

Graphical abstract
The PD-1/PD-L1 interaction has emerged as a significant target in cancer immunotherapy. Current medications include monoclonal antibodies, which have shown impressive clinical results in the treatment of several types of tumors. The cocrystal structure of human PD-1 and PD-L1 is expected to be a valuable starting point for the design of novel inhibitors, along with the recent crystal structures with monoclonal antibodies, small molecules, and macrocycles.
Keywords: antitumor agents, protein-protein interactions, PD-1, PD-L1, protein structures
1. Introduction: The PD-1/PD-L1 Pathway
The development of cancer is monitored by the immune system. Most tumors are eliminated through the process of immune surveillance. In this process, T-cells playamajor role; their activation stimulates an immune response against cancer cells. T-cell activation requires two signals: a specific peptide epitope of the antigen must be presented on the major histocompatibility complex (MHC) of an antigen-presenting cell (APC) and it must form a complex with the T-cell receptor. A second signal resulting from the interaction of costimulatory molecules of activation is necessary. In the absence of co-stimulatory molecules, T-cells enter the unresponsive state of clonal anergy.[1] Tumors tend to evade immune surveillance by down-regulating both MHC and costimulatory molecules and also up-regulating co-inhibitory molecules.[2] Mechanistic hallmarks by which tumors avoid immune surveillance are called immune checkpoints or coinhibitory pathways, and recently, they have emerged as a promising target for cancer immunotherapy.
Programmed death-1/ PD-1 (or CD279) is an immune checkpoint receptor and belongs to the B7-CD28 family of receptors.[3] Upon binding to either of its two ligands, PD-L1 (known also as CD274 or B7-H1) and PD-L2 (known also as CD273, B7-DC or PDCD1LG2), a co-inhibitory signal is delivered.[4] PD-1 is a 55-kDa monomeric type I surface transmembrane glycoprotein. The protein is composed of an extracellular IgV domain, a transmembrane domain, and an intracellular cytoplasmic domain, which contains two tyrosine-based immunoreceptor signaling motifs; the inhibitory motif (ITIM) and the switch motif (ITSM).[5–7] Both motifs can be phosphorylated upon PD-1 engagement and in turn recruit Src homology region 2 domain containing phosphatase-1 (SHP-1) and SHP-2.[8]The 40-kDa PD-L1 and the 25-kDa PD-L2 are both type I transmembrane proteins, containing extracellular IgV and IgC domains and a transmembrane domain. They lack an identifiable intracellular signaling domain.[9] The two ligands share 37% identity with each other, but differ significantly in their affinity for PD-1 and their tissue specific expression.
2. Antibodies: Approved and in Development
Currently, there are antibodies targeting both PD-1 and antibodies targeting PD-L1 under clinical investigation either as a monotherapy or in combinations with other immune checkpoint inhibitors, monoclonal antibodies (mAbs), chemotherapy motherapy, vaccines, or radiation. The first monoclonal antibodies targeting PD-1 approved by FDA in 2014 were pembrolizumab and nivolumab; both for the treatment of advanced melanoma. An overview of FDA-approved mAbs in this field is provided in Table 1. The current focus in clinical trials is to improve efficacy and patient response by searching for drug combinations, and thus close to 1,000 clinical trials are ongoing just for checkpoint inhibitors targeting programmed cell death protein 1 (PD-1) and its ligand PD-L1[10]
Table 1:
FDA-approved PD-1/PD-L1-directed monoclonal antibodies (www.fda.gov, last update 23/9/2017).
| mAb | Indication |
|---|---|
| Pembrolizumab (Keytruda) | melanoma, head and neck squamous cell carcinoma (HNSCC), non-small-cell lung cancer (NSCLC), Hodgkin lymphoma, urothelial carcinoma, solid tumors, gastric or gastroesophageal junction adenocarcinoma |
| Pembrolizumab (Keytruda) with peme- trexed and carboplatin |
non squamous non-small-cell lung cancer (NSCLC) |
| Nivolumab (Opdivo) | melanoma, non-small-cell lung cancer, renal cell carcinoma (RCC), squamous cell carcinoma of the head and neck (SCCHN), Hodgkin lymploma, urothelial carcinoma, colorectal cancer |
| Nivolumab (Opdivo) with ipilimumab (Yervoy)* |
melanoma |
| Atezolizumab (Tecentriq) | urothelial carcinoma, non-small-cell lung cancer (NSCLC), bladder cancer |
| Avelumab (Bavencio) | metastatic Merkel cell carcinoma, urothelial carcinoma |
| Durvalumab (Imfinzi) | urothelial carcinoma |
Ipilimumab is an anti-CTL4 mAb approved by the FDA in 2011 for melanoma
3. Biomarkers for PD-1/PD-L1 Checkpoint Blockade Immunotherapy
Following the clinical success of immune checkpoint inhibitors, the establishment of biomarkers in immunotherapy has emerged as an imperative need. Although dramatic survival benefits, mostly for patients with melanoma and less in other types of cancers, have been observed, a rather small percentage of patients currently respond to PD-1/PD-L1-directed treatments. Therefore, biomarkers play a crucial role in predicting a patientQs response, understanding the mechanisms of action, and avoiding immune-related adverse effects (irAEs). Cancer biomarkers have been successfully established in cases of KRAS mutation, HER2 expression, and estrogen receptor expression just to name a few. Currently, PD-L1 is under investigation as a predictive biomarker of response to PD-1/PD-L1 immunotherapy.
In a recent study, the tumor expression of PD-L1 was shown to be significantly different in different types of cancer. Over-expression of PD-L1 is correlated with better response to PD-1/PD-L1 inhibition in melanoma, non-small-cell lung cancer (NSCLC), and renal cell carcinoma (RCC).[11] A meta-analysis, including data from 20 clinical trials for melanoma, lung cancer, and genitourinary cancers showed that in the overall sample, a significant correlation was observed between PD-L1 expression and overall response rate (ORR), which was significantly higher in PD-L1 positive patients treated with nivolumab or pembrolizumab.[12] Notably, however, clinical response has also been demonstrated in patients with PD-L1-negative tumors.[13]
Moreover, although the upregulation of PD-L1 in selected solid tumors can be detected by immunohistochemistry (IHC) on both tumor and immune cells, confusion arises regarding the significance of this detection. PD-L1 is not present simultaneously on tumor and immune cells in all types of cancer.[14] The fact that the expression of PD-L1 is inducible complicates the situation even further. Therefore, it is possible for PD-L1 to be expressed heterogeneously even within a tumor.[11] So far, the methods used to evaluate PD-L1 status differ significantly. Interestingly however, in October 2015, following the accelerated approval of pembrolizumab for metastatic NSCLC, the FDA approved PD-L1 IHC 22C3 pharmDx (Dako North America) as the only predictive companion diagnostic for selecting NSCLC patients for pembrolizumab. The approval was based on an analysis showing that patients with at least 50% of their tumor cells expressing PD-L1 were most likely to respond to treatment. To observe PD-L1 expression in a spatially and temporally resolved manner, techniques other than immunohistochemistry, for example, the modern imaging technique positron emission tomography (PET), could be beneficial.
Currently, the data concerning the potential establishment of PD-L1 as a single biomarker remain controversial. Alternative biomarker approaches, such as the quantification of tumor infiltrating lymphocytes (TILs), the identification of tumor neoantigens, and the mutational load of the tumor biomarkers seem to offer a better correlation with the clinical outcomes.[15]
4. Is There A Need For Small Molecules And Other Approaches Beyond mAbs?
There are several arguments why it is desirable to search for alternatives to mAbs in immunoncology. Generally, the production cost of mAbs remains extremely high. Moreover, they are not orally bioavailable and their high molecular weight leads to poor diffusion, especially in large tumors. High-affinity antibodies bind tightly to the antigen on first encounter, meaning that they remain on the periphery of the tumor, which is far from ideal for targeting solid tumors. Furthermore, the Fc portion of IgG antibodies can interact with various receptors on the surface of different cell types, which affects their retention in the circulation.[16] mAbs are immunogenic and can lead to irAEs with deadly outcomes, albeit in rare cases. In general, adverse effects with anti-PD-1/ anti-PD-L1 mAbs are less severe than anti-CTLA-4 mAbs. The common side effects of mAbs against PD-1/PD-L1 as monotherapy are fatigue, dermatological toxicities, diarrhea, colitis, endocrine and hepatic toxicities, pneumonitis, neurological syndromes, and ocular toxicity. The reported grade 3–4 adverse effects range from 7–12% in cases of monotherapy.[17] Rare cases of deaths have been reported with pembrolizumab[18] and nivolumab.[19,20] It must be noted that the combination approaches seem to lead to elevated toxicity. For instance, the combination of ipilimumab and nivolumab showed notably increased toxicity compared to monotherapy with these mAbs.[17,21] The very long half-lives of PD-1- and PD-L1-directed mAbs can make irAEs difficult to treat. Small and medium-sized molecules (such as macrocycles) could potentially overcome these issues. The significance of protein–protein interactions (PPIs) is well-established, and although targeting PPIs with small molecules can be challenging, there are successful examples of small-molecule modulators of PPIs.[22]
5. Crystal Structures of PD-1/PD-L1 and PD-1/PD-L2
In 2008, the first high-resolution crystal structure com-plexes regarding this PPI were published. The complex of murine PD-1 and human PD-L1 (PDB ID: 3BIK)[23] and that of murine PD-1 and murine PD-L2 (PDB ID: 3BP5)[24] established the structural foundations of the PD-1/PD-L1 and PD-1/PD-L2 interactions. However, these structures do not allow assessment of the extent of plasticity in these interactions when starting from the apo-protein components of the complexes. The crystal structure of the extracellular domain of human PD-1 alone was determined in 2011 (PDB ID: 3RRQ).
Despite the fact that murine PD-1 binds in vitro to both murine and human PD-L1, and human PD-1 binds to the PD-L1 of each species, it should be taken into account that the protein sequence identity between murine and human PD-1 is only 64% and that between murine and human PD-L1 is 77%. This indicates likely differences in the details of the binding modes. This hypothesis was recently confirmed,[25] when the crystal structure of the human PD-1/human PD-L1 complex was reported (PDB IDs: 4ZQK, 5C3T), which indeed documents significant differences in the binding between murine and human PD-1 and the ligand (hPD-L1). This information also allowed the identification of features of three hotspot pockets in human PD-1/PD-L1 that are required for inhibition of this interaction.
PD-1 assumes a b-sandwich immunoglobulin-variable (IgV)-type topology, with Cys54 and Cys123 forming a characteristic disulfide bridge; however PD-1 lacks the second disulfide common to other family members (CD28, CTLA-4, and ICOS).
Similarly to PD-1, the interacting N-terminal domain of PD-L1 is characterized by the IgV-type topology. PD-1 and PD-L1 form a 1:1 complex within the crystal, in contrast to CTLA-4 complexes with its ligands, where both interacting partners form homodimers. The interaction of PD-1 and PD-L1 resembles that of IgV domains within antibodies and T-cell receptors, being mediated by the strands from the front faces of the interacting domains (GFCC0 b sheets).
In principle, the 2.45 & resolution of the reported crystal structure[25] provides a perfect starting point for the rational structure-based drug design (SBDD) of molecules against this PPI. However, the interface between the two proteins is rather large (ca. 1.700 Å2), hydrophobic, and flat, without deep binding pockets, which makes it a difficult target for small molecules. Moreover, the hydrophobic interface also increases the chances of discovering false positive hits considerably. Nonetheless, small-molecule interrupters of the PD-1/PD-L1 protein–protein interaction have been described recently (see below).
6. Cocrystal Structures with Monoclonal Antibodies
Recently, cocrystal structures of monoclonal antibodies targeting PD-1 or PD-L1 were reported, which shed light on their molecular interactions.
For pembrolizumab, an IgG4 antibody, a crystal structure with the full-length antibody was reported (PDB ID: 5DK3).[26] The complex of the pembrolizumab antigen-binding fragment (Fab) with hPD-1 (PDB ID: 5JXE)[27] revealed that the stoichiometry is 1:1. Furthermore, the structural superposition of this complex with hPD-1/hPD-L1 shows overlapping surface regions, thus indicating that the antibody can antagonize hPD-L1 by competing for binding to hPD-1. One more crystal structure of pembrolizumab with hPD-1 (PDB ID: 5B8C)[28] was obtained with higher resolution. It is in good agreement with the previous one and provides additional data regarding the interfacial water molecules at the binding interface, which have an impact on both the affinity and specificity of the interaction.
Moreover, a comparison of the crystal structure of the PD-1/nivolumab Fab complex (PDB ID: 5GGR) with that of PD-1/pembrolizumab (PDB ID: 5GGS)[29] indicated that the epitopes of both antibodies directly occupy part of the PD-L1 binding site and can thus outcompete PD-L1 for binding to PD-1.
Avelumab, an IgG1 antibody, utilizes both heavy (VH) and light chain (VL) to bind to the IgV domain of the PD-L1 in its complex with hPD-L1 (PDB ID: 5GRJ)[30] . The contribution of the light chain is greater than that of the heavy chain. Moreover, the binding epitope region of avelumab on hPD-L1 overlaps with the hPD-1 binding region, thus indicating that the partially overlapping pattern results in the blocking mechanism.
A crystal structure was also recently disclosed for the anti-PD-L1 mAB durvalumab (PDB ID: 5XJ4). In this case, both heavy and light chains contribute to the binding, resulting in steric clash that deters PD-L1 from binding to PD-1.[31]
A crystal structure of PD-L1 with BMS-936559 Fab, a fully human IgG4 antibody currently in clinical trials, showed that its epitope occupies a large part of the PD-1 binding site (PDB ID: 5GGT).[29]
Very recently, a co-crystal structure of atezolizumab, the first anti-PD-L1 approved by FDA, was solved (PDB ID: 5XXY). In this case, the binding involves extensive hydrogen bonding, hydrophobic interactions, and π-π stacking or cation-π interactions. Moreover, mutagenenesis studies revealed two hotspot residues of PD-L1 (E58, R113). Overall, atezolizumab competes with PD-1 for binding to the same surface site of PD-L1 (Figure 1).[32]
Figure 1.
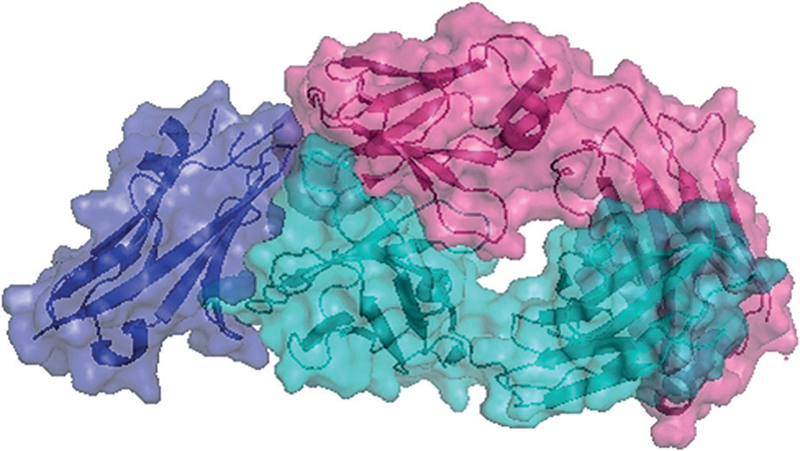
Complex of PD-L1 (purple) with atezolizumab Fab (cyan heavy chain, pink light chain; PDB ID: 5XXY).
In addition, in 2016 a cocrystal structure of an ultra-high-affinity engineered PD-1 mutant (HAC) with hPD-L1 was reported (PDB ID: 5IUS). This complex has a high degree of similarity with the hPD-1/hPD-L1. The main differences are observed in the β4-β5 loop. The high-affinity binding is driven by enthalpic gains, owning to the extensive polar contact network between the mutant and PD-L1.[33] In 2017, a second high-affinity mutant PD-1 was described that bears a single amino acid substitution (A132L). This leads to an increase in van der Waals interactions.[34]
Furthermore, a crystal structure of a PD-L1 nanobody (single domain antibody) was published (PDB ID: 5JDS). The nanobody KN035 competes with PD-1 for binding to PD-L1 mainly through a single surface loop of 21 amino acids.[35]
In general, the binding mode seems to differ between PD-1 and PD-L1 mAbs (Figure 2). A more thorough analysis of the structural biology of PD-1/PD-L1 was recently per-formed.[36]
Figure 2.
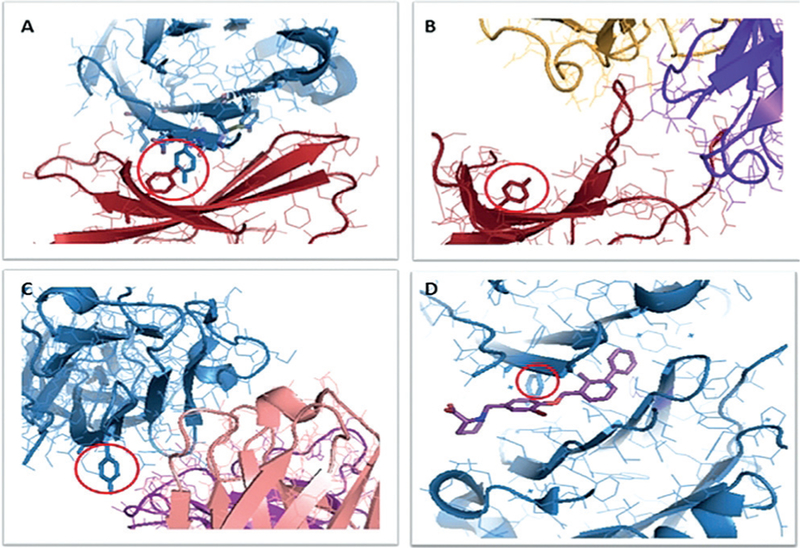
Different interaction modes of PD(L)-1. A) Complex of hPD-1 (red) with hPD-L1(blue; PDB ID: 4ZQK). The amino acids in the hotspots are shown as sticks models. The two residues in the red circle are Tyr68 of PD-1 (red) and Tyr123 of PD-L1 (blue). B) Complex of PD-1 (red) with nivolumab Fab (yellow light chain, purple heavy chain); PDB ID: 5GGR). C) Complex of PD-L1 (blue) with avelumab Fab (purple light chain, pink heavy chain; PDB 5GRJ). D) Complex (homodimer) of PD-L1 (blue) and BMS-08 (stick model, purple; PDB ID: 5J8O).
7. Cocrystal Structures with Small Molecules
Bristol-Myers Squibb (BMS) researchers have recently disclosed small molecules that bind to PD-L1 (Scheme 1).[37] The scaffold consists of a tri-aromatic structure, including a mono-ortho-substituted biphenyl substructure. Another phenyl ring is connected to the biphenyl and also contains a methylene amine moiety. The claimed biological activity of the reported compounds was established by a homogenous time-resolved fluorescence (HTRF) binding assay in which europium cryptate labeled antiIg was used. Typical examples are BMS-8, BMS-37, BMS-200, and BMS-202. No further in vitro or in vivo assays have been described to support the biological activity of compounds based on this scaffold.
Scheme 1.
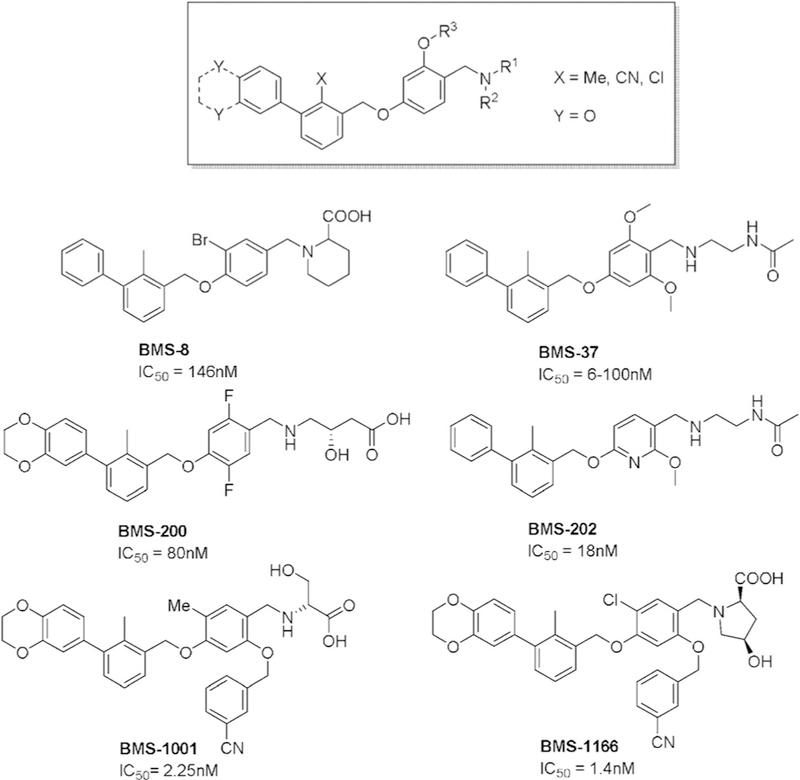
PD-1/PD-L1 inhibitors synthesized by BMS. IC50 values were established by HTRF binding assay.
The true nature of compounds BMS-202 and BMS-8 as PD-1/PD-L1 antagonists was recently rigorously demonstrat-ed by co-crystal structures with PD-L1 (PDB IDs: 5J89 and 5J8O, respectively).[38] The obtained crystals diffracted at 2.2 & resolution. Four protein molecules found in the asymmetric unit were organized into two dimers with one inhibitor molecule located at the interface of each dimer. The inhibitor inserts deep into a cylindrical, hydrophobic pocket created at the interface of two monomers within the dimer. The pocket is open to the solvent on one side of the dimer and restricted by the side chain of ATyr56 on the opposite side. Overall, the inhibitor–protein interaction is best described as bimodal, being spatially divided into hydrophobic and electrostatic parts according to the bimodal inhibitor design.
Furthermore, two novel crystal structures of BMS-37 and BMS-200 have been disclosed.[39] The crystals diffracted at 2.35 and 1.7 Å respectively (PDB IDs: 5N2D, 5N2F). NMR experiments indicated that both compounds bind to PD-L1 and induce its oligomerization in solution. Interestingly, the crystal structures revealed notable differences (Figure 3). The binding mode of compound BMS-37 follows the one already observed for BMS-8 and BMS-202. All of these are examples of the (2-methyl-3-biphenylyl)methanol scaffold. However, BMS-200, an example of a [3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methylphenyl]methanol scaffold, induced a conforma-tional change in ATyr56. The 2,3-dihydro-1,4-benzodioxinyl group forces the ATyr56 to take a different position, thus turning the previously observed deep hydrophobic cleft to a deep hydrophobic tunnel and making part of the compound accessible to solvent. Two novel crystal structures were reported for the optimized derivatives BMS-1001 (PDB ID: 5NIU) and BMS-1166 (PDB ID: 5NIX).[40] These derivatives in particular showed reduced unspecific cytotoxicity against tested cell lines. Furthermore, it was shown that both BMS-1001 and BMS-1166 have the potential to restore the activation of effector Jurkat T-cells, although less effectively than the monoclonal antibodies. More specifically, the immunomodulatory effects of BMS-1001 and BMS-1166 as EC50 values were 253 nm and 273 nm respectively, whereas for mAbs, the values were in the range 0.333–115 nm. Nevertheless, these data highlight the great potential of small molecules in this field.
Figure 3.
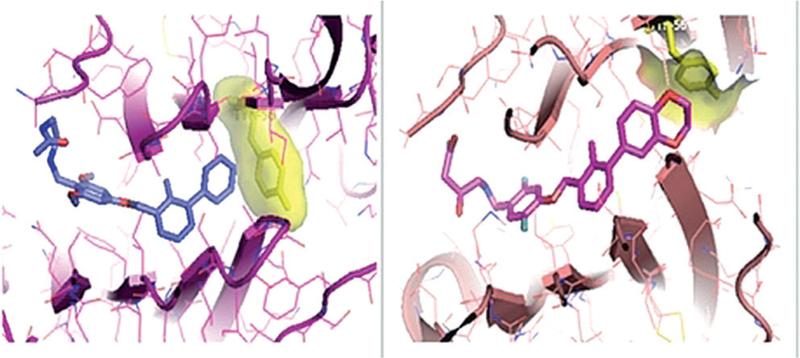
Binding mode of BMS-37 (left) and BMS-200 (right) to PD-L1. Yellow stick models show ATyr56.
Other small molecules have been claimed to antagonize the PD-1/PD-L1 protein–protein interaction, however their mode-of-action has not been rigorously proven so far. An overview of claimed PD-1/PD-L1 inhibitors from patents is provided here.[41]
8. Macrocycles
Several patents belonging to Bristol-Myers Squibb Com-pany claim macrocycles that show high affinity to PD-L1 at low concentrations.[42]
The majority of the described macrocycles contain either 14 or 13 amino acid residues (Scheme 2). In most of them, a sulfur atom is present and this is used as the starting point for the numbering of the amino acids. In another patent, this sulfur is replaced, either with oxygen or carbon.
Scheme 2.
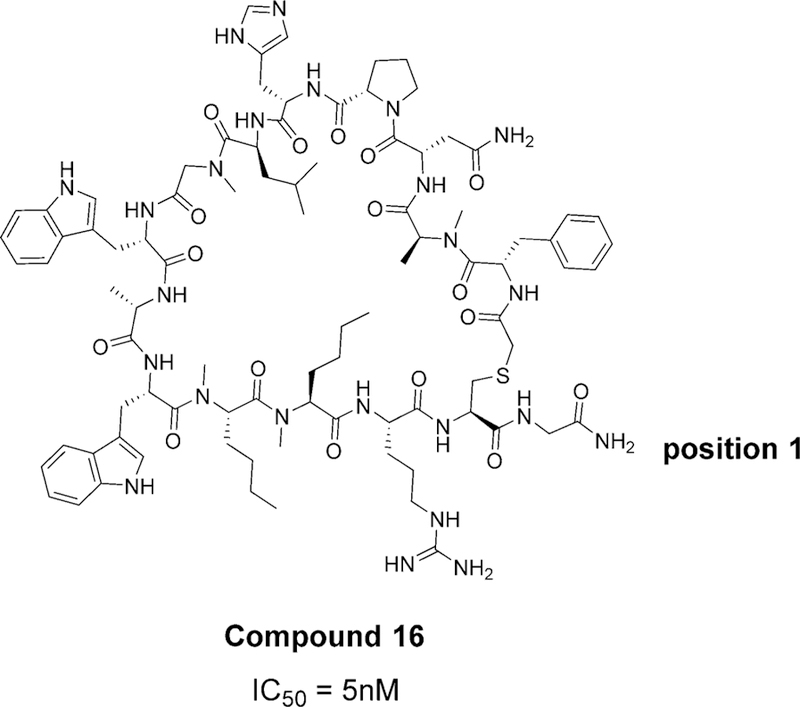
Example of a macrocycle with 14 amino acids (Bristol-Myers Squibb Company WO2014151634 A1 compound 16). The IC50 value was determined by HTRF assay. The numbering of amino acids starts from the position adjacent to the sulfur and continues clock-wise.
A comparison of the different structures with the 14-motif reveals that in most cases, the first amino acid is an unaltered neutral amide or bis-amide. Possible alterations include the addition of extra aromatic or aliphatic rings on the amide moiety to make this residue more hydrophobic. The second amino acid is frequently changed and varies from a hydrophobic isoleucine to polar amino acids, including aspartic acid, arginine, lysine, serine, or threonine. Amino acids 3 and 4 are mostly constant as hydrophobic moieties with butane chains. Moreover, the backbone nitrogen atoms in positions 3 and 4 are in almost all cases methylated. In position 5, a tryptophan is usually present or if altered it is towards a benzothiophene, a dihydropyrrole ring, or an indole ring bearing a carboxylic acid substitution. Morpholine or thiomorpholine also appear, but less frequently. A highly variable position among the patents is amino acid 6, which varies from polar (serine, lysine, tyrosine, aspartic acid, glutamic acid, glutamine) to hydrophobic (alanine, glycine). Position 7 is also highly constant as a tryptophan residue, whereas position 8 is almost always a proline or a hydroxylated proline. Isoleucine is usually found in position 9, but it could also vary towards polar residues (aspartic acid, glutamic acid, lysine, serine, asparagine, glutamine). Amino acid 10 also varies and usually it is a polar or basic residue (histidine, lysine, morpholine, hydroxypyrrole, serine, asparagine, glutamine). The next two amino acids are highly constant, with a proline in position 11 and an asparagine in position 12 in almost all cases. This is followed by a hydrophobic residue in position 13, usually an alanine or a proline is present. The final position 14 is always aromatic and the most common feature is tyrosine. In some cases, there are also halogens or methoxy substituents on the phenyl ring, but this seems to be less common than the tyrosine.
Regarding macrocycles with 13 amino acids, a sulfur bond is always included, as well as the two proline residues in positions 5 and 10. Most likely the latter are responsible for making beta turns in the macrocycles. The main difference from the 14-motif is that there are 5 phenyl rings present (positions 3, 4, 6, 7, 12, and 13) and not three (positions 5, 7, and 14). This feature makes these macrocycles more hydrophobic. Moreover, the tyrosine, which is the most common amino acid in the last position of the 14th motif, is always replaced with a phenyl ring with fluoro substituents in the 13-motif.
The BMS macrocyclic peptides that disrupt the PD-1/PD- L1 interaction were originally studied in HTRF assay.[43] Further studies were performed recently for these macro-cycles, including NMR, DSF, crystallography, and a cell assay, in order to determine their ability to restore T-cell function.[44] The analysis included peptide-57 (15-mer), peptide-71 (14-mer) and peptide-99 (13-mer). Peptide-57, peptide-71, and peptide-99 showed immumodulatory effects with EC50 values of 566 nm, 293 nm, and 6.30 mm, respectively in the cell assay, whereas for durvalumab and nivolumab, the values were 0.199 nm and 1.27 nm, respectively. Crystal structures were obtained for peptide-57 (PDB ID: 5O4Y) and peptide-71 (PDB ID: 5O45) in a peptide/PD-L1 ratio 1:1 (Scheme 3, Figure 4). The interaction is described as “face-on binding”. In both cases, there is a partial overlap with the PD-1 binding epitope and the binding is dominated by hydrophobic interactions and to a smaller extent polar interactions. Closer inspection of the interactions reveals significant differences between the peptides. For peptide-57, two significant pockets are occupied by bulky indole side chains, whereas for peptide-71 only one hydrophobic pocket is occupied by the side chain of phenylalanine. The polar interactions vary significantly between the two peptides, but in any case, the binding seems to be driven mainly by hydrophobic interactions. These novel crystal structures allow comparison of the binding mode with that of monoclonal antibodies and provide valuable structural information for drug design.
Scheme 3.
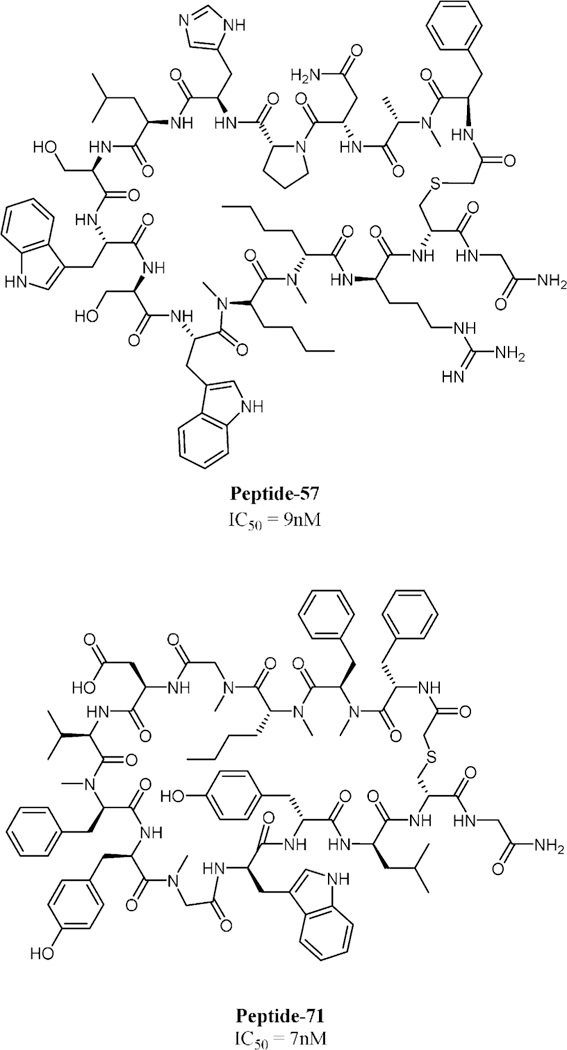
2D structures of peptide-57 (15-mer, up) and peptide-71 (14-mer, down). IC50 values were determined by HTRF assay.
Figure 4.
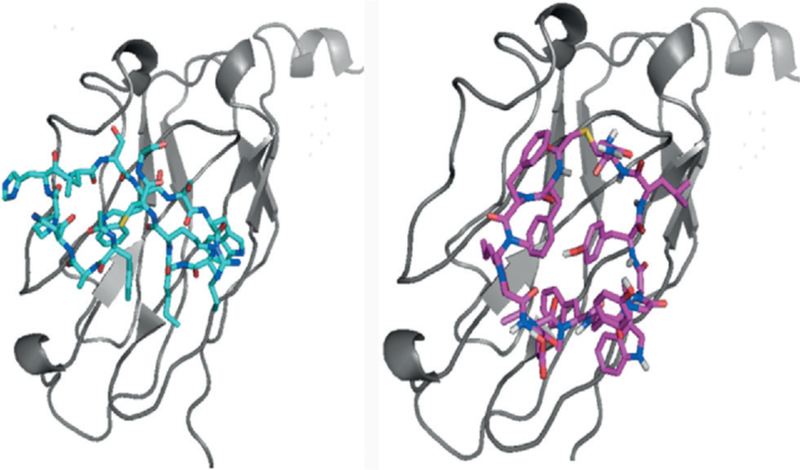
Binding of peptide-57 (15-mer, left) and peptide-71 (14-mer, right) to PD-L1.
9. Summary and Outlook
Immune checkpoint inhibitors represent an exciting new field in cancer treatment. Following the FDA approval of monoclonal antibodies targeting the PD-1/PD-L1 axis, a plethora of crystal structures was published, revealing the binding modes of antibodies, small molecules, and very recently, macrocycles. The crystal structures revealed significant differences, especially for small molecules that induced the dimerization of PD-L1. All these data taken together show significant hotspots and provide the missing pieces of structural information necessary for the rational design of small-molecule inhibitors, macrocycles, or middle-sized cyclic peptides that may have specific advantages compared to the already approved monoclonal antibodies. Importantly, some small molecules and macrocycles show activity comparable to approved mAbs in more complex cell based assays. Thus future developments in the area could result in drugs different from mAbs for specific cancer applications or different indications.
Acknowledgements
Our research is supported by grants from the National Institute of Health (NIH; 2R01GM097082–05), the European Lead Factory (IMI) under grant agreement number 115489, the Qatar National Research Foundation (NPRP6–065-3–012) to A.D.; and by Grants UMO-2012/06/A/ST5/00224 and UMO-2014/12/W/NZ1/00457 to T.A.H. from the National Science Centre, Poland. Moreover funding was received through ITN “Accelerated Early stage drug dIScovery” (AEGIS, grant agreement No. 675555), COFUND ALERT (grant agreement No. 665250), and KWF Kankerbestrijding grant (grant agreement No. 10504).
Biography

Markella Konstantinidou obtained her De-gree in Pharmacy (2012) and her M.Sc. in Medicinal Chemistry (2014) from the Aris-totle University of Thessaloniki under the supervision of Prof. D. Hadjipavlou-Litina. As a research assistant in the National Hellenic Research Foundation in Athens, she focused on the synthesis of dendrimers under the supevision of Dr. G. Heropoulos and Dr. B. Steele. In 2016, she joined the Drug Design Group in the University of Groningen as a PhD candidate under the guidance of Prof. A. Dçmling.

Tryfon Zarganes-Tzitzikas obtained his B.Sc. in Chemistry from the Aristotle University of Thessaloniki in 2010 and an M.Sc. degree under the guidance of Prof. J. Stephanidou-Stephanatou and Prof. K. Tsoleridis from the Aristotle University of Thessaloniki. In 2017 he obtained his Ph.D. in medicinal chemistry under the guidance of Prof. Alexander Dçmling in the Group of Drug Design at the University of Groningen, The Netherlands. Since 2014, he has been the co-founder and CSO of the drug synthesis company TelesisPharma B.V. in Groningen.

Katarzyna Magiera-Mularz received her Ph.D. degree in biochemistry from the Jagiellonian University, Krakow, Poland in 2016 under the supervision of Prof. T. Holak. Her PhD work focused on small-molecule inhibitors of the deubiquitinating protease USP2. As a postdoctoral fellow in the group of Prof. T. Holak, she continues her research on the proteins involved in tumorigenesis.

Prof. Tad A. Holak is the group leader of the Biological NMR Structure Group at the Max Planck Institute for Biochemistry, Martinsried (Germany). Since 2012, he has also been the head of the Chemical Biology and Drug Discovery Group at the Jagiellonian University (Krakow, Poland). His research focuses on discovering three-dimensional structures and structure-function properties of proteins using combination of multinuclear NMR spectroscopy, biochemistry and X-ray crystallography.

Prof. Alexander Dçmling has held the chair for Drug Design at the University of Groningen since 2011. He studied chemistry and biology at the Technische Universit t M4n-chen and obtained his Ph.D. under the guidance of Ivar Ugi. After a postdoctoral position under a Humboldt Fellowship in the group of Barry Sharpless, he founded the biotechnology company Morphochem and later Carmolex Inc. After his habilitation, he worked as a professor at the University of Pittsburgh in the School of Pharmacy. His interests are centered around multicomponent reaction (MCR) chemistry and its application to problems in drug design.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Markella Konstantinidou, Department of Drug Design, University of Groningen, A. Deusinglaan 1, 9713 AV, Groningen (The Netherlands).
Tryfon Zarganes-Tzitzikas, Department of Drug Design, University of Groningen, A. Deusinglaan 1, 9713 AV, Groningen (The Netherlands).
Katarzyna Magiera-Mularz, Jagiellonian University Ingardena 3, 30-060 Krakow (Poland).
Tad A. Holak, Jagiellonian University Ingardena 3, 30-060 Krakow (Poland), Max Planck Institute for Biochemistry, Am Klopferspitz 18a, 82152 Martinsried (Germany).
Alexander Dömling, Department of Drug Design, University of Groningen, A. Deusinglaan 1, 9713 AV, Groningen (The Netherlands).
References
- [1].Schwartz RH, Science 1990, 248, 1349–1356. [DOI] [PubMed] [Google Scholar]
- [2].Dçmling A, Holak TA, Angew. Chem. Int. Ed 2014, 53, 2286–2288; Angew. Chem. 2014, 126, 2318 – 2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen L, Nat. Rev. Immunol 2004, 4, 336–347. [DOI] [PubMed] [Google Scholar]
- [4].Keir ME, Butte MJ, Freeman GJ, Sharpe AH, Annu. Rev. Immunol 2008, 26, 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xia Y, Medeiros LJ, Young KH, Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ, Immunity 2007, 27, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Viricel C, Ahmed M, Barakat K, Mol J Graphics Modell 2015, 57, 131–142. [DOI] [PubMed] [Google Scholar]
- [8].Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL, Immunol 2004, 173, 945–954. [DOI] [PubMed] [Google Scholar]
- [9].Dong H, Zhu G, Tamada K, Chen L, Nat. Med 1999, 5, 1365–1369. [DOI] [PubMed] [Google Scholar]
- [10].Cavnar S, Valencia P, Brock J, Wallenstein J, Nat. Rev. Drug Discovery 2017, 16, 83–84. [DOI] [PubMed] [Google Scholar]
- [11].Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen, Pardoll DM, Topalian SL, Andres RA, Clin. Cancer Res 2014, 20, 5064–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Carbognin L, Pilotto S, Milella M, Vaccaro V, Brunelli M, Cali A*, Cuppone F, Sperduti I, Giannarelli D, Chilosi M, Bronte V, Scarpa A, Bria E, Tortora G, PLoS One 2015, 10, e0130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebb8 C, Charles J, Mihalcioiu C, Chiarion-Sileni V, Mauch C, Cognetti F, Arance A, Schmidt H, Schadendorf D, Gogas H, Lundgren-Eriksson L, Horak C, Sharkey B, Waxman IM, Atkinson V,Ascierto A, N. Engl. J. Med 2015, 372, 320–330. [DOI] [PubMed] [Google Scholar]
- [14].Meng X, Huang Z, Teng F, Xing L, Yu J, Cancer Treat. Rev 2015, 41, 868–876. [DOI] [PubMed] [Google Scholar]
- [15].Ma W, Gilligan BM, Yuan J, Li T, J. Hematol. Oncol 2016, 9, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chames P, Van Regenmortel M, Weiss E, Baty D, Br. J. Pharmacol 2009, 157, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, Postow MA, Wolchok JD, Ann. Oncol 2015, 26, 2375–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dermirtas S, El Aridi L, Acquitter M, Fleuret C, Plantin P, Ann. Dermatol. Venereol 2017, 144, 65–66. [DOI] [PubMed] [Google Scholar]
- [19].Jacob A, Unnikrishnan DC, Mathew A, Thyagarajan B, Patel S, Cancer Res J Clin. Oncol 2016, 142, 1869–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ,Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH,Anders A, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M, N. Engl. J. Med 2012, 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Davies M, Duffield EA, ImmunoTargets Ther 2017, 6, 51–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jin L, Wang W, Fang G, Annu. Rev. Pharmacol. Toxicol 2014, 54, 435–456. [DOI] [PubMed] [Google Scholar]
- [23].Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, Okazaki T, Honjo T, Minato N, Garboczi DN, Proc. Natl. Acad. Sci. USA 2008, 105, 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].LQzQr-MolnQr E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC, Proc. Natl. Acad. Sci. USA 2008, 105, 10483–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dçmling A, Dubin G, Holak TA, Structure 2015, 23, 2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, Kashi RS, Stickland C, Nat. Struct. Mol. Biol 2015, 22, 953–958. [DOI] [PubMed] [Google Scholar]
- [27].Na Z, Yeo SP, Bharath SR, Bowler MW, BalıkÅı E, Wang CI, Song H, Cell Res 2017, 27, 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Horita S, Nomura Y, Sato Y, Shimamura T, Iwata S, Nomura N, Sci. Rep 2016, 6, 35297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH, Lim H, Heo W, Park KY, Lee YJ, Ryu SE, Son JY, Lee JU, Heo S, Nat. Commun 2016, 7, 13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu K, Tan S, Chai Y, Chen D, Song H, Zhang CW, Shi Y, Liu J, Tan W, Lyu J, Gao S, Yan J, Qi J, Gao GF, Cell Res 2017, 27, 151–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tan S, Liu K, Chai Y, Zhang CW-H, Gao S, Gao GF, Qi J, Protein Cell 2017, DOI: https://10.1007/s13238-017-0412-8. [DOI] [PMC free article] [PubMed]
- [32].Zhang F, Qi X, Wang X, Wei D, Wu J, Feng L, Cai H, Wang Y, Zeng N, Xu T, Zhou A, Zheng Y, Oncotarget 2017, 8, 90215–90224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pascolutti R, Sun X, Kao J, Maute RL, Ring AM, Bowman GR, Kruse AC, Structure 2016, 24, 1719–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].LQzQr-MolnQr E, Scandiuzzi L, Basu I, Quinn T, Sylvestre E,Palmieri E, Ramagopal UA, Nathenson SG, Guha C, Almo SC, EBioMedicine 2017, 17, 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang F, Wei H, Wang X, Bai Y, Wang P, Wu J, Jiang X, Wang Y, Cai H, Xu T, Zhou A, Cell Discovery 2017, 3, 17004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zak KM, Grudnik P, Magiera K, Dçmling A, Dubin G, Holak TA, Structure 2017, 25, 1163–1174. [DOI] [PubMed] [Google Scholar]
- [37].a) Chupak LS, (Bristol-Myers Squibb Company), WO2015034820 A1, 2015;
- b) Chupak LS, (Bristol-Myers Squibb Company), WO2015160641, 2015.
- [38].Zak KM, Grudnik P, Guzik K, Zieba BJ, Musielak B, Dçmling A, Dubin G, Holak TA, Oncotarget 2016, 7, 30323–30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Guzik K, Zak KM, Grudnik P, Magiera K, Musielak B, Tçrner R, Skalniak L, Dçmling A, Dubin G, Holak TA, J. Med. Chem 2017, 60, 5857–5867. [DOI] [PubMed] [Google Scholar]
- [40].Skalniak L, Zak KM, Guzik K, Magiera K, Musielak B, Pachota M, Szelazek B, Kocik J, Grudnik P, Tomala M, Krzanik S, Pyrc K, Dçmling A, Dubin G, Holak TA, Onco-target 2017, 8, 72167–72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zarganes-Tzitzikas T, Konstantinidou M, Gao Y, Krzemien D, Zak K, Dubin G, Holak TA, Dçmling A, Expert Opin. Ther. Pat 2016, 26, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].a) Miller MM, (Bristol-Myers Squibb Company), WO2014151634, 2014;
- b) Miller MM, (Bristol-Myers Squibb Company), WO2016039749, 2016;
- c) Sun L-Q, (Bristol-Myers Squibb Company), WO2016057624 A1, 2016;
- d) Gillman KW, (Bristol-Myers Squibb Company), WO2016077518 A1, 2016;
- e) Mapelli C (Bristol-Myers Squibb Company), WO2016100285 A1, 2016;
- f) Sun L-Q, (Bristol-Myers Squibb Company), WO2016100608 A1, 2016;
- g) Miller MM, (Bristol-Myers Squibb Company), WO2016126646 A1, 2016;
- h) Boy KM, (Bristol-Myers Squibb Company), WO2016149351 A1, 2016.
- [43].Miller MM, (Bristol-Myers Squibb Company), US 20140294898 A1, 2014.
- [44].Magiera-Mularz K, Skalniak L, Zak KM, Musielak B, Rudzin´ska-Szostak E, Berlicki Ł, Kocik J, Grudnik P, Sala D, Zarganis-Tzitzikas T, Shaabani S, Dçmling A, Dubin G, Holak TA, Angew. Chem. Int. Ed 2017, 56, 13732–13735; Angew. Chem. 2017, 129, 13920 – 13923. [DOI] [PMC free article] [PubMed] [Google Scholar]