

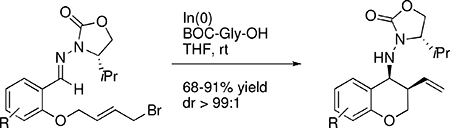
Abstract
The asymmetric intramolecular indium-mediated cyclization reaction delivers chromanes with excellent diastereoselectivity (68–91% yield, dr >99:1). The reaction was efficient for aryl substrates with both electron withdrawing or donating groups. Carboxylic acid additives were found to be necessary for optimal reaction.
Graphical Abstract
Chiral amine compounds display unique biological properties and this has inspired a great deal of effort in the development of asymmetric methods for their synthesis.i Furthermore, the increased need for enantiopure medicinal compounds and the rapid advancement for the field of asymmetric synthesis encourages the development of synthetic methods that utilize reagents and promoters of low relative toxicity.ii Indium has emerged as a metal of high potential in organic synthesis because of its unique properties. For example, indium metal is relatively unaffected by air or oxygen at ambient temperatures and can be handled safely without apparent toxicity. Allyl indium reagents are also tolerant of many functional groups and display low basicity and selective nucleophilicity, which permits excellent chemoselective transformations.iii Despite its extensive utility in intermolecular C-C bond formation, the corresponding intramolecular cyclizations are somewhat limited. Examples include the cyclization of enynes,iv cyclization of tethered propargyl bromides to carbonyl compounds,v Pd/In-mediated cyclization,vi atom-transfer cyclization,vii intramolecular allylindation of terminal alkynesviii and cyclization via hydrometallation of alkynes.ix As part of our continued effort to expand the synthetic utility of organoindium reagents, we disclose for the first time an efficient asymmetric indium-mediated intramolecular cyclization of chiral hydrazone derivatives and its application toward the synthesis of chromane antibiotics.
Hydrazones are relatively stable imine equivalents and are significantly less reactive than imines. Consequently, there are fewer reports of organometallic addition to hydrazones than to imines.x Due to their greater reactivity, imines are also prone to hydrolytic degradation and tend to be unstable during purification or prolonged storage. This is particularly problematic for aliphatic imines.xi This limits their utility as precursors for chiral amine compounds. Therefore, addition of carbon fragments to C=NX bonds (where X = stabilizing group) and related compounds are increasingly being used for the synthesis of chiral amines.xii Practical catalytic, enantioselective addition to C=N bonds are highly desirable.xiii Chiral auxiliaries offer a useful approach for chiral amine synthesis. The use of a stabilizing group allows a chiral auxiliary to be incorporated and cleaved without affecting the efficiency of the process.xiv,xv
We have reported several examples of highly efficient stereoselective intermolecular allylations of chiral hydrazones.xvi Considering the excellent success obtained in diastereoselective allylation we sought to develop an intramolecular variation of the reaction. Chromanes and related compounds have attracted interest due to their occurrence in many biological compounds. Vitamin E components,xvii cromakalim,xviii chromane antibiotics,xix to name a few, are examples of chromanes with desirable biological properties. We envisioned that an allylic bromide tethered hydrazone would provide the framework for entry into the core chromane structure.
Many indium-mediated allylation reactions are found to be optimal with at least 50% excess allyl halide (halide:In, 3:2). Thus the extension of this reaction to an intramolecular cyclization is not necessarily straightforward as it is inherently restricted to one equivalent of the allyl halide. Protic acid additives have been shown to improve such reactions.xx Thus, our initial studies scrutinized the influence of different Lewis acid and protic acid additives on the outcome of the cyclization. To investigate the intramolecular indium-mediated allylation reaction we first examined the cyclization of 1a which was easily prepared from the condensation of the corresponding aldehyde with chiral hydrazine followed by reaction with 1,4-dibromo-2-butene.xxi The results of our initial screen are summarized in Table 1. Treating 1a with two equivalents of indium metal in THF at room temperature was completely ineffective and the starting material was recovered intact (entry 1). Similarly, in the presence of protic acids HCl, H3PO4 and methane sulfonic acid no reaction ensued (entries 2–4). As we have previously shown,xvi indium(III) Lewis acids are effective in promoting the intermolecular allylation of hydrazones and the addition of In(OTf)3 and InBr3 was tested as well (entries 5 and 6). To our disappointment these reactions also failed affording only trace amounts of 2a. Furthermore, a mixture of at least three stereoisomers was produced with poor selectivity. However, when 6 equivalents of trifluoroacetic acid was added the reaction proceeded with good efficiency to produce 2a as a single isomer. The stereochemistry of the product was confirmed by the X-ray crystal structure of the N-trifluoracetate derivative 3 (see supporting information). The cis-stereochemistry was also observed in an analogous racemic cyclization.xx
Table 1.
Affect of additives on the cyclization of 1a.a
| entry | additive (equiv) | time (h) | yield (%)b | drc |
|---|---|---|---|---|
| 1 | none | 10 | 0 | — |
| 2 | aq HCl (6) | 10 | 0 | --- |
| 3 | H3PO4 (6) | 10 | 0 | --- |
| 4 | CH3SO3H (6) | 10 | 0 | --- |
| 5 | In(OTf) 3 (1.3) | 48 | <5 | --- |
| 6 | InBr3 (1.3) | 48 | <5 | --- |
| 7 | TFA (6) | 10 | 68 | >99:1 |
| 8 | BOC-L-Pro-OH (1.3) | 12 | 10 | >99:1 |
| 9 | BOC-L-Phe-OH (1.3) | 12 | 20 | >99:1 |
| 10 | BOC- Gly-OH (1.0) | 12 | 60 | >99:1 |
| 11 | BOC- Gly-OH (1.5) | 12 | 65 | >99:1 |
| 12 | BOC- Gly-OH (2) | 12 | 72 | >99:1 |
| 13 | BOC- Gly-OH (6) | 12 | 65 | >99:1 |
See experimental section for details.
Isolated yield.
Diastereomer ratio of the major isomer 2a shown vs. three other possible isomers. Determined by 1H NMR.
The observation that carboxylic acid had a pronounced effect on the cyclization of 1a led us to investigate other carboxylic acids as additives. As shown in Table 1, the addition of BOC-protected amino acids demonstrated a similar enhancement of reactivity and selectivity shown by TFA. The chiral amino acid derivatives of L-proline and L-phenylalanine provided complete control of the stereoselectivity however the yields were low (entries 8 and 9). No influence of the stereochemistry of the amino acid on the selectivity of the reaction was observed. We were pleased to discover that the simple glycine derivative was more effective and provided the highest observed yields (entires 10–13). Optimal reaction was obtained with two equivalents each of BOC-Gly-OH and indium metal affording a 72% isolated yield of 2a (entry 12). We have determined that both proton and carboxylate are required for best reaction as both simple protic acids (vide supra) as well as carboxylate salts (not shown) were ineffective. This may suggest that the carboxylate bridges between the hydrazone and the allylic indium species that would be formed upon reduction of the allyl bromide. However, this is only supposition and no further evidence has yet been obtained to conclude the role of acid. No reaction was observed in the absence of reducing In(0) metal.
Under the established optimal reaction conditions a variety of chiral hydrazones were efficiently converted into the corresponding cyclized products (Table 2). Substitution on the ring was well tolerated. Substrate 1d, having an electron- donating group para- to the allyl ether moiety and meta- to the hydrazone resulted in the highest yield (91%, entry 3). On the other hand, placing an electron-donating group meta- to the allyl ether moiety and para- to the hydrazone resulted in lower yield (68%, entry 4). In general, all reactions proceeded with good to excellent yield and complete stereocontrol to give the cis-products. Although the cyclization of aliphatic hydrazones was not attempted based on analogous intermolecular allylation reactions it should be feasible.xvi
Table 2.
Indium/carboxylic acid-mediated cyclization of chiral hydrazones.a
| entry | substrate | product | yield (%)b | ||
|---|---|---|---|---|---|
| 1 |  |
1b |  |
2b | 70 |
| 2 |  |
1c |  |
2c | 86 |
| 3 |  |
1d | 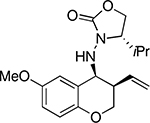 |
2d | 91 |
| 4 |  |
1e |  |
2e | 68 |
| 5 |  |
1f |  |
2f | 78 |
| 6 | 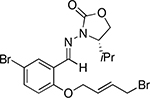 |
1g |  |
2g | 89 |
| 7 |  |
1h |  |
2h | 82 |
| 8 |  |
1i |  |
2i | 82 |
| 9 |  |
1j |  |
2j | 82 |
| 10 |  |
1k |  |
2k | 78 |
See experimental section for details.
Isolated yield.
The role of TFA in both promoting the reaction and controlling the stereochemical outcome is unknown but may involve multiple roles in templating and activating the transition state. Group 13 metal halides are well known to form bridged dimers which would likely be unable to cyclizae in the intramolecular allylation. The carboxyilic acid may help in breaking up aggregated organometallic intermediates. As illustrated in Figure 1, a chair transition state involving the allylic indium and imine moieties would afford the cis-aminochromane product. The acid may aid in activating the reaction through hydrogen bonding to the oxazolidinone carbonyl while simultaneously enhancing the nucleophilicity of the allylindium by donation from the acid carbonyl to the indium metal.
Figure 1.

Possible transition state for the carboxylic acid promoted In-mediated allylation.
To demonstrate the synthetic utility of this stereoselective intramolecular cyclization methodology, 2b was elaborated into a key intermediate (5, Scheme 1) for the synthesis of a chromane antibiotic (6) discovered by Pharmacia to have shown modest activity against S. aureus.xixa Reductive cleavage of the hydrazine was accomplished by acylating with trifluoroacetic anhydride followed by treatment with SmI2 to give 4 in good yield. Ozonolysis of the olefin and hydrolysis of the trifluoracetate provided amino alcohol 5 with good efficiency. Compound 5 was previously prepared by a nitrone cycladdition approach with poor stereoselectivity.xixa Spectroscopic data was identical to that reported in the literature.
Scheme 1.

Synthetic route to intermediate 5.
In conclusion, we have developed the first asymmetric indium-mediated intramolecular cyclization of hydrazones. This method proceeded with good chemical yield and excellent stereocontrol in the presence of simple carboxylic acids. The stereochemistry of the product was established by X-ray crystallography analysis. We have applied this method to the synthesis of the key intermediate of a chromane antibiotic via core structure 4. It should be pointed out that 4 possesses latent functionality in the olefin that could be utilized for further manipulation. Thus a variety of subsequent modifications could be envisioned to generate a diverse library of compounds with potential biological applications.
Experimental Section
Cyclization of hydrazone 1a to afford 2a.
To an oven-dried Schlenk flask under argon was added indium powder (133.2 mg, 1.16 mmol), hydrazone 1a (250 mg, 0.58 mmol) and dry THF (10.0 ml). BOC-Gly-OH (235.7 mg, 1.16 mmol) was added and the reaction mixture was stirred for 12 h at room temperature. The mixture was concentrated and purified by flash chromatography eluting initially with 20% ethyl acetate in hexanes and then 80% dichloromethane in hexanes. Compound 2a was obtained in 72% yield: 147.2 mg (0.42 mmol); mp 108–110 °C; [α]D20 = −97.6 (c = 0.3 in THF); IR (film): 1747, 2962 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 8.03 (d, J = 8.4 Hz, 1H), 7.72–7.68 (m, 2H), 7.49–7.45 (m, 1H), 7.31–7.27 (m, 1H), 7.05 (d, J = 8.8 Hz, 1H), 6.05–5.96 (m, 1H), 5.41–5.35 (m, 2H), 5.21 (d, J = 2.8 Hz, 1H), 4.70 (d, J = 10.0 Hz, 1H), 4.67 (d, J = 10.0 Hz, 1H), 4.31–4.27 (m, 1H), 3.60–3.57 (m, 1H), 3.11–3.07 (m, 1H), 2.93–2.87 (m, 1H), 2.15–2.11 (m, 1H), 1.87–1.81 (m, 1H), 0.65 (d, J = 6.8 Hz, 3H), 0.56 (d, J = 7.2, 3H); 13C NMR (100 MHz, CDCl3): δ = 158.5, 152.6, 134.0, 133.9, 130.0, 128.7, 128.5, 126.9, 123.4, 121.7, 119.0, 118.8, 113.0, 63.8, 63.2, 61.2, 49.1, 38.7, 27.8, 17.4, 15.8; HRMS Calc’d for C21H24N2O3 (M+Na)+: 375.1685; Found: 375.1684.
Preparation of 2,2,2-trifluoro-N-(2,3-dihydro-2-vinyl-1H-benzo[f]chromen-1-yl)acetamide (4).
To a solution of ent-3 (100 mg, 0.22 mmol) in MeOH under argon was added a solution of SmI2 (20 mL, 0.1 M in THF) and the reaction mixture was stirred for 45 min. The solution was opened to air and stirred for an additional 5 min. Concentration followed by flash chromatography (10:1 hexanes/ethyl acetate) gave 4 (63 mg, 90%) as a white solid; mp 166–168 °C; [α]D20 = −222.8 (c = 0.35 in THF); IR (film): 1693 cm−1; 1H NMR (400 MHz) δ = 7.77–7.72 (m, 2H), 7.65 (dd, J = 8, 0.8 Hz, 1H), 7.52–7.48 (m, 1H), 7.39–7.35 (m, 1H), 7.05 (d, J = 8 Hz, 1H), 6.33 (d, J = 4 Hz, 1H), 5.93–5.85 (m, 1H), 5.79 (dd, J = 8, 4 Hz, 1H), 5.30 (d, J = 4 Hz, 1H), 5.21 (d, J = 15 Hz, 1H), 4.41 (ddd, J = 15, 4, 1.6 Hz, 1H), 4.01 (t, J = 12 Hz, 1H), 3.10–3.04 (m, 1H); 13C NMR (100 MHz) δ = 156.9 (d, JCF = 40 Hz), 152.9, 132.7, 132.0, 131.0, 129.0, 128.5, 127.8, 120.1 (d, JCF = 144 Hz), 120.1, 110.8, 76.6, 64.0, 43.7, 40.4; HRMS Calc’d for C17H14F3NO2 (M+Na)+: 344.0874; Found: 344.0876.
Supplementary Material
Acknowledgement.
We are grateful to the National Science Foundation (NSF-CHM-0616485) and the NDSU NIH Center for Protease Research (NCRR-P20-RR15566) for financial support of this project.
Footnotes
Supporting Information Available: Experimental procedures, characterization data and 1H and 13C NMR spectra for all new compounds. Crystallographic data for 3a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- i (a).Friestad GK; Mathies AK Tetrahedron 2007, 63, 2541–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kobayashi S; Ishitani H Chem. Rev 1999, 99, 1069. [DOI] [PubMed] [Google Scholar]; (c) Bloch R Chem. Rev 1998, 98, 1407. [DOI] [PubMed] [Google Scholar]; (d) Enders D; Reinhold U Tetrahedron: Asymmetry 1997, 8, 1895. [Google Scholar]; (e) Denmark SE; Nicaise OJ-C Chem. Commun 1996, 999. [Google Scholar]
- ii (a).Li C-J Chem. Rev 1993, 93, 2023 [Google Scholar]; (b) Noyori R Green Chem., 2003, 5, G37 [Google Scholar]; (c) Lindstrom UM; Chem. Rev 2002, 102, 2751. [DOI] [PubMed] [Google Scholar]; (d) Yanada R; Obika S; Nishimori N; Yamauchi M; Takemoto Y Tetrahedron Lett. 2004, 45, 2331 [Google Scholar]; (e) Li C-J Acc. Chem. Res 2002, 35, 533. [DOI] [PubMed] [Google Scholar]
- iii (a).Alonso F; Beletskaya IP; Yus M Chem. Rev 2002, 102, 4009. [DOI] [PubMed] [Google Scholar]; (b) Ranu BC; Dutta J; Guchhait SK J. Org. Chem 2001, 66, 5624. [DOI] [PubMed] [Google Scholar]
- iv.Lee PH; Kim S; Lee K; Seomoon D; Kim H; Lee S; Kim M; Han M; Noh K; Livinghouse T Org. Lett 2004, 6, 4825. [DOI] [PubMed] [Google Scholar]
- v.Kang H-Y Kim Y-T; Yu Y-K; Cha JW; Cho YS; Koh HY Synlett 2004, 45. [Google Scholar]
- vi (a).Kang S-K; Lee S-W; Jung J; Lim Y J. Org. Chem 2002, 67, 4376. [DOI] [PubMed] [Google Scholar]; (b) Nguyen VC; Kim Y-T; Yu Y-K; Kang H-Y Bull. Korean Chem. Soc 2005, 26, 711. [Google Scholar]
- vii (a).Yanada R; Nishimori N; Matsumura A; Fujii N; Takemoto Y Tetrahedron Lett. 2002, 43, 4585 [Google Scholar]; (b) Cook GR; Hayashi R; Org. Lett 2006, 8, 1045. [DOI] [PubMed] [Google Scholar]
- viii (a).Nair V; Ros S; Jayan CN; Pillai BS Tetrahedron 2004, 60, 1959 [Google Scholar]; (b) Salter MM; Sardo-Inffiri S Synlett 2002, 2068. [Google Scholar]
- ix.Takami K; Yorimitsu H; Oshima K Org. Lett 2002, 4, 2993. [DOI] [PubMed] [Google Scholar]
- x (a).Kobayashi S; Hamada T; Manabe K; J. Am. Chem. Soc 2002, 124, 5640. [DOI] [PubMed] [Google Scholar]; (b) Friestad GK; Ding H Angew. Chem. Int. Ed 2001, 40, 4491. [DOI] [PubMed] [Google Scholar]; (c) Burk MJ; Feaster JE; J. Am. Chem. Soc 1992, 114, 6266 [Google Scholar]; (d) Kadota J; Park J-Y; Yamamoto Y Chem. Commun 1996, 841. [Google Scholar]
- xi.Jacobsen MF; Ionita L; Skrydstrup T J. Org. Chem 2004, 69, 4792. [DOI] [PubMed] [Google Scholar]
- xii (a).Friestad GK; Shen Y; Ruggles EL Angew. Chem. Int. Ed 2003, 42, 5061. [DOI] [PubMed] [Google Scholar]; (b) Friestad GK; Qin GK,J J. Am. Chem. Soc 2001, 123, 9922. [DOI] [PubMed] [Google Scholar]
- xiii (a).Saaby S; Fang X; Gathergood N; Jørgensen KA; Angew. Chem., Int. Ed 2000, 112, 4280. [DOI] [PubMed] [Google Scholar]; (b) Saaby S; Bayo P; Aburel PS; Jørgensen KA J. Org. Chem 2002, 67, 4352. [DOI] [PubMed] [Google Scholar]; (c) Johannsen M Chem. Commun 1999, 2233. [Google Scholar]
- xiv (a).Maryanoff BE; Zhang H,-C; Cohen JH; Turchi IJ; Maryanoff CA Chem. Rev 2004, 104, 1431. [DOI] [PubMed] [Google Scholar]; (b) Weinreb SM; Top. Curr. Chem 1997, 190, 131 [Google Scholar]; (c) Weinreb SM; Orr RK Synthesis 2005, 1205. [Google Scholar]
- xv (a).Friestad GK Eur. J. Org. Chem 2005, 3157 [Google Scholar]; (b) Enders D; Reinhold U Tetrahedron: Asymmetry 1997, 8, 1895 [Google Scholar]; (c) Davis FA; Zhou P; Chen B-C Chem. Soc. Rev 1998, 27, 13 [Google Scholar]; (d) Bloch R Chem.Rev 1998, 98, 1407. [DOI] [PubMed] [Google Scholar]; (e) Ellman JA; Owens TD; Tang TP; Acc. Chem. Res 2002, 35, 984. [DOI] [PubMed] [Google Scholar]; (f) Job A; Janeck CF; Bettray W; Peters R; Enders D Tetrahedron 2002, 58, 2253 [Google Scholar]; (g) Zhou P; Chen B; Davis FA; Tetrahedron 2004, 60, 8003 [Google Scholar]; (h) Friestad GK Eur. J. Org. Chem 2005, 3157 [Google Scholar]; (i) Ogawa C; Sugiura M; Kobayashi S; J. Org. Chem 2002, 67, 5359. [DOI] [PubMed] [Google Scholar]
- xvi.Cook GR; Maity BC; Kargbo R Org. Lett, 2004, 6, 1741. [DOI] [PubMed] [Google Scholar]
- xvii (a).Couladouros EA; Moutsos VI; Lampropoulou M; Little JL; Hyatt JA J. Org. Chem 2007, 72, 6735. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Shen HC, Surivet JP, Angew. Chem., Int. Ed 2003, 42, 3943. [DOI] [PubMed] [Google Scholar]; (c) Tietze LF; Sommer KM; Zinngrebe J; Stecker F Angew. Chem., Int. Ed 2004, 43, 2. [DOI] [PubMed] [Google Scholar]
- xviii.Rovnyak GC; Ahmed SZ; Ding CZ; Dzwonczyk S; Ferrara FN; Humphreys WG; Grover GJ; Santafianos D; Atwal KS; Baird AJ; McLaughlin LG; Normandin DE; Sleph PG; Traeger SC J. Med. Chem 1997, 40, 24. [DOI] [PubMed] [Google Scholar]
- xix (a).Zhao Q; Han F; Romero DL J. Org. Chem 2002, 67, 3317. [DOI] [PubMed] [Google Scholar]; (b) Broggini G; Folcio F; Sardone N; Sonzogni M; Zeechi G; Tetrahedron: Asymmetry 1996, 7, 797. [Google Scholar]
- xx.Kang H-Y; Yu Y-K Bull. Korean Chem. Soc 2004, 25, 1627. [Google Scholar]
- xxi.See supporting information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.