produced upstream of HldD or WaaC, respectively. In E. coli, ΔhldE and ΔwaaC mutants have a phenotype of increased bacterial auto‐aggregation and stronger cell surface hydrophobicity compared to wt bacteria 23. In S. flexneri, deletion mutants are much more invasive than wt (Fig 4B), making the comparison of TIFA oligomerization between all strains inappropriate. For this reason, only mutants that had the same range of infectivity were considered for quantification. Data confirmed that none of the single‐complemented mutants induced early TIFA‐GFP oligomerization (Fig 4C). In contrast, although infectivity of the double‐complemented mutant was slightly reduced compared to ΔhldE bacteria (Fig 4B), it significantly led to TIFA‐GFP oligomerization (Fig 4C). The weak impact of HldA and HldC complementation on bacterial infectivity compared to the difference observed between ΔhldE and wt (Fig 4B) suggested that the LPS of these bacteria was still significantly altered and that complementation was only partial. This observation was correlated with the rather modest restoration of inflammatory signaling previously observed with the double‐complemented mutant (Fig 3B–D), and may be explained by low expression or weak enzymatic activities of HldA and HldC in these bacteria. In line with lysate data (Fig 3B), infection with ΔhldD and ΔwaaC mutants induced oligomerization at 30 min (Fig 4C). Altogether, these results showed that βHBP is not sufficient to trigger early signaling in S. flexneri‐infected cells and revealed the critical role of ADP‐heptose in this process. In order to better understand the dynamics of the inflammatory response during infection, TIFA‐GFP oligomerization and IL‐8 production were analyzed 4 h postinfection. In agreement with a role of βHBP in late signaling, significant oligomerization was visible upon infection with pHldA‐complemented bacteria (Fig 4D). However, no IL‐8 was observed in response to this infection (Figs 4E and EV3). We hypothesized that the level of TIFA‐GFP oligomerization was too low to elicit IL‐8 production within 4 h. As expected, infection with pHldC‐complemented bacteria failed to trigger TIFA‐GFP oligomerization (Fig 4D) and IL‐8 production (Figs 4E and EV3), confirming the strict requirement of βHBP bacterial synthesis. In line with data obtained at 30 min (Fig 4A and C), infection with the double‐complemented mutant induced significant oligomerization of TIFA and IL‐8 production. This latter is more likely induced by ADP‐heptose than βHBP since, within 4 h of infection, the single pHldA‐complemented mutant was unable to produce IL‐8 (Figs 4E and EV3). Finally, infection with ΔhldD and ΔwaaC mutants showed oligomerization and IL‐8 production (Figs 4D and E, and EV3). Altogether, these results show that ADP‐heptose is a PAMP that causes inflammation during S. flexneri infection.
Figure 4. ADP‐heptose sensing induces inflammation in Shigella flexneri infection.
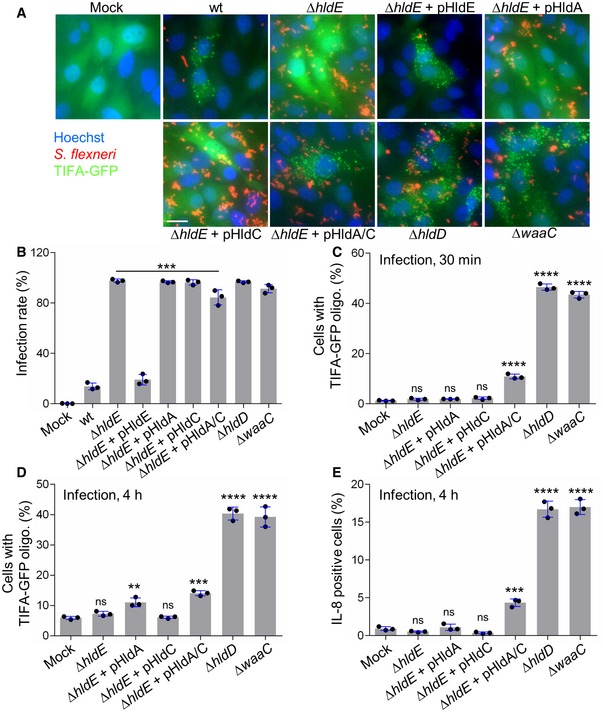
- Representative images of the formation of TIFA oligomers in HeLa cells infected for 30 min with dsRed‐expressing wt or indicated mutants of S. flexneri (MOI 50). pHldA/C means plasmid‐encoded HldA and HldC. Fluorescence intensity was adjusted between strains to optimize visualization of bacteria. Scale bar, 20 μm.
- Infectivity of indicated S. flexneri strains.
- TIFA‐GFP oligomerization after 30 min of infection with selected mutants of the LPS biosynthesis pathway (MOI 10).
- TIFA‐GFP oligomerization after 4 h of infection with the S. flexneri mutants shown in (C).
- IL‐8 production 4 h postinfection (MOI 5).
Figure EV3. IL‐8 measurements after infection with selected mutants of the LPS biosynthesis pathway.
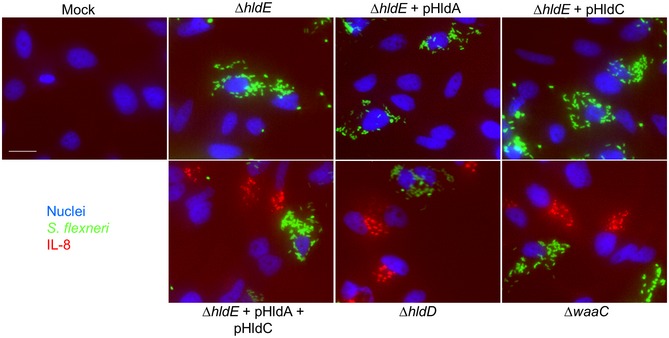
Cells were infected with selected mutants of the LPS biosynthesis pathway at MOI 5, and IL‐8 accumulation was visualized after 4 h. Hoechst, Shigella flexneri, and IL‐8 are shown in blue, green, and red, respectively. Scale bar, 20 μm.
ADP‐heptose sensing is ALPK1‐dependent
We previously reported that S. flexneri‐induced TIFA oligomerization is under the control of the atypical kinase ALPK1 3. To check that this finding was consistent with the newly identified role of ADP‐heptose as a PAMP in S. flexneri infection, we investigated ADP‐heptose‐induced TIFA oligomerization in ALPK1‐depleted cells. For this purpose, HeLa cells expressing TIFA‐GFP were transfected with control or ALPK1‐targeting siRNAs. After 48 h, they were transfected again with an empty vector or a siRNA‐resistant ALPK1 cDNA construct as previously described 3. Cells were then treated for 30 min with digitonin and synthetic ADP‐heptose or infected with wt S. flexneri. As in the case of infection, ADP‐heptose‐induced TIFA‐GFP oligomerization was abolished in ALPK1‐depleted cells (Fig 5A). As expected, it was restored upon transfection of siRNA‐resistant ALPK1, showing that the mechanism of ADP‐heptose sensing was ALPK1‐dependent. In line with these results, multiplex cytokine analysis revealed that the secretion of IL‐8, interleukin‐6 (IL‐6), and interleukin‐4 (IL‐4) was significantly enhanced in response to S. flexneri infection or ADP‐heptose and that this induction was also largely ALPK1‐dependent (Fig 5B). Altogether, these results strongly support the role of ADP‐heptose as a PAMP in S. flexneri infection, and show that the mechanism of ADP‐heptose sensing is ALPK1‐dependent.
Figure 5. ADP‐heptose sensing is ALPK1‐dependent.
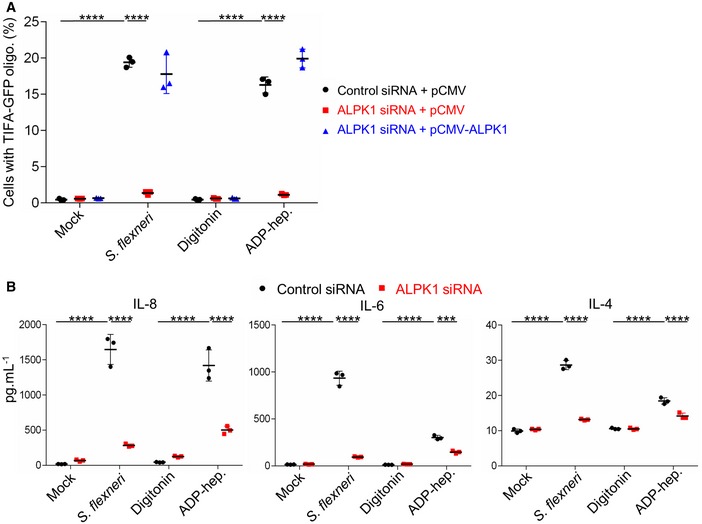
- ADP‐heptose‐induced TIFA‐GFP oligomerization depends on ALPK1. After siRNA transfection, HeLa cells were transfected with empty pCMV or pCMV‐ALPK1 as indicated. They were then infected with wt Shigella flexneri (MOI 10) or treated with digitonin and ADP‐heptose (10−5 M) for 30 min. As control, cells were left untreated (mock) or treated with digitonin.
- ADP‐heptose‐induced cytokine secretion is ALPK1‐dependent. HeLa cells were transfected with control or ALPK1‐targeting siRNAs and infected with wt S. flexneri (MOI 2) or treated with ADP‐heptose (10−5 M). As control, cells were left untreated (mock) or treated with digitonin. Cytokines were measured in cell culture supernatants after 8 h.
In conclusion, our data rule out a major contribution of βHBP in S. flexneri infection and identify ADP‐heptose as a new potent bacterial PAMP that can be detected down to a concentration of 10−10 M. The delay in inflammatory signaling observed in response to βHBP suggested that this bacterial metabolite is not directly recognized by host defense mechanisms. At the time of finalizing this manuscript, Zhou et al 24 confirmed this hypothesis by showing that βHBP must be converted into ADP‐heptose 7‐phosphate by host adenylyltransferase enzymes of the NMNAT family to induce inflammatory signaling. Although βHBP is a bacteria‐derived factor triggering an immune response, its classification as a genuine PAMP is challenged by the fact that it is not directly sensed via the interaction with a cognate pathogen recognition receptor. The role of ADP‐heptose in S. flexneri infection questions our previous results implicating βHBP 3. They were based on the ability of the ΔgmhB mutant to induce TIFA oligomerization. We now hypothesize that this mutant has only a partial phenotype. Indeed, the observation that, unlike ΔhldE, ΔhldD, and ΔwaaC, the ΔgmhB mutant is not hyper‐invasive 3 suggested that its LPS is only partially altered and that ADP‐heptose synthesis still occurs in these bacteria. A partial phenotype of the ΔgmhB mutant was observed in E. coli, and the existence of a yet unidentified phosphatase activity that compensates for the absence of GmhB was proposed 22. Given that the ΔgmhB‐based argument was also used in other studies 12, 13, the direct contribution of βHBP was likely more broadly overestimated. The identification of ADP‐heptose as a PAMP in S. flexneri infection opens up new avenues for understanding how inflammation is regulated at the level of the intestinal epithelium in shigellosis. The mechanism of ADP‐heptose sensing will have to be thoroughly characterized. We show in our study that it is ALPK1‐dependent. This result is consistent with the study by Zhou et al 24 who elegantly reported that the kinase ALPK1 is indeed the cytosolic immune receptor for both ADP‐L‐β‐d‐heptose and ADP‐D‐β‐d‐heptose. More work is required to identify the respective contribution of these two metabolites during S. flexneri infection, understand how they are delivered in the cytoplasm of host cells, and characterize the spatio‐temporal dynamics of ALPK1 activation. In particular, it will be interesting to investigate whether they are injected in infected cells by the type III secretion apparatus of bacteria, and whether they diffuse via gap junctions to trigger TIFA oligomerization and cytokine production in uninfected bystander cells as previously described 3. Alternative and non‐exclusive mechanisms implicating the release of ADP‐heptose from damaged bacteria upon rupture of the internalization vacuole or the contribution of a cellular transporter will also have to be investigated. Given that ADP‐heptose is broadly present in bacteria, our study offers also new perspectives to elucidate the molecular mechanisms regulating innate immunity in infections by other important human pathogens, including H. pylori, S. typhimurium, and Pseudomonas aeruginosa. Finally, beyond infections, it will contribute to characterize new molecular cross‐talks between bacteria of the microbiota, the intestinal epithelium, and the immune system that control intestinal homeostasis.
Materials and Methods
Cell culture and transfections
HeLa cells (American Type Culture Collection) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS and 2 mM GlutaMAX‐1 (complete growth medium) at 37°C under 5% CO2. HeLa cells stably expressing TIFA‐GFP were obtained after transfection of pEGFP‐C1 plasmid encoding TIFA‐GFP and geneticin selection at 1 mg ml−1. Reverse transfection of siRNAs was carried out using RNAiMAX according to the manufacturer's instructions (Invitrogen Thermo Fisher). HeLa cells, seeded in 96‐well plates (8,000 cells/well), were reverse‐transfected with 20 nM siRNA and used 72 h after transfection. As a control, cells were transfected with a non‐targeting sequence (4390843) from Ambion (Thermo Fisher). As previously described for ALPK1 depletion 3, cells were transfected with a validated ALPK1 siRNA (s37074) from Ambion (Thermo Fisher). Validation data are available on the manufacturer's website. To demonstrate knockdown specificity, an ALPK1 rescue experiment was performed as previously described 3. Briefly, 48 h after siRNA transfection, cells were transfected with a siRNA‐resistant ALPK1 cDNA construct (pCMV‐ALPK1) or an empty vector (pCMV) using FuGENE 6 (Roche).
Bacterial strains and generation of mutants
Wild‐type (wt), ΔhldE, and ΔwaaC M90T S. flexneri strains have been previously described 3. The ΔhldD mutant was generated by allelic exchange using a modified protocol of lambda red‐mediated gene deletion 25. Briefly, the kanamycin cassette of the pkD4 plasmid was amplified by PCR with the primers listed in Table 1. The purified PCR product was electroporated into the wt strain expressing the genes for lambda red recombination from the pKM208 plasmid. Recombinants were selected on TSB plates containing 50 μg ml−1 of kanamycin. Single colonies were screened by PCR. S. flexneri M90T expressing the hldA gene from N. meningitidis was previously described 3. Shigella flexneri M90T expressing the hldC gene from N. meningitidis was generated as follows. hldC was amplified by PCR from a bacterial lysate with the primers listed in Table 1. After gel purification (Macherey–Nagel), the PCR product was digested with EcoRI and HindIII, and ligated into an EcoRI/HindIII‐digested pHSG396 plasmid, a derivative of pUC type of plasmid harboring a chloramphenicol resistance cassette (Clontech). The ligation product was used to transform Top10 E. coli. pHSG396‐HldC was purified and used to electroporate ΔhldE and ΔhldE expressing hldA strains of S. flexneri M90T. All strains were transformed with pCLR7 and constitutively expressed dsRed protein (kind gift from D. Bumann, Biozentrum, Basel, Switzerland).
Table 1.
Table listing the primers used in this study
| Primer | Sequence (5′ > 3′) |
|---|---|
| Shigella flexneri ΔhldD | |
| Forward | AGCATCCTTGCACCTCTATGTAAAGGGCTGAAGGGATTCGGATGTGATGGTATGATTACAGACATTCGTGTCTGAGATTGTCTCTGACTCCATAATTCGAAGGTTACAGTTATGATCTGTGTAGGCTGGAGCTGCTTCG |
| Reverse | ACATCGATTATCGCCTGGGGATAGCGCGCCTGGAGCGTGCGATAGAGACTTTGCGACATCATCATGTCGCCAACCCAAGACGGGCCGATCACCAGTATTTTCATGCAGAGCTCTTATGCCATATGAATATCCTCCTTAG |
| Neisseria meningitidis hldC | |
| Forward | CCCAAGCTTAATGGTTGACGCTTGGTCTGTCCCT |
| Reverse | CCGGAATTCTCATTTTCCGCCCTCTGCCGCACG |
Preparation of bacterial lysates
Bacteria from overnight cultures were washed in PBS and resuspended at 1010 bacteria ml−1. Once transferred into cryotubes, they underwent through two freeze–thaw cycles in liquid nitrogen. Bacterial debris were removed by centrifugation at 16,000 g for 20 min. Lysates were stored at −80°C.
Permeabilized cell assay
Cells were seeded in 96‐well plates the day before experiment. They were washed in permeabilization buffer containing 100 mM KCl, 3 mM MgCl2, 50 mM HEPES, 0.1 mM DTT, 85 mM sucrose, 0.2% BSA, and 0.1 mM ATP. Cells were then incubated with digitonin (2.5 μg ml−1) alone or digitonin plus bacterial lysates, βHBP, or ADP‐heptose for 30 min in this same buffer. To monitor TIFA‐GFP oligomerization at 30 min, they were fixed with 4% PFA. To monitor TIFA‐GFP and IL‐8 production at 6 h, they were washed to remove digitonin and incubated for 5.5 h in DMEM supplemented with 1% FCS.
Infections
For S. flexneri infection experiments, bacteria were used in exponential growth phase. Cells, seeded in 96‐well plates, were infected at indicated MOIs in DMEM supplemented with 10 mM HEPES, 2 mM GlutaMAX‐1, and 1% FCS. In infection experiments using only wt S. flexneri, bacteria were coated with poly‐l‐lysin prior infection. After adding bacteria, plates were centrifuged for 3 min at 300 g and placed at 37°C for the indicated time periods. For 30‐min infection experiments, cells were fixed by adding 4% PFA after 30 min. For 4‐h infection experiments, cells were washed after 30 min. Extracellular bacteria were killed by adding gentamicin (100 μg ml−1), and infection was stopped after 3.5 h by adding 4% PFA. For infection rate measurements, cells were infected with bacteria for 30 min, washed, treated with gentamycin, incubated for 3.5 h, and fixed with 4% PFA. Infected cells were quantified with automated image analysis (see below).
IL‐8 measurements
The production of IL‐8 was measured by immunofluorescence 6 h after cell treatments or 4 h postinfection. In order to trap IL‐8 intracellularly, monensin (50 μM) was added 3 h before fixation. After fixation in 4% PFA, cells were stained with an anti‐human IL‐8 antibody in 0.2% saponin in PBS (BD Pharmingen, San Jose, USA) and an Alexa Fluor 647‐conjugated secondary antibody (Invitrogen, Carlsbad, USA). IL‐8 was quantified by automated image analysis (see below). In parallel, cells were stained with Hoechst (Invitrogen) to visualize nuclei.
Cytokine multiplex assay
The secretion of cytokines was measured in the supernatant of cells infected for 8 h with S. flexneri (MOI 2) or treated with digitonin and ADP‐heptose (10−5 M) by using Cytokine Human Magnetic 10‐Plex Panel for the Luminex (Life Technologies). For infection, cells were washed after 30 min and extracellular bacteria were killed by adding gentamicin (100 μg ml−1). As a control, cells were left untreated or treated with digitonin alone. Measurements for IL‐1β, IL‐2, TNF‐α, GM‐CSF, IFN‐γ, IL‐10, and IL‐5 were below detection limit.
Automated microscopy and image analysis
Images were acquired with an ImageXpress Micro (Molecular Devices, Sunnyvale, USA). Each data point corresponds to triplicate wells, and more than nine images were taken per well. Image analysis was performed using the custom module editor (CME) of MetaXpress. For IL‐8, measurements were performed as previously described 3. Briefly, cell nuclei were identified by the “Autofind blobs” function. Each nucleus was extended by six pixels to define the cell mask in which IL‐8 signals were quantified. They were detected with the “Keep marked object” function of CME based on minimal/maximal size requirements and intensity threshold. For the oligomerization of TIFA, foci were detected by applying the “Find blobs” function based on size and intensity above background parameters. Cell nuclei were identified with the “Find round objects” module. Cells were defined by extending each nucleus by 10 pixels with the “Grow objects without touching” function, and TIFA punctates were quantified within this mask. For infection measurements, nuclei and cells were segmented as above. Bacteria were detected by the “Find blobs” function based on size and intensity above background parameters. For IL‐8, TIFA oligomerization, and infection measurements, the fraction of cells respectively containing IL‐8, TIFA oligomers, or bacteria was extracted and used for quantification.
Chemical synthesis of βHBP
General information
Thin layer chromatography (TLC) was performed on precoated slides of silica gel 60 F254 (Merck). Detection was effected when applicable, with UV light, and/or by charring with orcinol (35 mM) in 4 N aqueous (aq.) sulfuric acid and ethanol (95:5). Flash chromatography was performed by elution from columns of silica gel 60 (particle size 0.040–0.063 mm). Analytical reversed‐phase high‐performance liquid chromatography (RP‐HPLC) was performed by elution from a C18 XBridge Waters Amide column (4.6 × 250 mm), using CH3CN in 5 mM aq. ammonium formate (0→3%) at 0.5 ml min−1 and detecting at 230 nm. Nuclear magnetic resonance (NMR) spectra were recorded at 30°C for solutions in CDCl3 or D2O (400 MHz for 1H, 100 MHz for 13C). Residual CHCl3 (7.28 ppm for 1H and 77.0 ppm for 13C) and HOD (4.79 ppm) were used as internal references for solutions in CDCl3 and D2O, respectively. Proton‐signal assignments were made by first‐order analysis of the spectra and analysis of 2D 1H‐1H correlation maps (COSY). Of the two magnetically non‐equivalent geminal protons at C‐7, the one resonating at lower field is denoted H‐7a, and the one at higher field is denoted H‐7b. Carbon‐signal assignments were supported by 2D 13C‐1H correlation maps (HSQC) and analysis of the DEPT and 13C spectra. Interchangeable assignments are marked with an asterisk. Electrospray Ionization time‐of‐flight (ESI‐TOF) mass spectra were recorded in the positive‐ion mode using a 1:1 CH3CN/water containing 0.1% formic acid ESI‐TOF spectrometer solution. Anhydrous (anhyd.) dichloromethane (DCM), 1,2‐dichloroethane (DCE), tetrahydrofuran (THF), N,N‐dimethylformamide (DMF), and toluene (Tol), sold on molecular sieves (MS), were used as such. Reactions requiring anhyd. conditions were performed under an Argon atmosphere.
Synthetic protocols and analytical data
Phenyl 2,3,4‐tri‐ O ‐benzyl‐1‐thio‐α‐ d ‐mannopyranoside (3). To a solution of thioglycoside 2 19 (15.0 g, 55.1 mmol, 1 equiv.) and imidazole (11.3 g, 165.2 mmol, 3 equiv.) in anhyd. THF (110 ml) was added triisopropylsilyl chloride (TIPSCl; 12.4 ml, 58.8 mmol, 1.05 equiv.) dropwise for 15 min at 0°C. The reaction mixture was stirred at 0°C for 20 min before warming up to room temperature for 2.5 h, at which time TLC showed the disappearance of the starting material (DCM/MeOH 8:2, Rf = 0.71) and the presence of a less polar compound (Tol/EtOAc 3:7, Rf = 0.6). An additional amount of TIPSCl (0.08 ml, 0.40 mmol, 0.05 equiv.) was added, and after 5 h, the reaction was then quenched with aq. saturated (sat.) NH4Cl. The phases were separated, the aq. layer was extracted with EtOAc, and the organic phases were washed with brine. The organic phase was dried over Na2SO4, filtered, and evaporated under reduced pressure, and the crude was used as such in the next step.
To a solution of the crude triol (23.6 g, 55.1 mmol, 1 equiv.) and benzyl bromide (BnBr, 23.6 ml, 198.4 mmol, 3.6 equiv.) in anhyd. DMF (306 ml) was added NaH (60% in mineral oil, 13.2 g, 330.6 mmol, 6 equiv.) portionwise for 15 min at 0°C. The reaction mixture was allowed to warm up to room temperature (rt) overnight, at which time TLC showed the disappearance of the starting material (Tol/EtOAC 3:7, Rf = 0.6) and the presence of a less polar compound (cyclohexane/EtOAc 9:1, Rf = 0.75). The reaction was then quenched with MeOH at 0°C, and volatiles were evaporated under reduced pressure. EtOAc and H2O were added, and the aq. layer was extracted with EtOAc. The organic phases were washed successively with aq. sat. NH4Cl and brine, dried over Na2SO4, filtered, and evaporated under reduced pressure, and the crude was used as such in the next step.
To a solution of the silyl ether (38.5 g, 55.1 mmol, 1 equiv.) in anhyd. THF (306 ml) was added tetrabutylammonium fluoride (TBAF, 110 ml, 1 M in THF, 110 mmol, 2 equiv.) at rt. The reaction mixture was stirred for 3 h at which time TLC showed the disappearance of the starting material (Tol/EtOAc 7:3, Rf = 0.9) and the presence of a more polar compound (Tol/EtOAc 7:3, Rf = 0.6). The reaction was quenched with aq. sat. NH4Cl. The phases were separated, the aq. layer was extracted with EtOAc, and the organic phases were washed successively with aq. sat. NH4Cl and brine, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude was purified by column chromatography on silica gel using cyclohexane/EtOAc (100:0–80:20) to afford alcohol 3 as a yellow oil (28.0 g, 93% over three steps). (ESI+)‐HRMS: Calcd for C33H38NO5S [M+NH4]+ m/z: 560.2471, Found: 560.2485. Other analytical data were similar to those published 14.
Phenyl 2,3,4‐tri‐ O ‐benzyl‐6,7‐dideoxy‐1‐thio‐α‐ d ‐ manno ‐hept‐6‐enopyranoside (4) [ 20 ]. To a solution of alcohol 3 (15.0 g, 276.4 mmol, 1 equiv.) in anhyd. DCM (2.0 ml) stirred at 0°C were added successively DMSO (9.8 ml, 138.2 mmol, 5 equiv.), DIPEA (14.5 ml, 82.9 mmol, 3 equiv.), and SO3·Py (13.2 g, 82.9 mmol, 3 equiv.). The reaction mixture was stirred at 0°C for 15 min and was then allowed to warm up to rt for 4 h. At this time, the reaction was quenched with aq. sat. NH4Cl. The phases were separated, the aq. layer was extracted with DCM, and the organic phases were washed successively with aq. sat. NH4Cl and brine, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude was used as such in the next step.
To a suspension of Ph3PMeBr (52.3 g, 146.5 mmol, 5.3 equiv.) in anhyd. THF (420 ml) stirred at 0°C was added n‐BuLi (33.2 ml, 82.9 mmol, 3 equiv.) dropwise. The yellow solution was warmed up to rt for 1 h. Then, a solution of the obtained aldehyde (14.9 g, 276 mmol, 1 equiv.) in anhyd. THF (10 ml) was added dropwise at −78°C. The reaction mixture was allowed to warm up to rt overnight, at which time TLC showed the disappearance of the starting material (Tol/EtOAc 8:2, Rf = 0.65) and the presence of a less polar compound (cyclohexane/EtOAc 8:2, Rf = 0.9). The reaction was quenched with aq. sat. NH4Cl. The phases were separated, the aq. layer was extracted with EtOAc, and the organic phases were washed successively with aq. sat. NH4Cl and brine, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude was purified by column chromatography on silica gel using Tol/EtOAc (100:0–95:5) to afford heptoside 4 as a yellow oil (13.6 g, 85%). (ESI+)‐HRMS: Calcd for C34H38NO4S [M+NH4]+ m/z: 556.2521, Found: 556.2527. Other analytical data were similar to those published 14.
2,3,4,6‐Tetra‐ O ‐benzyl‐ d ‐ glycero ‐α‐ d ‐ manno ‐heptopyranose (5). To a solution of phenyl 2,3,4,6‐tetra‐O‐benzyl‐1‐thio‐d‐glycero‐α‐d‐manno‐heptopyranoside 14 (1.0 g, 1.51 mmol, 1 equiv.) in a 9:1 mixture of acetone/H2O (15 ml) was added N‐bromosuccinimide (NBS; 537 mg, 3.02 mmol, 2 equiv.) at 0°C under Ar and in the dark. The orange mixture was stirred at 0°C for 15 min and then was warmed up to rt. After stirring for 1.5 h, an additional amount of NBS (1 equiv.) was added to the colorless solution at 0°C. The mixture was stirred overnight at rt, at which time TLC showed the disappearance of the starting material (Tol/EtOAc 8:2, Rf = 0.7) and the presence of a more polar compound (Tol/EtOAc 8:2, Rf = 0.2). The reaction was quenched with aq. sat. Na2S2O3. The phases were separated, the aq. layer was extracted with EtOAc, and the organic phases were washed successively with aq. sat. NaHCO3 and brine, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude was purified by column chromatography on silica gel using Tol/EtOAc (100:0–50:50) to afford heptopyranose 5 as a white wax (822 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 7.38–7.17 (m, 20H, H‐Ph), 5.18 (brs, 1H, H‐1), 4.92 (d, J = 11.1 Hz, 1H, H‐CH 2Ph), 4.77 (d, J = 12.2 Hz, 1H, H‐CH 2Ph), 4.71 (d, J = 12.0 Hz, 1H, H‐CH 2Ph), 4.64 (s, 2H, H‐CH 2Ph), 4.64 (d, J = 12.3 Hz, 1H, H‐CH 2Ph), 4.59 (d, J = 11.1 Hz, 1H, H‐CH 2Ph), 4.55 (d, J = 12.0 Hz, 1H, H‐CH 2Ph), 4.13 (brd, J = 8.6 Hz, 1H, H‐5), 4.02–3.95 (m, 2H, H‐3, H‐4), 3.80 (tapp, J = 2.2 Hz, 1H, H‐2), 3.80–3.73 (m, 2H, H‐6, H‐7a), 3.72–3.65 (m, J = 14.3, 7.6 Hz, 1H, H‐7b), 3.45 (brs, 1H, OH). 13C NMR (100 MHz, CDCl3) δ 138.3 (Cq‐Ph), 138.3 (Cq‐Ph), 138.2 (Cq‐Ph), 138.1 (Cq‐Ph), 128.6–127.6 (20C‐Ph), 92.5 (C‐1), 79.9 (C‐3*), 78.1 (C‐6), 75.1 (C‐2), 74.7 (C‐4*), 74.7 (C‐CH2Ph), 72.8 (C‐CH2Ph), 72.7 (C‐5), 72.1 (C‐CH2Ph), 71.8 (C‐CH2Ph), 61.8 (C‐7). (ESI+)‐HRMS: Calcd for C35H42O7N [M+NH4]+ m/z: 588.2961, Found: 588.2979.
d ‐ Glycero ‐β‐ d ‐ manno ‐heptopyranose 1,7‐bisphosphate (βHBP). To a solution of dibenzyl 2,3,4,6‐tetra‐O‐benzyl‐7‐di(benzyloxy)phosphoryl‐d‐glycero‐β‐d‐manno‐heptopyranosyl phosphate (70 mg, 64 μmol, 1 equiv.) in a 3:1 mixture of 1,4‐dioxane/H2O (4 ml) was added Pd(OH)2/C (20%, 20 mg) at rt. The reaction mixture was stirred for 16 h under H2 (1 atm). The reaction mixture was degassed, filtered on 0.2‐μm filter, and washed with H2O. The filtrate was adjusted to pH = 7.0 with Na+ Dowex 50 and stirred for 30 min. The resin was filtered off, and the filtrate was lyophilized to give βHBP in its sodium salt form as a hygroscopic solid (28 mg, 94%). (ESI−)‐HRMS: Calcd for C7H15O13P2 [M−H]− m/z: 368.9988, Found: 368.9974. tr (RP‐HPLC): 3.69 min. Other analytical data were similar to those published 14 (see 1H NMR spectrum shown in Fig EV1).
Chemical synthesis of ADP‐heptose
ADP‐L‐β‐d‐heptose was chemically synthesized as previously described 26. Final purification of synthetic ADP‐heptose (1 mg) was performed by anion‐exchange chromatography on Bio‐Rad Macro‐Prep® High Q Support (HCOO− form, column 100 × 15 mm). Elution was performed by a stepwise gradient [0.01 M to 0.25 M TEAB (pH 7)]. Fractions containing ADP‐heptose (detection by thin layer chromatography on PEI‐cellulose, 0.25 M TEAB, Rf 0.64, and on HPTLC cellulose F, MeOH‐H2O‐0.2 M TEAB, 10:2:1, Rf 0.2, visualization under UV light) were pooled and diluted with deionized sterile water to a volume of 10 ml. To remove residual TEAB, the pH of solution was adjusted to 3 with Dowex 50 (H+‐form) at 0°C, the resin was removed by filtration, the filtrate was neutralized to pH 7 by addition of a diluted solution of Et3N (pH 8) at 0°C, and the solution was concentrated to a volume of 3 ml and lyophilized to furnish 0.87 mg of ADP‐heptose (monotriethylammonium salt). Analytical data were in full accordance with those previously published 26.
Author contributions
CA, LAM, DG‐W, and A‐SD designed research. JC, LAM, and LT synthesized βHBP. AZ synthesized ADP‐heptose. DG‐W, A‐SD, and HR performed research and analyzed data. CA, LAM, JC and AZ wrote the manuscript. All authors discussed the results and commented on the article.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We gratefully acknowledge financial support from the Agence Nationale pour la Recherche (grants no ANR‐14‐ACHN‐0029‐01 and ANR‐17‐CE15‐0006 including postdoctoral fellowships to DGW and JC), from Fondation ARC pour la Recherche sur le Cancer (grant no ARC—PJA20171206187), and from Institut Pasteur including a postdoctoral fellowship to JC (grant GPH Vaccinology‐FlexBiVac). We thank Catherine Guerreiro (UCB) for her assistance with HPLC analysis and Frédéric Bonhomme (CNRS UMR3523) for assistance with HRMS measurements and NMR spectroscopy. We also thank K. Bailly from the Cochin Cytometry and Immunobiology Facility for her technical help.
EMBO Reports (2018) 19: e46943
References
- 1. Gaudet RG, Sintsova A, Buckwalter CM, Leung N, Cochrane A, Li J, Cox AD, Moffat J, Gray‐Owen SD (2015) INNATE IMMUNITY. Cytosolic detection of the bacterial metabolite HBP activates TIFA‐dependent innate immunity. Science 348: 1251–1255 [DOI] [PubMed] [Google Scholar]
- 2. Tettelin H, Saunders NJ, Heidelberg J, Jeffries AC, Nelson KE, Eisen JA, Ketchum KA, Hood DW, Peden JF, Dodson RJ et al (2000) Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 287: 1809–1815 [DOI] [PubMed] [Google Scholar]
- 3. Milivojevic M, Dangeard A‐S, Kasper CA, Tschon T, Emmenlauer M, Pique C, Schnupf P, Guignot J, Arrieumerlou C (2017) ALPK1 controls TIFA/TRAF6‐dependent innate immunity against heptose‐1,7‐bisphosphate of gram‐negative bacteria. PLoS Pathog 13: e1006224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Valvano MA, Marolda CL, Bittner M, Glaskin‐Clay M, Simon TL, Klena JD (2000) The rfaE gene from Escherichia coli encodes a bifunctional protein involved in biosynthesis of the lipopolysaccharide core precursor ADP‐L‐glycero‐D‐manno‐heptose. J Bacteriol 182: 488–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Taylor PL, Sugiman‐Marangos S, Zhang K, Valvano MA, Wright GD, Junop MS (2010) Structural and kinetic characterization of the LPS biosynthetic enzyme D‐alpha, beta‐D‐heptose‐1,7‐bisphosphate phosphatase (GmhB) from Escherichia coli . Biochemistry 49: 1033–1041 [DOI] [PubMed] [Google Scholar]
- 6. Kasper CA, Sorg I, Schmutz C, Tschon T, Wischnewski H, Kim ML, Arrieumerlou C (2010) Cell‐cell propagation of NF‐κB transcription factor and MAP kinase activation amplifies innate immunity against bacterial infection. Immunity 33: 804–816 [DOI] [PubMed] [Google Scholar]
- 7. Takatsuna H, Kato H, Gohda J, Akiyama T, Moriya A, Okamoto Y, Yamagata Y, Otsuka M, Umezawa K, Semba K et al (2003) Identification of TIFA as an adapter protein that links tumor necrosis factor receptor‐associated factor 6 (TRAF6) to interleukin‐1 (IL‐1) receptor‐associated kinase‐1 (IRAK‐1) in IL‐1 receptor signaling. J Biol Chem 278: 12144–12150 [DOI] [PubMed] [Google Scholar]
- 8. Li J, Lee GI, Van Doren SR, Walker JC (2000) The FHA domain mediates phosphoprotein interactions. J Cell Sci 113(Pt 23): 4143–4149 [DOI] [PubMed] [Google Scholar]
- 9. Huang C‐CF, Weng J‐H, Wei T‐YW, Wu P‐YG, Hsu P‐H, Chen Y‐H, Wang S‐C, Qin D, Hung C‐C, Chen S‐T et al (2012) Intermolecular binding between TIFA‐FHA and TIFA‐pT mediates tumor necrosis factor alpha stimulation and NF‐κB activation. Mol Cell Biol 32: 2664–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ea C‐K, Sun L, Inoue J‐I, Chen ZJ (2004) TIFA activates IkappaB kinase (IKK) by promoting oligomerization and ubiquitination of TRAF6. Proc Natl Acad Sci USA 101: 15318–15323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zimmermann S, Pfannkuch L, Al‐Zeer MA, Bartfeld S, Koch M, Liu J, Rechner C, Soerensen M, Sokolova O, Zamyatina A et al (2017) ALPK1‐ and TIFA‐dependent innate immune response triggered by the Helicobacter pylori type IV secretion system. Cell Rep 20: 2384–2395 [DOI] [PubMed] [Google Scholar]
- 12. Gall A, Gaudet RG, Gray‐Owen SD, Salama NR (2017) TIFA signaling in gastric epithelial cells initiates the cag type 4 secretion system‐dependent innate immune response to Helicobacter pylori infection. MBio 8: e01168‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stein SC, Faber E, Bats SH, Murillo T, Speidel Y, Coombs N, Josenhans C (2017) Helicobacter pylori modulates host cell responses by CagT4SS‐dependent translocation of an intermediate metabolite of LPS inner core heptose biosynthesis. PLoS Pathog 13: e1006514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liang L, Vincent SP (2017) Synthesis of d‐glycero‐d‐manno‐heptose 1,7‐bisphosphate (HBP) featuring a β‐stereoselective bis‐phosphorylation. Tetrahedron Lett 58: 3631–3633 [Google Scholar]
- 15. Sauvageau J, Bhasin M, Guo CX, Adekoya IA, Gray‐Owen SD, Oscarson S , Guazzelli L, Cox A (2017) Alternate synthesis to d‐glycero‐β‐d‐manno‐heptose 1,7‐biphosphate. Carbohydr Res 450: 38–43 [DOI] [PubMed] [Google Scholar]
- 16. Inuki S, Aiba T, Kawakami S, Akiyama T, Inoue J‐I, Fujimoto Y (2017) Chemical synthesis of d‐glycero‐d‐manno‐heptose 1,7‐bisphosphate and evaluation of its ability to modulate NF‐κB activation. Org Lett 19: 3079–3082 [DOI] [PubMed] [Google Scholar]
- 17. Borio A, Hofinger A, Kosma P, Zamyatina A (2017) Chemical synthesis of the innate immune modulator – bacterial d‐glycero‐β‐d‐manno‐heptose‐1,7‐bisphosphate (HBP). Tetrahedron Lett 58: 2826–2829 [Google Scholar]
- 18. Andaloussi M, Atamanyuk D, Gerusz V, Moreau F, Oxoby M, Tikad A, Vincent S (2012) New heptose derivatives and biological applications thereof. Patent WO2012073214A2
- 19. Crich D, Chandrasekera NS (2004) Mechanism of 4,6‐O‐benzylidene‐directed beta‐mannosylation as determined by alpha‐deuterium kinetic isotope effects. Angew Chem Int Ed Engl 43: 5386–5389 [DOI] [PubMed] [Google Scholar]
- 20. Brimacombe JS, Kabir AKM (1986) Convenient syntheses of l‐glycero‐d‐manno‐heptose and d‐glycero‐d‐manno‐heptose. Carbohydr Res 152: 329–334 [Google Scholar]
- 21. Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli . Nat Chem Biol 5: 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kneidinger B, Marolda C, Graninger M, Zamyatina A, McArthur F, Kosma P, Valvano MA, Messner P (2002) Biosynthesis pathway of ADP‐l‐glycero‐β‐d‐manno‐Heptose in Escherichia coli . J Bacteriol 184: 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakao R, Ramstedt M, Wai SN, Uhlin BE (2012) Enhanced biofilm formation by Escherichia coli LPS mutants defective in Hep biosynthesis. PLoS One 7: e51241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou P, She Y, Dong N, Li P, He H, Borio A, Wu Q, Lu S, Ding X, Cao Y et al (2018) Alpha‐kinase 1 is a cytosolic innate immune receptor for bacterial ADP‐heptose. Nature 561: 122–126 [DOI] [PubMed] [Google Scholar]
- 25. Datsenko KA, Wanner BL (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zamyatina A, Gronow S, Oertelt C, Puchberger M, Brade H, Kosma P (2000) Efficient chemical synthesis of the two anomers of ADP‐L‐glycero‐ and D‐glycero‐D‐manno‐heptopyranose allows the determination of the substrate specificities of bacterial heptosyltransferases. Angew Chem Int Ed Engl 39: 4150–4153 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File